

Abstract
Alkaptonuria is a rare, autosomal recessive disorder of tyrosine degradation due to deficiency of the third enzyme in the catabolic pathway. As a result, homogentisic acid (HGA) accumulates and is excreted in gram quantities in the urine, which turns dark upon alkalization. The first symptoms, occurring in early adulthood, involve a painful, progressively debilitating arthritis of the spine and large joints. Cardiac valvular disease and renal and prostate stones occur later. Previously suggested therapies have failed to show benefit, and management remains symptomatic. Nitisinone, a potent inhibitor of the second enzyme in the tyrosine catabolic pathway, is considered a potential therapy; proof-of-principle studies showed 95% reduction in urinary HGA. Based on those findings, a prospective, randomized clinical trial was initiated in 2005 to evaluate 40 patients over a 36-month period. The primary outcome parameter was hip total range of motion with measures of musculoskeletal function serving as secondary parameters. Biochemically, this study consistently demonstrated 95% reduction of HGA in urine and plasma over the course of 3 years. Clinically, primary and secondary parameters did not prove benefit from the medication. Side effects were infrequent. This trial illustrates the remarkable tolerability of nitisinone, its biochemical efficacy, and the need to investigate its use in younger individuals prior to development of debilitating arthritis.
Keywords: Alkaptonuria, Ochronosis, Nitisinone, Homogentisic acid
Introduction
Sir Archibald Edward Garrod first described alkaptonuria (AKU, MIM 203500) in 1902 and coined the term “inborn error of metabolism.”[1-3] Today, alkaptonuria is widely recognized as an autosomal recessive disorder that results from deficiency of homogentisic acid dioxygenase, the third enzyme in the tyrosine degradation pathway (Fig. 1); consequently, homogentisic acid (HGA) accumulates in urine, plasma, cartilage and connective tissues.[4-6] In fact, alkaptonuria is named for the massive amount of HGA that accumulates in the urine, causing it to darken upon standing or upon exposure to alkaline conditions. Through ancillary pathways, HGA is oxidized to benzoquinones, which polymerize and bind to cartilage and connective tissues.[7-11] This causes an early-onset destructive arthritis of the spine and large joints, as well as valvular heart disease.[5, 12] The orthopedic complications of alkaptonuria begin with vertebral disk narrowing in the thirties and progress to large joint destruction in the forties and fifties.[13] Kidney stones and, in men prostate stones also occur.[5, 13]
Figure 1. Tyrosine catabolic pathway.
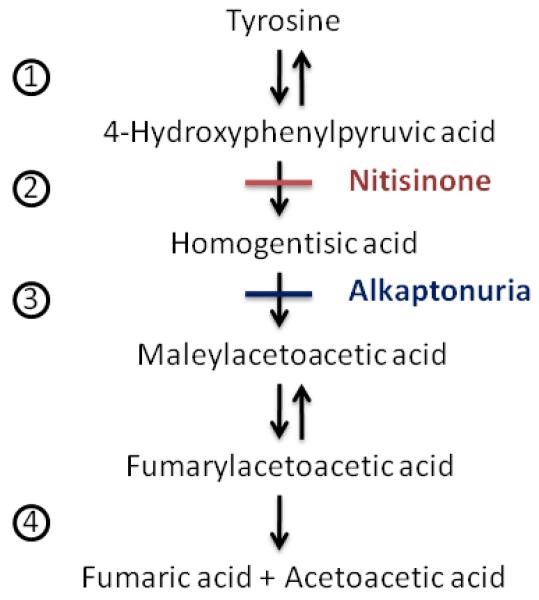
Designated enzymes: 1 = Tyrosine aminotransferase, 2 = 4-Hydroxyphenylpyruvic acid dioxygenase, 3 = Homogentisate 1,2-dioxygenase, 4 = Fumarylacetoacetic acid hydrolase. Nitisinone inhibits the 2nd enzyme, 4-hydroxyphenylpyruvic acid dioxygenase. Deficiency of the 3rd enzyme, homogentisate 1,2-dioxygenase causes alkaptonuria.
While the clinical features of alkaptonuria continue to be elucidated, there has been little advance in therapy; the mainstay of treatment remains symptomatic. In 1998, nitisinone (NTBC; 2-(2-nitro-4-fluromethylbenzoyl)-1, 3-cyclohexanedione), a potent inhibitor of para-hydroxyphenylpyruvic acid oxygenase, the second enzyme in the tyrosine catabolic pathway, was suggested as a potential treatment to block production of the offending HGA molecule (Fig. 1).[14] Indeed, after receiving new drug approval for the treatment of hereditary tyrosinemia in 2002, nitisinone (OrfadinR) was given to two alkaptonuria patients to establish proof of this priniciple.[13] In these two women, urinary HGA load was reduced by 95%, preparing the way for additional clinical trials. A second study established appropriate dosing guidelines and evaluated safety.[15] These studies provided the foundation for a three-year trial designed to confirm biochemical efficacy, investigate clinical outcomes, and further evaluate the safety of nitisinone in alkaptonuria patients.
