

Abstract
Parathyroid hormone-related protein (PTHrP) increases the growth and osteolytic potential of prostate cancer cells, making it important to control PTHrP expression. PTHrP expression is suppressed by 1,25-dihydroxyvitamin D3 (1,25D). The aim of this study was to identify the pathways via which 1,25D exerts these effects. Our main findings are that 1,25D regulates PTHrP levels via multiple pathways in PC-3 and C4-2 (human prostate cancer) cell lines, and regulation is dependent on VDR expression. The human PTHrP gene has three promoters (P); PC-3 cells preferentially utilize P2 and P3, while C4-2 cells preferentially utilize P1. 1,25D regulates PTHrP transcriptional activity from both P1 and P3. The 1,25D-mediated decrease in PTHrP mRNA levels also involves a post-transcriptional pathway since 1,25D decreases PTHrP mRNA stability. 1,25D also suppresses PTHrP expression directly at the protein level by increasing its degradation. Regulation of PTHrP levels is dependent on VDR expression, as using siRNAs to deplete VDR expression negates the 1,25D-mediated downregulation of PTHrP expression. These results indicate the importance of maintaining adequate 1,25D levels and VDR status to control PTHrP levels.
Keywords: parathyroid hormone-related protein; prostate cancer; 1,25-dihydroxyvitamin D3; vitamin D receptor; negative vitamin D response element
1 Introduction
Prostate cancer is the second-leading cause of cancer-related death in men in the United States (Jemal et al., 2007). The most common site of prostate cancer metastasis is the bone (Bubendorf et al., 2000; Rana et al., 1993). Histologic evidence shows that these metastases form a heterogeneous mixture of osteolytic and osteoblastic lesions (Berruti et al., 1996; Guise et al., 2006; Keller et al., 2001). The metastatic process requires that cells acquire new capabilities, including an increased ability to migrate and invade surrounding tissues to reach the vasculature and lymphatics (Hanahan and Weinberg, 2005). This process is accompanied by neoangiogenesis (Eccles, 2005).
Androgens play a pivotal role in the development and physiologic function of the prostate, as well as in prostate cancer. However, additional factors such as growth factors, neuroendocrine peptides and cytokines are also involved in prostate physiology and pathology (Deftos, 2000). Several studies have demonstrated a role for parathyroid hormone-related protein (PTHrP) in the pathogenesis and progression of prostate carcinoma and its tendency to metastasize to the bone (Hall et al., 2005, 2006). Both normal and neoplastic prostate epithelial cells express PTHrP (Iwamura et al., 1994; Kramer et al., 1991). PTHrP enhances prostate cancer cell proliferation, survival, migration, invasion and anchorage-independent cell growth in vitro (Bhatia et al., 2009; Dougherty et al., 1999; Tovar Sepulveda and Falzon, 2002), and increases growth of prostate and colon cancer cells in a nude mouse model (Bhatia et al., 2009; Shen et al., 2007). The in vivo effects of PTHrP are accompanied by increased angiogenesis and decreased apoptosis (Bhatia et al., 2009). Moreover, PTHrP overexpression in prostate cancer cells decreases the latency and increases the severity of bone lesions. PTHrP also changes the bone lesion profile from predominantly osteoblastic to osteolytic (Bhatia et al., 2009). PTHrP expression also directly correlates with prostate cancer differentiation. In well-differentiated prostate cancer, expression is predominantly confined to the basal layer. As the cancer progresses to poorly-differentiated, intense cytoplasmic and nuclear PTHrP immunoreactivity is observed (Bhatia et al., 2009). These studies underlie the critical role of PTHrP in prostate cancer.
There are limited options for the treatment of metastatic prostate cancer. Although prostate cancers initially respond to androgen ablation therapy, they eventually become androgen independent. Biological response modifiers such as vitamin D analogs may be effective in slowing prostate cancer progression. Epidemiological studies have shown that vitamin D deficiency is linked with increased prostate cancer incidence (reviewed in Bouillon et al., 2008). 1,25-Dihydroxyvitamin D3 (1,25D), the hormonally active form of vitamin D, regulates cell proliferation, differentiation, apoptosis, immune responses and angiogenesis in many cancer cell types (DeLuca, 2004; Holick, 2003; Mantell, 2000). 1,25D also regulates PTHrP expression in a number of human cell types, including prostate cancer cell lines (El Abdaimi et al., 1999; Haq et al., 1993; Tovar Sepulveda et al., 2006).
The human PTHrP gene is complex, with nine exons spanning more than 15 kb of genomic DNA (Broadus and Stewart, 1994). PTHrP transcription may initiate at three promoters (P). P1 and P3 are canonical TATA promoters, and P2 is a high GC-element promoter (Vasavada et al., 1993). Promoter usage is cell type-specific (Cataisson et al., 2002; Hamzaoui et al., 2007; Luparello et al., 1999). The regulation of PTHrP expression by 1,25D has focused predominantly on suppression of PTHrP mRNA levels at the transcriptional level, mediated via a negative vitamin D response element (nVDRE) located within P1 (Abe et al., 1998; Nishishita et al., 1998; Tovar Sepulveda and Falzon, 2003). In this study, we asked whether alternative pathways independent of the nVDRE within P1 contribute to transcriptional regulation of the PTHrP gene by 1,25D, and whether post-transcriptional and post-translational components are also involved. Since VDR levels may be altered in cancer cells, we also investigated the effect of 1,25D on PTHrP levels in cells with decreased VDR expression. The C4-2 and PC-3 cell lines were used as model systems. The C4-2 cell line is a second-generation LNCaP subline that is androgen-independent and metastasizes to the lymph node and bone when injected orthotopically into nude mice (Thalmann et al., 1994; Wu et al., 1994). C4-2 cells produce mixed lytic/blastic lesions (Vinholes et al., 1996). The androgen-independent PC-3 cell line was initiated from a bone metastasis and produces predominantly lytic lesions. PTHrP plays both proliferative and metastatic roles in prostate cancer. Thus, identifying the pathways via which its expression is suppressed by 1,25D may lead to new therapeutic approaches.
2. Materials and Methods
2.1. Materials
1,25D was kindly provided by Dr. M. Uskokovic (Hoffmann La-Roche, Inc., Nutley, New Jersey), and was dissolved in ethanol at 10−3 M. Fetal bovine serum (FBS) and dialyzed FBS were obtained from Atlanta Biologicals (Norcross, GA) and HyClone (Logan, UT), respectively. Tissue culture supplies were purchased from Gibco (Carlsbad, CA). Antibodies for Western blot analysis were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The small interfering RNAs (siRNAs) targeting the VDR and the corresponding non-target control (NTC) siRNA sequences were purchased from Dharmacon (Lafayette, CO).
2.2. Cell culture
PC-3 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were grown at 37 °C in a humidified 95% O2/5% CO2 atmosphere in RPMI-1640 medium supplemented with 8% FBS and L-glutamine. C4-2 cells were purchased from UroCor, Inc. (Oklahoma City, OK), and were grown under the same conditions in RPMI-1640 supplemented with 10% FBS and L-glutamine. At 48 h before treatment with 1,25D, conventional FBS was replaced with dialyzed FBS in order to minimize the exposure of the cells to endogenous steroids present in the serum.
2.3. Plasmid constructs
Constructs containing regions from P1, P2 or P3 from the human PTHrP gene, cloned in the luciferase reporter plasmid pGL-2 (Promega, Madison, WI), were obtained from Dr. Z. Bouizar. These constructs have been described (Cataisson et al., 2002). These constructs, as well as the empty vector control, were transfected into PC-3 and C4-2 cells using Lipofectamine Plus (Invitrogen, Carlsbad, CA).
2.4. Cell transfections and 1,25D treatment
To ask if 1,25D alters transcription from the PTHrP gene, we measured promoter activity of each of the three PTHrP promoters in the presence and absence of 1,25D. Cells were plated in 24-well plates at 1 × 105 cells/well in medium containing dialyzed FBS (HyClone). After 48 h, the cells were transfected with the promoter constructs using Lipofectamine Plus, per the manufacturer’s instructions (Invitrogen). Cells transfected with the empty vector were used as controls. Cells were co-transfected with a construct expressing Renilla Luciferase for standardization purposes. After 4 h, the transfection medium was removed and replaced with fresh medium containing 1,25D (10−9 to 10−7 M). Ethanol was used as the vehicle control (final volume 0.01% v/v). After 24 h, cell lysates were prepared and promoter activity was assayed using the Dual Luciferase assay kit (Promega). Empty vector control values were subtracted from the respective firefly and Renilla luciferase values. The firefly luciferase activity corresponding to each PTHrP promoter was normalized to Renilla luciferase activity, and the fold differences were plotted as the firefly/Renilla ratio.
For siRNA transfections, cells were plated as described above. After 48 h, they were transfected by electroporation with ON-Target plus siRNAs directed against the VDR (100 nM; Dharmacon). To eliminate the potential for off-target effects, two independent siRNAs were used. As a control, cells were transfected with ON-Target-plus non-target control (NTC) siRNAs.
To determine the effect of 1,25D on PTHrP protein levels, PC-3 and C4-2 cells were plated in 100 mm plates at 5 × 105 cells/plate in medium containing dialyzed FBS (HyClone). When the cells were ~70% confluent, they were treated with 1,25D (10−9 to 10−7 M) or ethanol (vehicle control). Cell extracts were processed for Western blot analysis after 48 h of treatment.
2.5. Analysis of PTHrP and VDR mRNA levels
Total RNA was extracted using the RNAqueous® isolation kit (Ambion Inc., Austin, TX), per the manufacturer’s protocol. RNA concentrations were determined by spectrophotometry. PTHrP, CYP24a1, and VDR mRNA levels were analyzed by reverse transcription/real-time PCR as described (Shen et al., 2007a). The following TaqMan inventoried products were used: PTHrP, Hs00174969_m1; VDR Hs01045840_m1; CYP24a1, Hs00167999_m1; and the pre-developed 18S rRNA primers (VIC™-dye labeled probe, TaqMan® assay reagent, P/N 4319413E), and were obtained from Applied Biosystems, as was the universal PCR master mix reagent kit (P/N 4304437).
2.6. Western blot analysis
Cells were grown to 70–80% confluence in 100 mm plates. To prepare whole cell extracts, cells were washed twice with cold PBS on ice and lysed in RIPA buffer containing a Protease Inhibitor cocktail and Phosphatase Inhibitor cocktails A and B (Santa Cruz Biotechnology). Nuclear extracts were prepared using the NE-PER Nuclear Protein Extraction Kit (Pierce Biotechnology Inc.), according to the manufacturer’s instructions. Briefly, cells were washed with ice-cold PBS containing phosphatase inhibitors. After scraping the cell monolayer and centrifugation, the pellet was resuspended and incubated in 1 × hypotonic buffer for 15 min on ice. Detergent was then added. After centrifugation, the pellet (nuclear fraction) was resuspended in complete lysis buffer and stored at −80 °C. Protein concentrations were estimated using the Bio-Rad protein assay. Protein levels were analyzed by Western blot analysis as previously described (Shen et al., 2007a). GAPDH was used as loading control. The signals were detected using the SuperSignal West Pico Substrate kit (Pierce Biotechnology Inc., Rockford, IL). Densitometric analysis was performed using the Alpha Innotech Image Analysis system (Alpha Innotech Corporation, San Leandro, CA).
2.7. Manipulation of VDR levels
Protein synthesis was inhibited by treatment with the protein synthesis inhibitor cycloheximide (CHX; Sigma) at a final concentration of 150 µg/mL. After 6 h, the medium containing CHX was replaced with fresh medium containing dialyzed FBS and 1,25D (10−7 M) or ethanol (vehicle control). The cells were harvested after periods ranging from 3 h to 72 h. Cell extracts were processed for Western blot analysis. Densitometric analysis was performed as described above.
To determine the effect of 1,25D on the stability of the VDR, cells were pre-treated with CHX for 30 min. 1,25D (10−8M) or ethanol (vehicle control) was then added and cells were harvested after 4 h and 8 h. To address the involvement of the proteasome pathway in VDR degradation, CHX-pretreated cells were treated with the proteasomal inhibitor MG132 (50 µM) or DMSO (vehicle control) in the presence of 1,25D or ethanol and were harvested after 4 h and 8 h. Nuclear extracts were then prepared using the NE-PER Nuclear Protein Extraction Kit (Pierce Biotechnology Inc., Rockford, IL).
2.8. Determination of PTHrP half-life after 1,25D treatment
To ask whether 1,25D regulates PTHrP mRNA levels at the post-transcriptional level, the PTHrP mRNA half-life (t½) was measured in the presence and absence of 1,25D. PC-3 and C4-2 cells were plated in 100 mm plates at 5 × 105 cells/plate in medium containing dialyzed FBS. After 24 h in culture, the cells were treated with 1,25D (10−7 M) or ethanol (vehicle control) for 48 h. The transcriptional inhibitor 5,6-dichlororibofuranosylbenzimidazole (DRB; Sigma) was then added to a final concentration of 25 µg/ml (Alizadeh et al., 2000; Dhawan et al., 1991; Misquitta et al., 2002) and incubation continued in the presence of 1,25D. Cells were harvested at time 0 and after 1, 1.5, 3, 6, 9, and 12 h for RNA isolation. DMSO was used as vehicle control for DRB.
To ask whether 1,25D regulates PTHrP protein levels via a post-translational pathway, the protein t½, was measured in the presence and absence of 1,25D. Cells were plated in 100 mm plates at 106 cells/plate in medium containing dialyzed FBS. When the cells were ~70% confluent, they were treated with 1,25D (10−7 M) or ethanol (vehicle control). After 48 h, CHX was added to a final concentration of 150 µg/mL. The cells were harvested after 0.5, 1, 2, 4 and 6 h and cell extracts were processed for Western blot analysis. Densitometric analysis was performed as described in Section 2.6.
2.9. Statistics
Numerical data are presented as the mean ± standard error of the mean (S.E.M). The data were analyzed by one-way analysis of variance (ANOVA) followed by a Tukey-Kramer multiple comparisons post-test to determine the statistical significance of differences. All statistical analyses were performed using INSTAT Software (GraphPad Software, Inc., San Diego, CA).
3. Results
3.1. PTHrP promoter activity in C4-2 and PC-3 cells
PTHrP expression is regulated in a cell type-specific manner; promoter usage varies among different cell lines. The relative activities of P1-, P2- and P3-driven luciferase reporters in C4-2 and PC-3 cells was measured after transfecting the cells with each of these promoter constructs. In C4-2 cells, maximal P1 activity was ~45-fold higher than P2 and P3 activity (Fig. 1). In PC-3 cells, P2 and P3 showed comparable activity; P1 activity was significantly lower (Fig. 1). However, the difference between the activities of P1 and P3 vs. that of P2 was only ~4-fold in these cells (Fig. 1), suggesting that all three promoters are relatively active in PC-3 cells. The promoter activity was also dependent on the concentration of transfected DNA, reaching a peak between 0.4 and 0.8 µg of transfected DNA in both cell lines (Fig. 1).
Figure 1. PTHrP promoter usage in C4-2 and PC-3 cells.
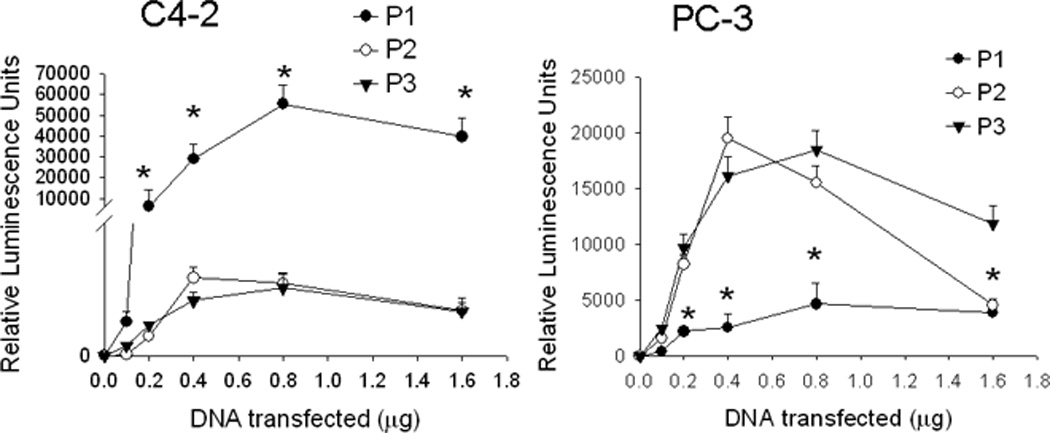
Cells were transfected with the indicated amounts of promoter-driven luciferase reporter construct DNA for promoters 1, 2 and 3 (P1, P2 or P3). Luciferase activity was measured after 24 h. Control (empty vector) values were subtracted from the respective firefly and Renilla luciferase values. Values were normalized to Renilla luciferase activity, and are expressed as Relative Luminescence Units. P = promoter. Each point is the mean ± SEM of three experiments. * = Significantly different from the P2 and P3 values (P < 0.001).
3.2. 1,25D regulates PTHrP expression via promoters P1 and P3
Previous studies showing regulation of PTHrP mRNA levels by 1,25D used a nVDRE sequence located at −546 to −517 within P1 (Nishishita et al., 1998; Tovar Sepulveda and Falzon, 2003). Regulation via this nVDRE was cell type-specific, and was observed in C4-2, but not PC-3 cells (Tovar Sepulveda and Falzon, 2003). Here we report that, when a 1.25 kb P1 sequence extending from −917 to +338 relative to the transcriptional start-site was used, 1,25D downregulated P1 activity in both C4-2 and PC-3 cells (Fig. 2). As shown in Fig. 1, this same P1 sequence was transcriptionally active in both PC-3 and C4-2 cells. We also show that 1,25D downregulated P3 activity in both PC-3 and C4-2 cells, although the magnitude of the 1,25D-mediated decrease in P3 activity was lower than that observed for P1 (Fig. 2). This effect on P1 and P3 activity was evident with 1,25D concentrations of 10−9 M and above (Fig. 2). 1,25D had no effect on P2 activity at any of the concentrations tested (10−7 to 10−9 M; Fig. 2).
Figure 2. Effect of 1,25D on PTHrP promoter activity mediated via P1, P2 or P3 in C4-2 and PC-3 cells.
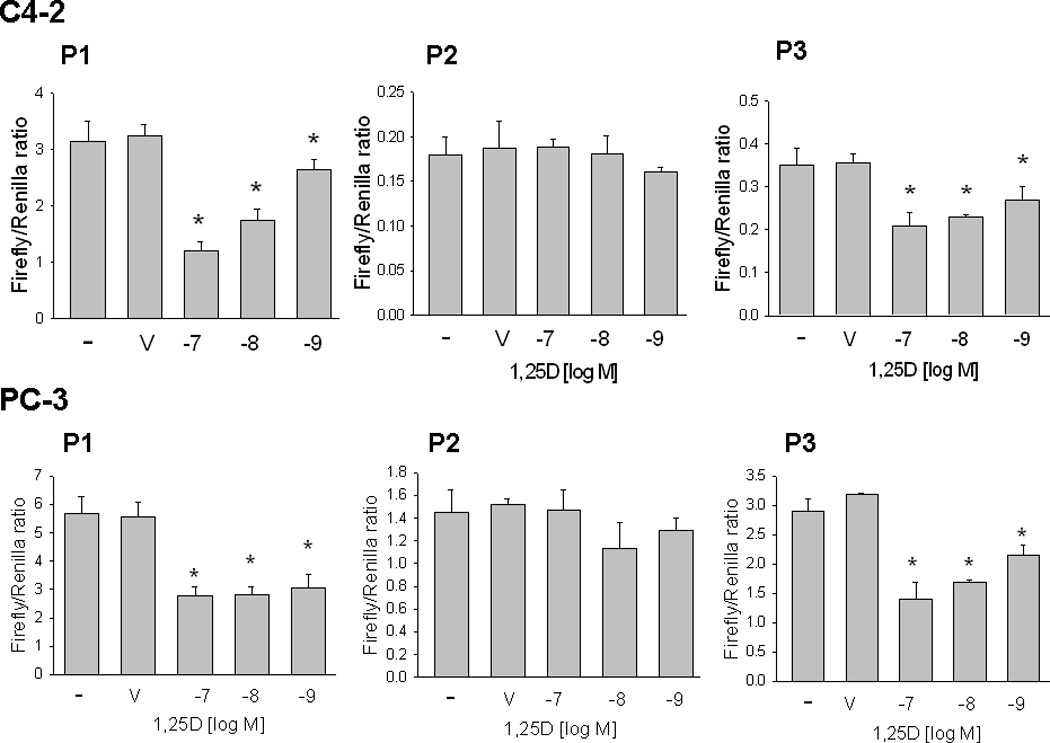
Cells were transfected with the indicated promoter-driven luciferase reporter constructs plus Renilla luciferase construct, then treated with the indicated concentration of 1,25D or with ethanol (V, vehicle control). Luciferase activity was measured after 24 h. Empty vector control values were subtracted from the respective firefly and Renilla luciferase values. Values were then normalized to Renilla luciferase activity, and are expressed as the Firefly/Renilla ratio. P = promoter. Each bar is the mean ± SEM of three experiments. * = Significantly different from the vehicle control (P < 0.001).
3.3. 1,25D decreases PTHrP mRNA stability
To determine if the decrease in PTHrP mRNA levels after 1,25D treatment is due to changes in mRNA stability, RNA synthesis was blocked using 5,6-dichlororibofuranosylbenzimidazole (DRB), and PTHrP mRNA levels were analyzed by reverse transcription/real-time PCR at different time-points after inhibition. These experiments were performed in PC-3 cells because C4-2 cells express low levels of PTHrP mRNA and therefore PTHrP mRNA levels at the later time points after DRB treatment were undetectable or below the level of accurate measurement. Treatment with 1,25D decreased the PTHrP mRNA t½ by ~ 2.0-fold (Fig. 3, A and B).
Figure 3.
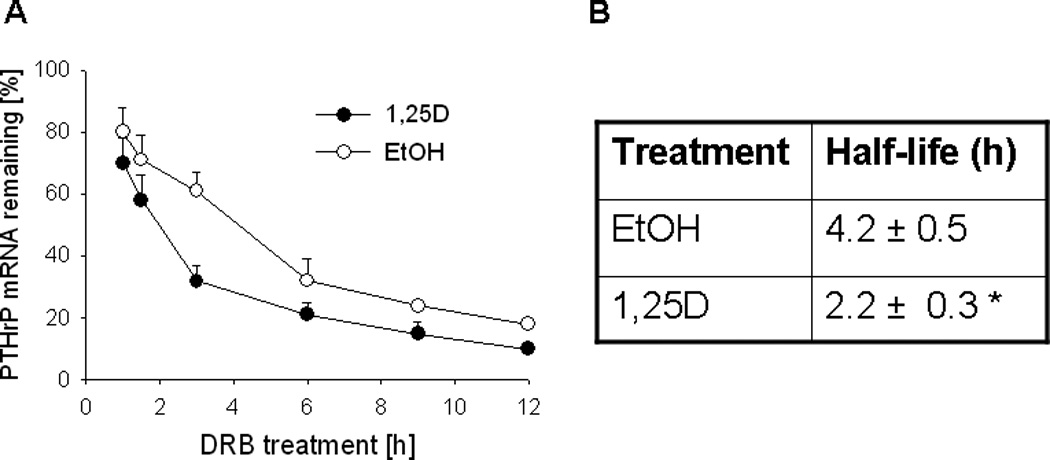
(A) Decay profile of PTHrP mRNA in PC-3 cells treated with 1,25D or ethanol The cells were treated with 1,25D (10−7 M) or ethanol (EtOH, vehicle control) for 48 h, followed by the transcriptional inhibitor 5,6-dichlororibofuranosylbenzimidazole (DRB). PTHrP mRNA levels were measured by reverse transcription/real-time PCR at the indicated time points after addition of DRB, and are expressed as a percentage of the value obtained at the time of DRB addition (0 h), set at 100%. Each point represents the mean ± S.E.M. of three independent experiments. (B) Comparison of the PTHrP mRNA half-life in 1,25D- and ethanol-treated cells. The values were obtained from the data presented in (A). * = Significantly different from the vehicle control value (P < 0.001).
3.4. 1,25D regulates PTHrP at the protein level
Experiments determining the effects of 1,25D on PTHrP protein levels were generated using both PC-3 and C4-2 cells. Unless otherwise noted, data are shown for PC-3 cells, and similar results were obtained for C4-2 cells. In this and previous studies, we show that 1,25D decreases PTHrP mRNA levels. Western blot analysis was carried out to determine whether this decrease is reflected in changes at the protein level. PTHrP is detected as one major band of ~ 18 kDa in both PC-3 and C4-2 cells (Fig. 4, A and C). Minor bands are also evident in PC-3, but not C4-2, cells (Fig. 4, A and C). Treating PC-3 and C4-2 cells with 1,25D for 48 h produced a concentration-dependent decrease in PTHrP protein levels (Fig. 4, A and B). Treatment for 72 h did not increase the effect of 1,25D at any of the concentrations tested (data not shown).
Figure 4. Effect of 1,25D on PTHrP protein levels, analyzed by Western blot analysis. (A and B) PTHrP levels in PC-3 and C4-2 cells treated with the indicated concentrations [log[(M)] of 1,25D.
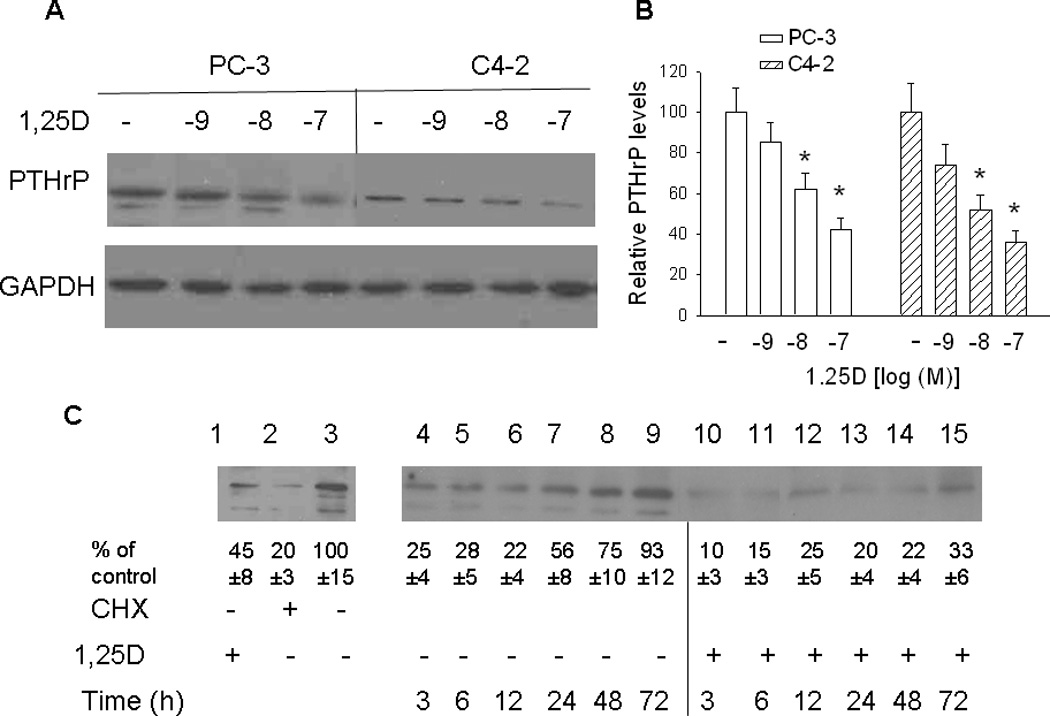
After 48 h, lysates were prepared for Western blotting. (A) Representative Western blot. (B) The relative PTHrP levels were obtained after densitometric scanning of the Western blots and normalization to GAPDH. The control value (−, ethanol treated cells) was set at 100%. In PC-3 cells, only the major band was analyzed. In C4-2 cells, only one band was evident. Each bar represents the mean ± S.E.M. of three independent experiments. * = Significantly different from the vehicle control (P < 0.001). (C) Recovery of PTHrP expression in PC-3 cells treated with 1,25D or ethanol. Lane 1, PTHrP levels in cells treated with 10−7 M 1,25D for 72 h; lane 2, PTHrP levels in cells treated with the protein synthesis inhibitor cycloheximide (CHX) for 6 h; lane 3, PTHrP levels in control (ethanol-treated) cells. Lanes 4–15. PC-3 cells were pre-treated with CHX for 6 h, then with 10−7 M 1,25D (+ 1,25D lanes) or with ethanol (vehicle control; − 1,25D lanes). At the indicated time points, lysates were prepared for Western blotting. The relative PTHrP levels were obtained after densitometric scanning of the Western blots and normalization to GAPDH. The control value (−, ethanol treated cells) was set at 100%. The mean and S.E.M. values represent data from three independent experiments.
1,25D also sustained the decrease in PTHrP levels observed after inhibition of protein synthesis using CHX followed by release of the protein synthesis block. For these experiments, cells were pre-treated with CHX for 6 h, then treated with 1,25D (10-7 M). Ethanol was used as vehicle control. Treatment with CHX significantly decreased PTHrP levels (Fig. 4C, lane 2 vs. lane 3). PTHrP levels recovered to the pre-CHX treatment levels within 72 h in ethanol-treated cells (Fig. 4C, lanes 4 to 9). In contrast, when cells were treated with 1,25D after removal of CHX, PTHrP levels reached only ~ 20% of the levels in untreated cells within 48 h of CHX removal (Fig. 4C, lane 3 vs. lanes 10 to 14). After removal of CHX, protein levels return to normal within ~ 8 h (Ignowski, and Schaffer, 2004). Reduced PTHrP levels at the earlier time-points (3 h and 6 h) may therefore be due to the lag in build-up of PTHrP protein. The lower PTHrP levels in 1,25D-treated vs. untreated cells may be due to residual VDR activity in CHX-treated cells. Differences in protein levels between 1,25D-treated and -untreated cells at early time-points may also be due to the transcriptional effects of 1,25D on PTHrP, since such effects are evident 3 h after treatment with 1,25D (Karmali et al., 1999). In addition, any residual VDR is stabilized after addition of 1,25D (Masuyama and MacDonald, 1998; see also Section 3.5 below). At later time-points, when protein synthesis has recovered, the effects of 1,25D may be due to transcriptional/post-transcriptional repression as well as a direct effect on protein levels, as shown in Fig. 6.
Figure 6. Effect of 1,25D on the PTHrP protein half-life.
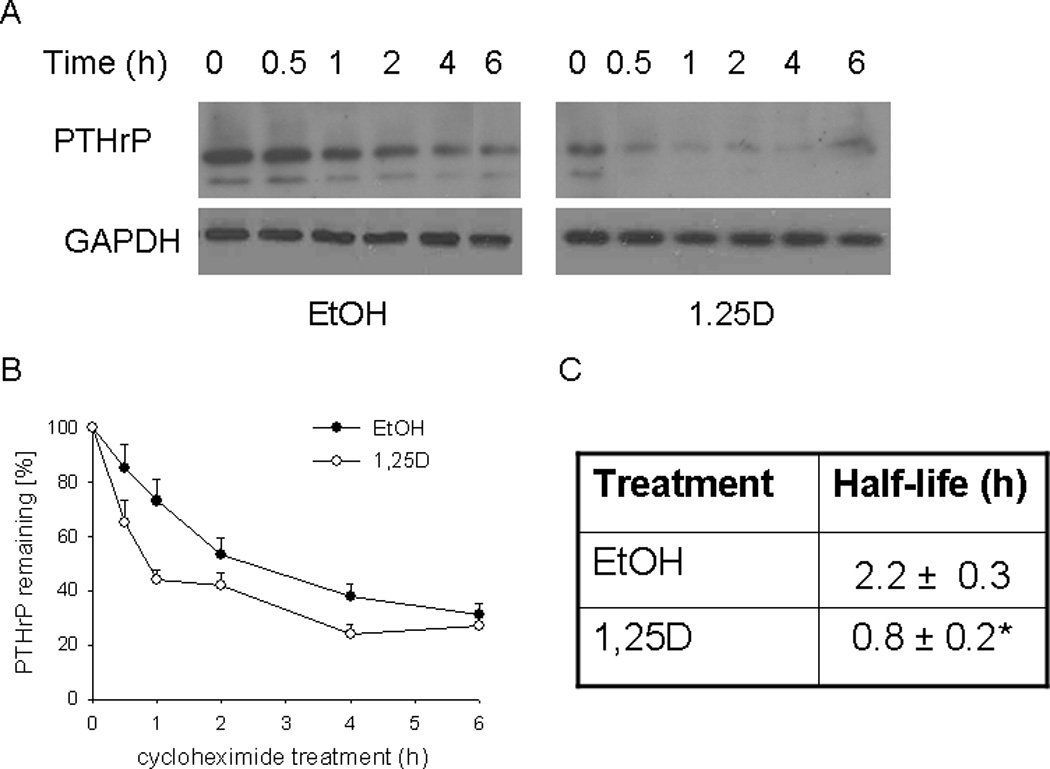
PC-3 cells were treated with 1,25D (10−7 M) or ethanol (EtOH, vehicle control) for 48 h. The protein synthesis inhibitor CHX (150 µg/ml) was then added. At the indicated time points, lysates were prepared for Western blotting. (A) Representative Western blot showing decay of PTHrP levels in 1,25D- or ethanol-treated cells co-treated with CHX. (B) Densitometric analysis of the decay of PTHrP levels after CHX treatment in cells pre-treated with 1,25D or ethanol. Each point is the mean ± SEM of data from three independent experiments. The 0 time-point (at the time of CHX addition) is set at 100%. (C) Comparison of halflife of PTHrP in 1,25D- and ethanol-treated cells. The values were obtained from the data presented in (B). * = Significantly different from the vehicle control value (P < 0.001).
3.5. Regulation of PTHrP is dependent on VDR expression
Here we investigated the role played by the VDR in downregulation of PTHrP expression by 1,25D. We first established that inhibition of protein synthesis after CHX treatment does result in depletion of the VDR in the cells, and that the VDR is stabilized in the presence of its ligand 1,25D. For this purpose, cells were pre-treated with CHX for 30 min, then treated with 1,25D or ethanol in the presence of CHX. In 1,25D-treated cells, VDR expression was still evident after 4 h of CHX treatment (Fig. 5A). In the absence of 1,25D, VDR levels were negligible at time points ≥ 4h of CHX treatment (Fig. 5A), indicating more rapid degradation. VDR degradation involves ubiquitination (Masuyama and MacDonald, 1998). Here we also established that VDR depletion in cells treated with CHX involves degradation via the proteasome pathway, since inclusion of the proteasome inhibitor MG132 prevented VDR degradation, and comparable VDR levels were evident in the presence and absence of 1,25D (Fig. 5A). DMSO (vehicle control) had no effect on VDR levels (data not shown).
Figure 5.
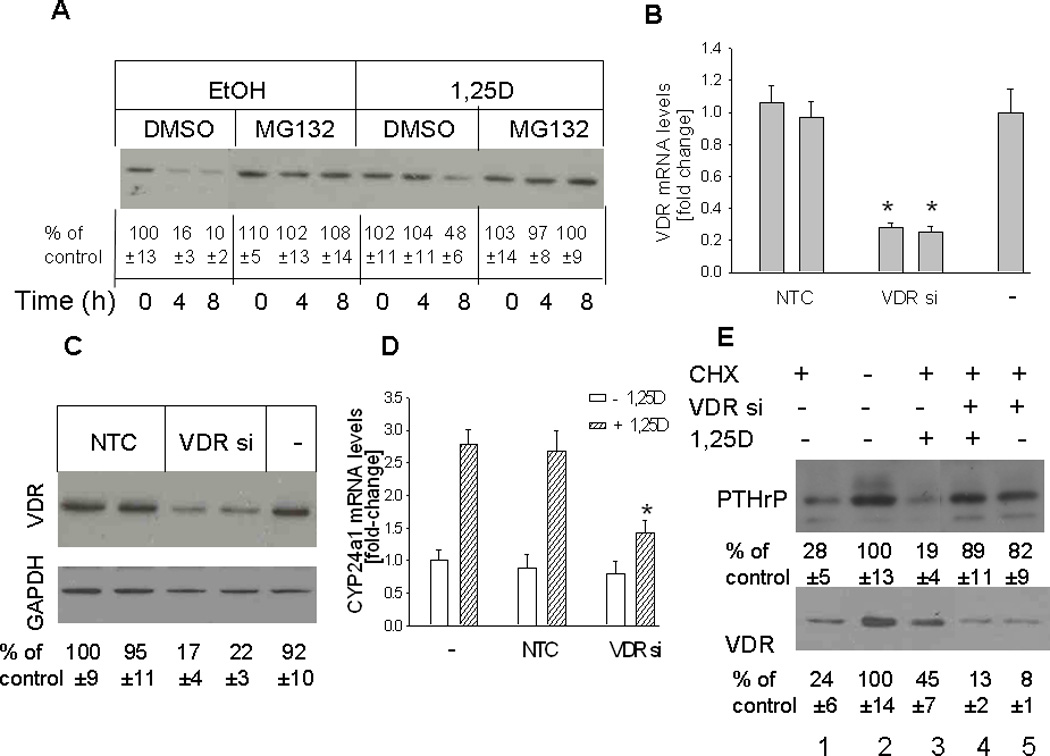
(A) Time course for VDR depletion in cells treated with 1,25D or ethanol (vehicle control) Cells were pretreated with the protein synthesis inhibitor cycloheximide (CHX) for 30 min, then with 1,25D (10 −7M) in the presence or absence of the proteasome inhibitor MG132 (50 µM). Ethanol (EtOH) and DMSO were used as vehicle controls for 1,25D and MG132, respectively. Nuclear extracts were prepared at the indicated time points for Western blotting. (B–D) Effects of suppressing VDR expression on levels of the VDR (B,C) and of the 1,25D target gene CYP24a1 (D). VDR expression was suppressed by transfection with siRNA targeting the VDR. (B, D) VDR and CYP24a1 mRNA levels were analyzed by reverse transcription/real-time PCR analysis. Each bar is the mean ± SEM of three experiments for each of two independent non-target control siRNAs (NTC), VDR-targeting siRNAs (VDR si), or non-transfected control (−). (C) Western blot analysis for VDR levels in VDR siRNA-transfected cells. (E) Recovery of PTHrP expression in cells with suppressed VDR expression treated with 1,25D or ethanol. PC-3 cells transfected with an siRNA targeting the VDR (VDR si +) were pre-treated with the protein synthesis inhibitor cycloheximide (CHX) for 6 h, then with 1,25D (10−7 M) (+ 1,25D lanes) or with ethanol (vehicle control; − 1,25D lanes). − VDR si lanes were transfected with NTC siRNA. After 48 h, lysates were prepared for Western blotting. In (A), (C) and (E), the relative PTHrP levels were obtained after densitometric scanning of the Western blots and normalization to GAPDH. The control value (−, ethanol treated cells) was set at 100%. In (A), (C) and (E), the mean and S.E.M. values represent data from three independent experiments.
To link VDR expression with downregulation of PTHrP expression, PC-3 cells were transfected with siRNA targeting the VDR. To eliminate the potential for off-target effects, two independent VDR-specific siRNAs were used. Transfection with either of the two siRNAs caused an ~ 80% decrease in VDR mRNA and protein levels (Fig. 5, B and C). As shown in Fig. 5D, depletion of the VDR also reversed the 1,25D-mediated increase in mRNA levels of CYP24a1, a classic VDR target gene (Tashiro et al., 2007), indicating that suppression of VDR expression interferes with 1,25D signaling. Transfection with the NTC siRNA had no effect on VDR mRNA or protein levels, or on CYP24a1 mRNA levels, when compared to parental controls (Fig. 5, B to D).
The effect of 1,25D on PTHrP levels in cells with suppressed VDR expression was investigated using the conditions established in Fig. 5, A and B–D. Cells transfected with siRNA targeting the VDR or with NTC siRNA were pretreated with CHX for 6 h, followed by 1,25D. Both PTHrP and VDR levels were decreased in cells treated with CHX (Fig. 5E, lanes 1 vs. 2). As also shown in Fig. 5A, VDR levels were decreased to a higher extent in control cells vs. cells treated with 1,25D (Fig. 5E, lanes 1 vs. 3). VDR levels were negligible in cells transfected with VDR siRNA, both in the presence and absence of 1,25D (Fig. 5E, lanes 4 and 5). After CHX treatment, recovery of PTHrP expression was comparable in cells with suppressed VDR expression treated with 1,25D (Fig. 5E, lane 4) and in cells with suppressed VDR expression treated with ethanol (Fig. 5E, lane 5), indicating that VDR expression is necessary for suppression of the recovery of PTHrP expression to occur. Under both these treatment conditions, PTHrP levels were comparable to those in control cells (Fig. 5E, lane 2).
3.6. 1,25D decreases PTHrP protein half-life
We next asked whether 1,25D affects the stability of PTHrP at the protein level. Data are shown for PC-3 cells; similar results were obtained for C4-2 cells (not shown). To determine the PTHrP protein t½, cells were treated with 1,25D or ethanol for 48 h, and then exposed to CHX to inhibit new protein synthesis. In control cells, the decrease in PTHrP levels predominantly reflects the degradation of the protein. Based on this profile, the t½ of PTHrP was ~ 2.2 h (Fig. 6). In cells pre-treated with 1,25D, the rate of decrease in PTHrP levels was significantly faster, such that the t½ was decreased by ~ 3-fold (Fig. 6). 1,25D had no effect on the t½ of GAPDH (Fig. 6), indicating that the observed effects on the t½ of PTHrP are not due to a generic global effect of 1,25D. These data indicate that 1,25D also regulates PTHrP at the post-translational level.
4. Discussion
PTHrP contributes both to the pathogenesis and progression of prostate cancer, where it increases prostate cancer cell proliferation, survival, migration and invasion (Bhatia et al., 2009; Dougherty et al., 1999; Tovar Sepulveda and Falzon, 2002). The protein also plays a role in the preferred metastasis of prostate cancer to bone (Hall et al., 2005, 2006), and is involved in both the initial osteolytic phase and the ensuing osteoblastic phase of metastasis (Hall et al., 2006). Therefore, targeting PTHrP production in prostate cancer may prove therapeutically beneficial. Since the options for treating metastatic prostate cancer are currently limited, there remains a need to develop well-tolerated alternative treatments to slow progression. The natural hormone 1,25D and its analogs inhibit proliferation and induce apoptosis of numerous cancer cell types, including those derived from prostate cancer. 1,25D also inhibits tumor cell migration, metastasis and angiogenesis (Stewart and Weigel, 2004).
The PTHrP gene has nine exons and three promoters, located upstream of exons I, III and IV (Vasavada et al., 1993; Southby et al., 1995). Only two exons (exons V and VI) are present in all PTHrP transcripts. Alternative promoter usage and 3’ splicing allows the production of mRNAs specifying each of the three PTHrP isoforms (139, 141 and 173 amino acids). Both promoter usage and 3’ splicing are cell type-specific (Cataisson et al., 2002; Hamzaoui et al., 2007; Luparello et al., 1999; Southby et al., 1995). Moreover, exon II may be included or excluded from transcripts originating from P1 (Mangin et al; 1990; Glatz et al., 1994). The splicing pathways generating the 139 and 141 amino acid PTHrP isoforms are predominantly utilized (Southby et al., 1995). This differential regulation of the PTHrP gene in different cell lines may give rise to splice variant protein products of comparable size but representing transcripts originating from different promoters as well as different 3’ splicing patterns. This may explain why no noticeable difference in protein size is observed in Western blots from PC-3 and C4-2 cells, which utilize different promoters, and why one predominant band and minor bands are observed in PC-3 cells, where P1 and P2 activity is comparable.
While it has been shown that 1,25D downregulates PTHrP expression via a transcriptional pathway involving the nVDRE within P1 of the human PTHrP gene (Nishishita et al., 1998; Tovar Sepulveda and Falzon, 2003), alternative pathways via which 1,25D may regulate PTHrP levels have not been fully investigated. Here we show that 1,25D suppresses PTHrP levels at both the mRNA and protein levels, and that transcriptional regulation involves both P1 and P3. We show comparable activity from P2 and P3 in PC-3 cells; P1 activity is significantly lower in these cells. Conversely, P2 and P3 activities are significantly lower than P1 activity in C4-2 cells. Methylation of cytosine residues within CpG dinucleotide sequences in DNA plays an important role in the regulation of gene activity (Bird, 1986). Selective promoter activity in the PC-3 and C4-2 cell lines may be mediated via differential promoter methylation patterns. In fact, Holt et al. (1993) have shown cell type-specific CpG methylation within the promoter regions of the PTHrP gene, and that methylation correlated with transcript levels.
Previous studies have established the presence of a nVDRE within P1 of the human PTHrP gene (Nishishita et al., 1998; Tovar Sepulveda and Falzon, 2003). A 30 bp sequence encompassing the P1 nVDRE conferred 1,25D responsiveness in C4-2 and LNCaP cells, but not in PC3 cells (Tovar Sepulveda and Falzon, 2003). Conversely, here we report a decrease in promoter activity from a 1.25 kb P1 sequence encompassing the nVDRE in both PC-3 and C4-2 cells. Transcription of 1,25D-sensitive genes involves other accessory proteins that form part of the transcription machinery in addition to the VDR. After interaction with the nVDRE, the VDR interacts with these accessory proteins to modulate transcription. The additional DNA sequence may favor the interaction of these accessory proteins with the VDR/nVDRE, and therefore strengthen the VDR/nVDRE interaction, allowing downregulation of PTHrP expression. Favorable interaction of these accessory proteins in PC-3 cells, where P1 activity is low, may be more crucial than in C4-2 cells, where P1 activity is strong. Alternatively, the 1.25 kb P1 sequence may include more than one nVDRE. The previously identified nVDRE at (−546 to −517) may not be functional in PC-3 cells, and the 1,25D responsiveness of the 1.25 kb P1 sequence in PC-3 cells may be conferred through other presently unidentified nVDRE(s). Downregulation of P3 activity also contributes to the observed decrease in PTHrP mRNA levels after 1,25D treatment. Future studies will identify other nVDREs within the 1.25 kb P1 region, and as well as nVDRE(s) within P3.
The t½ of PTHrP mRNA has been reported to range from < 1 h to ~ 10 h (Cataisson et al., 2002; Schordan et al., 2004; Sellers et al., 2002, 2004). Here we report that the PTHrP mRNA t½ in PC-3 cells is ~ 4 h, a value comparable to that in the lung squamous carcinoma cell line, HARA (Sellers et al., 2004). This range of t½ for PTHrP mRNA is dependent on the particular PTHrP transcript expressed in a cell type-specific manner (Sellers et al., 2004). Treatment with 1,25D decreased the PTHrP mRNA stability, indicating post-transcriptional regulation of PTHrP by 1,25D. 1,25D induces differentiation in prostate cells through an effect on the cell cycle, involving accumulation of cells in G0/G1 and a decrease in the number of cells in the G2/M plus S phase (Blutt et al, 1997; Zhuang and Burnstein; 1998; Jensen et al., 2001; Washington et al., 2011). Multiple stimuli regulate mRNA stability in a cell, including changes in cell proliferation and differentiation (Guhaniyogi and Brewer, 2001). Since 1,25D regulates these processes in prostate cancer cells, it is possible that its effects on the PTHrP mRNA t½ may be related to its cell-cycle effects. Sequences within the 3’UTR regulate PTHrP mRNA stability in squamous cell carcinoma cells treated with TGF-β (Sellers et al., 2002). RNA decay is mediated via multiple pathways, including deadenylation, 5’ → 3’ decay, and 3’ → 5’ decay (Garneau et al., 2007). Multiple heterogenous nuclear ribonucleoproteins (hnRNPs) have been identified that play a role in mRNA decay (Garneau et al., 2007). Several signaling pathways have been found to impinge on mRNA stability, including p38 MAPK, ERK, JNK and Wnt/β-catenin (Garneau et al., 2007). These pathways alter mRNA decay by altering the phosphorylation of these RNA-binding proteins. Since the activity of these kinases can be altered in a cell cycle-dependent manner, the 1,25D-mediated alterations of the cell cycle may alter the PTHrP mRNA t½ through an effect on RNA-binding proteins. In addition, steroid hormones, including 1,25D, regulate levels of small non-coding RNAs, including microRNA (miRNA) and siRNA. Both miRNA and siRNA in turn regulate mRNA cleavage and degradation (Nothnick, 2010; Suzuki 2009). Cytoplasmic processing bodies (P-bodies) are the scaffolding centers of miRNA function (Parker and Sheth, 2007). Future studies will address whether P-body formation is altered after 1,25D treatment, and whether P-bodies play a role in the regulation of PTHrP by 1,25D. Coupled with its effects on the transcriptional activity of the PTHrP gene, enhanced PTHrP mRNA decay by 1,25D may also contribute to the potential therapeutic value of 1,25D treatment in prostate cancer. In fact, downregulation of PTHrP expression at the post-transcriptional level by the von Hippel-Lindau tumor suppressor protein, which functions as a gatekeeper for Clear Cell Renal Carcinoma, is accompanied by improved prognosis (Massfelder et al., 2004).
1,25D also decreases PTHrP directly at the protein level. While downregulation of PTHrP mRNA levels contributes to the observed decrease in protein levels, 1,25D regulates PTHrP protein levels directly via a direct post-translational effect, as shown by a decrease in the protein t½. 1,25D also alters the protein t½ of p27Kip1, although in this case 1,25D exerts a stabilizing effect (Li et al., 2004). These effects of 1,25D are thought to be mediated through p27 hypophosphorylation as well as regulation of Skp2 ubiquitin ligase levels (Li et al., 2004). PTHrP is a substrate for the ubiquitin proteolytic system (Meerovitch et al., 1997, 1998), although the specific E3 ligase responsible for mediating proteasomal degradation has not been identified. It is possible that the 1,25D-mediated alteration in PTHrP stability involves effect(s) on component(s) of the proteasome pathway.
There is considerable evidence supporting a link between vitamin D deficiency and an increased incidence of prostate cancer (reviewed in Polek and Weigel, 2002; Schwartz, 2009). 1,25D and non-hypercalcemic 1,25D analogs such as seocalcitol (EB1089) inhibit proliferation and induce cell differentiation and apoptosis (Bhatia et al., 2009; Haq et al., 1993; Mathiasen, 1993). EB1089 also decreases C4-2 cell migration, invasion, and anchorage-independent cell growth in vitro, and decreases prostate cancer xenograft growth and bone metastasis in vivo (Bhatia et al., 2009). Multiple Phase I to III studies have utilized intervention with 1,25D and analogs in prostate cancer, with varying levels of success (summarized in Schwartz, 2009). It has been concluded that treatment with 1,25D analogs may be more beneficial in recurrent disease rather than in advanced cancer (Schwartz, 2009). We previously showed that regulation of PTHrP expression by 1,25D plays a role in the anti-proliferative, anti-migratory and anti-invasive effects of 1,25D in C4-2 cells (Bhatia et al., 2009, Shen et al., 2007a). This study delineates the pathways via which 1,25D suppresses PTHrP levels, leading to the observed beneficial effects of 1,25D.
The physiologic consequences of impaired signaling of the 1,25D/VDR axis have been investigated in animal models. LPB-Tag mice (model for prostate cancer) (Kasper et al., 1998) crossed with VDR-knockout mice show significantly higher prostate cancer cell proliferation compared to LPB-Tag mice with wild-type VDR expression (Mordan-McCombs, 2010). Tumor angiogenesis in the TRAMP (transgenic adenocarcinoma mouse prostate) model is also elevated in VDR knock-out mice (Chung et al., 2009). Stable transfection of the VDR into cancer cells that express low levels of this receptor results in increased growth inhibition by 1,25D (Zhuang et al., 1997). VDR depletion also increases the severity of mammary gland and skin carcinogenesis (Welsh, 2004; Christados et al., 1996). Vdr−/− mice also show hyperproliferation and increased mitotic activity in the descending colon (Kallay et al., 2001). These studies underlie the importance of VDR expression in cancer. PTHrP expression in prostate cancer is linked with increased in vivo tumor growth and metastasis (Bhatia et al., 2009). We previously showed that one of the pathways via which 1,25D exerts its anti-proliferative effects is through downregulation of PTHrP expression (Shen et al., 2007a). Here we show that the VDR is required for the 1,25D-mediated suppression of PTHrP expression. A number of prostate cancer cell lines and primary cultures of prostate cancer cells are resistant to the growth-inhibitory effects of 1,25D (Peehl and Feldman, 2003). This loss of response to 1,25D may be due to decreased VDR levels in these cells. This may lead to loss of regulation of PTHrP and other 1,25D-responsive genes, resulting in enhanced cancer growth. Therefore maintenance of VDR levels may prove to be therapeutically beneficial, as can be achieved through the use of proteasome inhibitors. Future studies will investigate whether PTHrP plays a role in the observed increased prostate cancer cell proliferation and angiogenesis in VDR knockout mice, and compare the effects of EB1089 in VDR−/− and control mice.
In conclusion, these studies show that 1,25D regulates PTHrP expression at the transcriptional, post-transcriptional, and post-translational levels. Regulation of PTHrP levels is dependent on VDR expression by the cells, in that suppressing VDR expression with siRNA inhibits the 1,25D-mediated downregulation of PTHrP expression. Since PTHrP plays a major role in prostate cancer growth and metastasis, these studies underlie the importance of maintaining adequate 1,25D and VDR levels to control PTHrP expression, as well as the need for further investigation of the effect of 1,25D in prostate cancer.
Highlights.
>1,25D downregulates PTHrP mRNA and protein levels. > 1,25D regulates PTHrP mRNA levels via a transcriptional pathway. > 1,25D also alters the mRNA half-life of PTHrP. > 1,25D also regulates PTHrP via a post-translational pathway.
Acknowledgements
We thank Dr. Milan Uskokovic, Hoffman La Roche, for supplying 1,25-dihydroxyvitamin D3, Dr. Zhor Bouizar, INSERM, for supplying the PTHrP promoter constructs, and Dr. David Konkel for critically editing the manuscript. This work was supported by NIH grant CA83940.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe M, Akeno N, Ohida S, Horiuchi N. Inhibitory effects of 1,25-dihydroxyvitamin D3 and 9-cis-retinoic acid on parathyroid hormone-related protein expression by oral cancer cells (HSC-3) J. Endocrinol. 1998;156:349–357. doi: 10.1677/joe.0.1560349. [DOI] [PubMed] [Google Scholar]
- Alizadeh M, Gelfman GM, Bench SR, Hjelmeland LM. Expression and splicing of FGF receptor mRNAs during ARPE-19 cell differentiation in vitro. Invest. Ophthalmol. Vis. Sci. 2000;41:2357–2362. [PubMed] [Google Scholar]
- Berruti A, Piovesan A, Torta M, Raucci CA, Gorzegno G, Paccotti P, Dogliotti L, Angeli A. Biochemical evaluation of bone turnover in cancer patients with bone metastases: Relationship with radiographic appearances and disease extension. Br. J. Cancer. 1996;73:1581–1587. doi: 10.1038/bjc.1996.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Saini MK, Shen X, Bi LX, Qiu S, Weigel NL, Falzon M. EB1089 inhibits the PTHrP-enhanced bone metastasis and xenograft growth of human prostate cancer cells. Mol. Cancer Ther. 2009;8:1787–1798. doi: 10.1158/1535-7163.MCT-09-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Blutt SE, Allegretto EA, Pike JW, Weigel NL. 1,25-Dihydroxyvitamin D3 and 9-cis-retinoic acid act synergistically to inhibit the growth of LNCaP prostate cells and cause accumulation of cells in G1. Endocrinology. 1997;138:1491–1497. doi: 10.1210/endo.138.4.5063. [DOI] [PubMed] [Google Scholar]
- Bouillon R, Bischoff-Ferrari H, Willett W. Vitamin D and Health: perspectives from mice and man. J. Bone Min. Res. 2008;23:974–979. doi: 10.1359/jbmr.080420. [DOI] [PubMed] [Google Scholar]
- Broadus AE, Stewart AF. Parathyroid hormone-related protein structure, processing and physiological actions. In: Bilezikian JP, editor. The Parathyroids. New York: Raven Press; 1994. [Google Scholar]
- Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TS, Mihatsch MJ. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum. Path. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- Cataisson C, Gordon J, Roussiére M, Abdalkhani A, Lindemann R, Dittmer J, Foley J, Bouizar Z. Ets-1 activates parathyroid hormone-related protein gene expression in tumorigenic breast epithelial cells. Mol. Cell. Endo. 2002;204:155–168. doi: 10.1016/s0303-7207(02)00298-8. [DOI] [PubMed] [Google Scholar]
- Christados S, Raval-Pandya M, Wernyj RP, Yang W. Genomic mechanisms involved in the pleiotropic actions of 1,25-dihydroxyvitamin D3. Biochem. J. 1996;316:361–371. doi: 10.1042/bj3160361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung I, Han G, Seshadri M, Gillard BM, Yu WD, Foster BA, Trump DL, Johnson CS. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009;69:967–975. doi: 10.1158/0008-5472.CAN-08-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deftos LJ. Prostate carcinoma: production of bioactive factors. Cancer. 2000;88:3002–3008. doi: 10.1002/1097-0142(20000615)88:12+<3002::aid-cncr16>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- De Luca HF. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004;80:1689S–1696S. doi: 10.1093/ajcn/80.6.1689S. [DOI] [PubMed] [Google Scholar]
- Dhawan J, Lichtler AC, Rowe DW, Farmer SR. Cell adhesion regulates pro-alpha 1(I) collagen mRNA stability and transcription in mouse fibroblasts. J Biol Chem. 1991;266:8470–8475. [PubMed] [Google Scholar]
- Dougherty KM, Blomme EAG, Koh AJ, Henderson JE, Pienta KJ, Rosol TJ, McCauley LK. Parathyroid hormone-related protein as a growth regulator of prostate carcinoma. Cancer Res. 1999;59:6015–6022. [PubMed] [Google Scholar]
- Eccles SA. Targeting key steps in metastatic tumour progression. Curr. Opin. Genetics Devel. 2005;15:77–86. doi: 10.1016/j.gde.2004.12.001. [DOI] [PubMed] [Google Scholar]
- El Abdaimi K, Papavasilou V, Rabbani SA, Rhim JS, Goltzmann D, Kremer R. Reversal of hypercalcemia with the vitamin D analogue EB1089 in a human model of squamous cancer. Cancer Res. 1999;59:3325–3328. [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nature Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Glatz JA, Heath JK, Southby J, O’Keeffe LM, Kiriyama T, Moseley JM, Martin TJ, Gillespie MT. Dexamethasone regulation of parathyroid hormone-related protein (PTHrP) expression in a squamous cancer cell line. Mol. Cell. Endo. 1994;101:295–306. doi: 10.1016/0303-7207(94)90246-1. [DOI] [PubMed] [Google Scholar]
- Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene. 2001;265:11–23. doi: 10.1016/s0378-1119(01)00350-x. [DOI] [PubMed] [Google Scholar]
- Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, Vessella R, Corey E, Padalecki S, Suva L, Chirgwin JM. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006;12(20 Suppl.):6213s–6216s. doi: 10.1158/1078-0432.CCR-06-1007. [DOI] [PubMed] [Google Scholar]
- Hall CL, Kang S, MacDougals OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J. Cell. Biochem. 2006;97:661–672. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- Hall CL, Bafico A, Dia J, Aaronson SA, Keller ET. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65:7554–7560. doi: 10.1158/0008-5472.CAN-05-1317. [DOI] [PubMed] [Google Scholar]
- Hamzaoui H, Risk-Rabin M, Gordon J, Offutt C, Bertherat J, Bouizar Z. PTHrP P3 promoter activity in breast cancer cell lines: Role of Ets1 and CBP (CREB binding protein) Mol. Cell. Endo. 2007;268:75–84. doi: 10.1016/j.mce.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2005;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Haq M, Kremer R, Goltzman D, Rabbani SA. A vitamin D analogue (EB1089) inhibits parathyroid hormone-related peptide production and prevents the development of malignancy-associated hypercalcemia in vivo. J. Clin. Invest. 1993;91:2416–2422. doi: 10.1172/JCI116475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holick MF. Vitamin D: a millennium perspective. J. Cell. Biochem. 2003;88:296–307. doi: 10.1002/jcb.10338. [DOI] [PubMed] [Google Scholar]
- Holt EH, Vasavada RC, Bander NH, Broadus AE, Philbrick WM. Region-specific methylation of the parathyroid hormone-related peptide gene determines its expression in human renal carcinoma cell lines. J. Biol. Chem. 1993;268:20639–20645. [PubMed] [Google Scholar]
- Ignowski JM, David V, Schaffer DV. Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnology and Bioengineering. 2004;86:827–834. doi: 10.1002/bit.20059. [DOI] [PubMed] [Google Scholar]
- Iwamura M, Wu R, Abrahamsson PA, di Sant’Agnese PA, Cockett AT, Deftos LJ. Parathyroid hormone-related protein is expressed by prostatic neuroendocrine cells. Urology. 1994;43:667–674. doi: 10.1016/0090-4295(94)90182-1. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics. CA Cancer J. Clin. 2007;57:1873–1882. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Jensen SS, Madsen MW, Lukas J, Binderup L, Bartek J. Inhibitory effects of 1α,25-dihydroxyvitmain D3 on the G1-S phase-controlling machinery. Mol. Endo. 2001;15:1370–1380. doi: 10.1210/mend.15.8.0673. [DOI] [PubMed] [Google Scholar]
- Kallay E, Pietschmann P, Toyokuni S, Bajna E, Hahn P, Mazzucco K, Bieglmayer C, Kato S, Cross HS. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis. 2001;22:1429–1435. doi: 10.1093/carcin/22.9.1429. [DOI] [PubMed] [Google Scholar]
- Karmali R, Nus-de-Wolf N, Beyer I, Hendy GN, Bergmann P. 1,25-Dihydroxyvitmain D3 inhibits parathyroid hormone-related peptide mRNA expression in fetal rat long bones in culture. In Vitro Cell. Dev. Biol. 1999;35:296–298. doi: 10.1007/s11626-999-0074-9. [DOI] [PubMed] [Google Scholar]
- Kasper S, Sheppard PC, Yan Y, Pettigrew N, Borowsky AD, Prins GS, Dodd JC, Duckworth RJ, Matusik RJ. Development, progression and androgen-dependence of prostate tumors in probasin-large-T-antigen transgenic mice: a model for prostate cancer. Lab Invest. 1998;78:319–333. [PubMed] [Google Scholar]
- Keller ET, Zhang J, Cooper CR, Smith PC, McCauley LK, Pienta KJ, Taichman RS. Prostate carcinoma skeletal metastases: cross-talk between tumor and bone. Cancer Metastasis Rev. 2001;20:333–349. doi: 10.1023/a:1015599831232. [DOI] [PubMed] [Google Scholar]
- Kramer S, Reynolds FJ, Jr, Castillo M, Valenzuela DM, Thorikay M, Sorvillo JM. Immunological identification and distribution of parathyroid hormone-like protein polypeptides in normal and malignant tissues. Endocrinol. 1991;128:1927–1937. doi: 10.1210/endo-128-4-1927. [DOI] [PubMed] [Google Scholar]
- Li P, Li C, Zhao X, Zhang X, Nicosia SV, Bai W. p27Kip1 stabilization and G1 arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. J. Biol. Chem. 2004;279:25260–25267. doi: 10.1074/jbc.M311052200. [DOI] [PubMed] [Google Scholar]
- Luparello C, Schilling T, Cirincione R, Pucci-Minafra I. Extracellular matrix regulation of PTHrP and PTH/PTHrP receptor in a human breast cancer cell line. FEBS Letters. 1999;463:265–269. doi: 10.1016/s0014-5793(99)01635-x. [DOI] [PubMed] [Google Scholar]
- Mangin M, Ikeda K, Dreyer BE, Broadus AE. Identification of an upstream promoter of the human parathyroid hormone-related peptide gene. Mol. Endocrinol. 1990;4:851–858. doi: 10.1210/mend-4-6-851. [DOI] [PubMed] [Google Scholar]
- Mantell DJ, Ownes PE, Bundred NJ, Mawer EB, Canfiels AE. 1α,25-Dihydroxyvitamin D3 inhibits angiogenesis in vitro and in vivo. Circ. Res. 2000;87:214–220. doi: 10.1161/01.res.87.3.214. [DOI] [PubMed] [Google Scholar]
- Massfelder T, Lang H, Schordan E, Lindner V, Rothhut S, Welsch S, Simon-Assmann P, Barthelmebs M, Jacqmin D, Helwig J-J. Parathyroid hormone-related protein is an essential growth factor for human clear cell renal carcinoma and a target for the von Hippel-Lindau tumor suppressor gene. Cancer Res. 2004;64:180–188. doi: 10.1158/0008-5472.can-03-1968. [DOI] [PubMed] [Google Scholar]
- Masuyama H, MacDonald PN. Proteasome-mediated degradation of the vitamin D receptor (VDR) and a putative role for SUG1 interaction with the AF-2 domain of VDR. J. Cell. Biochem. 1998;71:429–440. [PubMed] [Google Scholar]
- Mathiasen IS, Colston KW, Binderup L. EB1089: a novel vitamin D analogue, has strong antiproliferative and differentiation inducing effects of cancer cells. J. Steroid Biochem. Mol. Biol. 1993;46:365–371. doi: 10.1016/0960-0760(93)90226-m. [DOI] [PubMed] [Google Scholar]
- Meerovitch K, Wing S, Goltzman D. Preproparathryoid hormone-related protein, a secreted peptide, is a substrate for the ubiquitin proteolytic system. J. Biol. Chem. 1997;272:6706–6713. doi: 10.1074/jbc.272.10.6706. 1997. [DOI] [PubMed] [Google Scholar]
- Meerovitch K, Wing S, Goltzman D. Preproparathyroid hormone-related protein is associated with the chaperone protein BiP and undergoes proteasome-mediated degradation. J. Biol. Chem. 1998;273:21025–21030. doi: 10.1074/jbc.273.33.21025. [DOI] [PubMed] [Google Scholar]
- Misquitta CM, Mwanjewe J, Nie L, Grover AK. Sarcoplasmic reticulum Ca2+ pump mRNA stability in cardiac and smooth muscle: role of the 3’-untransloated region. Am. J. Physiol. Cell Physiol. 2002;283:C560–C568. doi: 10.1152/ajpcell.00527.2001. [DOI] [PubMed] [Google Scholar]
- Mordan-McCombs S, Brown T, Wang W-LW, Gaupel A-C, Welsh J, Tenniswood M. Tumor progression in the LPB-Tag transgenic model of prostate cancer is altered by vitamin D receptor and serum testosterone levels. J. Steroid Biochem. Mol. Biol. 2010;121:368–371. doi: 10.1016/j.jsbmb.2010.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishishita T, Okazaki T, Ishikawa T, Igarashi T, Hata K, Ogata E, Fujita T. A negative vitamin D response DNA element in the human parathyroid hormone-related peptide gene binds to vitamin D receptor along with Ku antigen to mediate negative gene regulation by vitamin D. J. Biol .Chem. 1998;273:10901–10907. doi: 10.1074/jbc.273.18.10901. [DOI] [PubMed] [Google Scholar]
- Nothnick WB, Healy C, Hong X. Steroidal regulation of uterine miRNAs is associated with modulation of the miRNA biogenesis components Exportin-5 and Dicer1. Endocrinology. 2010;37:265–273. doi: 10.1007/s12020-009-9293-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol. Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Peehl DM, Feldman D. The role of vitamin D and retinoids in controlling prostate cancer progression. Endocrine-Related Cancer. 2003;10:131–140. doi: 10.1677/erc.0.0100131. [DOI] [PubMed] [Google Scholar]
- Polek TC, Weigel NL. Vitamin D and Prostate cancer. J. Andrology. 2002;23:9–17. doi: 10.1002/j.1939-4640.2002.tb02596.x. [DOI] [PubMed] [Google Scholar]
- Rana A, Chisholm GD, Khan M, Sekharjit SS, Merrick MV, Ellton RA. Patterns of bone metastasis and their prognostic significance in patients with carcinoma of the prostate. Br. J. Urol. 1993;72:933–936. doi: 10.1111/j.1464-410x.1993.tb16301.x. [DOI] [PubMed] [Google Scholar]
- Schordan E, Welsch S, Rothhut S, Lambert A, Barthelmers M, Helwig J-J, Massfelder T. Role of parathyroid hormone–related protein in the regulation of stretch-induced renal vascular smooth muscle cell proliferation. J. Amer. Soc. Nephrol. 2004;15:3016–3025. doi: 10.1097/01.ASN.0000145529.19135.EF. [DOI] [PubMed] [Google Scholar]
- Schwartz GG. Vitamin D and intervention trials in prostate cancer: from theory to therapy. Ann. Epidemiol. 2009;19:96–102. doi: 10.1016/j.annepidem.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Sellers RB, Capen CC, Rosol TJ. Messenger RNA stability of parathyroid hormone-related protein regulated by transforming growth factor-beta 1. Mol. Cell. Endo. 2002;25:37–46. doi: 10.1016/s0303-7207(01)00752-3. [DOI] [PubMed] [Google Scholar]
- Sellers RS, Luchin AI, Richard V, Brena RM, Lima D, Rosol TJ. Alternative splicing of parathyroid hormone-related protein mRNA: expression and stability. J. Mol. Endo. 2004;33:227–241. doi: 10.1677/jme.0.0330227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Rychahou PG, Evers BM, Falzon M. PTHrP increases xenograft growth and promotes integrin α6β4 expression and Akt activation in colon cancer. Cancer Letters. 2007;258:241–252. doi: 10.1016/j.canlet.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Mula RVR, Li J, Weigel NL, Falzon M. PTHrP contributes to the anti-proliferative and integrin α6β4-regulating effects of 1,25-dihydroxyvitamin D3. Steroids. 2007a;72:930–938. doi: 10.1016/j.steroids.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southby J, O’Keefe LM, Martin TJ, Gillespie MT. Alternative promoter usage and mRNA splicing pathways for parathyroid hormone-related protein in normal tissues and tumours. Brit. J. Cancer. 1995;72:702–707. doi: 10.1038/bjc.1995.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart LV, Weigel NL. Vitamin D and prostate cancer. Exp. Biol. Med. (Maywood) 2004;229:277–284. doi: 10.1177/153537020422900401. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kelleher AD. Transcriptional regulation by promoter targeted RNAs. Current Topics Med. Chem. 2009;9:1079–1087. doi: 10.2174/156802609789630875. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Ishii C, Ryoji M. Role of distal upstream sequence in vitamin D-induced expression in human CYP24 gene. Biochem. Biophys. Res. Commun. 2007;358:259–265. doi: 10.1016/j.bbrc.2007.04.103. [DOI] [PubMed] [Google Scholar]
- Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- Tovar Sepulveda VA, Falzon M. Parathyroid hormone-related protein enhances PC-3 prostate cancer cell growth via both autocrine/paracrine and intracrine pathways. Reg. Pept. 2002;105:109–120. doi: 10.1016/s0167-0115(02)00007-1. [DOI] [PubMed] [Google Scholar]
- Tovar Sepulveda VA, Falzon M. Prostate cancer cell type-specific regulation of the human PTHrP gene via a negative VDRE. Mol. Cell. Endocrinol. 2003;204:51–64. doi: 10.1016/s0303-7207(03)00148-5. [DOI] [PubMed] [Google Scholar]
- Tovar Sepulveda VA, Weigel NL, Falzon M. Prostate cancer cell type-specific involvement of the VDR and RXR in regulation of the human PTHrP gene via a negative VDRE. Steroids. 2006;71:102–115. doi: 10.1016/j.steroids.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Vasavada JJ, Broadus AE, Philbrick WM. Identification and characterization of a GC-rich promoter of the human parathyroid hormone-related peptide gene. Mol. Endocrinol. 1993;7:273–282. doi: 10.1210/mend.7.2.8469240. [DOI] [PubMed] [Google Scholar]
- Vinholes J, Coleman R, Eastell R. Effects of bone metastases on bone metabolism: Implications for diagnosis, imaging and assessment of response to cancer treatment. Cancer Treat. Res. 1996;22:289–331. doi: 10.1016/s0305-7372(96)90021-3. [DOI] [PubMed] [Google Scholar]
- Washington MN, Kim J-S, Weigel NL. 1α25-dihydroxyvitamin D3 inhibits C4-2 prostate cancer cell growth via a retinoblastoma protein (Rb)-independent G1 arrest. Prostate. 2011;71:98–110. doi: 10.1002/pros.21226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J. Vitamin D and breast cancer: insights from animal models. Amer. J. Clin. Nutr. 2004;80:1721S–1724S. doi: 10.1093/ajcn/80.6.1721S. [DOI] [PubMed] [Google Scholar]
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int. J. Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- Zhuang S-H, Burnstein Antiproliferative effect of 1α,25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin-dependent kinase 2 activity and persistent G1 accumulation. Endocrinology. 1998;139:1197–1207. doi: 10.1210/endo.139.3.5770. [DOI] [PubMed] [Google Scholar]
- Zhuang SH, Schwartz GG, Cameron D, Burnstein KL. Vitamin D receptor content and transcriptional activity do not fully predict antiproliferative effects of vitamin D in human prostate cancer cell lines. Mol. Cell. Endo. 1997;126:83–90. doi: 10.1016/s0303-7207(96)03974-3. [DOI] [PubMed] [Google Scholar]