1. Methods
2.1 Patient evaluations
Forty patients were enrolled in a prospective, randomized, IRB-approved protocol (clinicaltrials.gov # NCT00107783) between April, 2005 and April, 2009. Written, informed consent was obtained. Study length was 36 months from the time the fortieth patient was enrolled; since the enrollment period was 11 months, some patients participated for nearly 4 years. Twenty patients were randomized to the control group, receiving neither placebo nor surrogate medication. Twenty patients were randomized to the treatment group and received nitisinone 2 mg orally, once daily. Nitisinone (Orfadin®) is manufactured by Swedish Orphan Pharmaceuticals and was supplied to the patients through IND #71,780. The patients could not be masked to their treatment because their urine color revealed whether or not they were receiving nitisinone.
Participants were between the ages of 38 and 68 years at the study onset. Individuals were excluded if both hips had been replaced or they had significant, uncontrolled medical issues, a history of substance abuse, or psychiatric disease that would interfere with patient compliance.
All patients were evaluated initially and returned to the National Institutes of Health Clinical Center every four months for follow-up evaluation. Comprehensive blood and urine testing was performed at the initial visit and all subsequent visits. Urine studies included microscopic urinalysis and 24-hour collections for quantitative homogentisic acid, creatinine clearance, urine protein, glucose, minerals and electrolytes. Blood studies included plasma amino acids, plasma homogentisic acid and nitisinone levels, complete blood counts with differential and platelets, chemistry-20 panel, lipid panel, prothrombin and partial thromboplastin times, thyroid function testing, erythrocyte sedimentation rate, high sensitivity CRP, sex hormones, osteocalcin, calcitonin, and parathyroid hormone.
Initial evaluation included plain radiographs of the spine, chest, shoulders, hips and knees. A renal ultrasound and non-contrast CT of the abdomen and pelvis was performed to detect genitourinary stones. A baseline MRI/MRA of the brain was obtained. Bone densitometry using DXA (Hologic QDR4500, Bedford, MA) was performed to measure the central vertebral body bone mineral density. The values were compared to the Hologic reference data. An electrocardiogram and transthoracic echocardiogram assessed baseline cardiac function and extent of valvular disease. Baseline pulmonary function testing was performed. Plain radiographs, renal ultrasound, CT of the abdomen and pelvis, electrocardiogram and echocardiogram were repeated annually to monitor for adverse events. Magnetic resonance imaging and MRA of the brain, bone densitometry, and pulmonary function tests were repeated at the 3 year visit also to monitor for safety. These parameters were not primary or secondary outcome parameters. Echocardiograms and bone densitometry results are reported in the exploratory outcomes section.
An extensive musculoskeletal evaluation was performed at the initial visit and each subsequent visit. Evaluations included measurement of joint total range of motion (ROM) using goniometry of the shoulders, hips, and knees, Schober’s test of spine flexion, functional reach assessment, timed get up and go measurement and 6 minute walk tests. Full ophthalmologic evaluations were completed at each visit. Pain management was reviewed by the pain and palliative care service during each admission and was tailored to each individual’s needs.
Upon enrollment, and annually thereafter, patients recorded three-day food diaries that were reviewed by a registered dietician, coded, and analyzed using Nutrition Data System for Research software version 2008 developed by the Nutrition Coordinating Center (NCC), University of Minnesota, to quantify total protein, phenylalanine, and tyrosine intake. Averages of each category were calculated for the control and nitisinone treatment groups at baseline. Collective averages from all subsequent visits for each group were also calculated.
A major concern related to nitisinone use is the occurrence of a corneal keratopathy. Children with hepatorenal tyrosinemia treated with nitisinone are routinely placed on a protein-restricted diet in an attempt to prevent corneal changes; compliance is variable. For our protocol, participants were allowed to continue their regular diet. There was no protein restriction for either the control or nitisinone treated groups, since adult compliance with such a diet was considered unlikely, published reports show no correlation between plasma tyrosine concentrations and the occurrence of corneal changes,[16-18] and every case of nitisinone-related keratopathy was reversible upon stopping nitisinone treatment.[16, 17]
2.2 Outcome parameters and statistical considerations
The primary outcome parameter was total (internal + external) hip range of motion in the worse hip, i.e., the hip with the greatest loss of rotation at baseline. Secondary outcome parameters included Schober’s test of spinal flexion, functional reach, timed get up and go, and 6 minute walk tests.
Primary and secondary data were analyzed using repeated measures with random coefficients. The model contains terms for treatment*time effects, along with baseline age*time and baseline worse hip rotation*time effects, complete with random intercepts and slopes. One individual died one month after enrolling in the study, and two individuals had their second hip replaced during the course of the study. For statistical considerations, hip range of motion following death or hip replacement was recorded as zero. Two individuals receiving nitisinone were removed from treatment during the course of the study. Both individuals’ data were analyzed in the treatment group under an intent-to-treat approach.
2.3 Biochemical evaluations
Urine HGA was measured using the assay described by Lustberg et al. in 1971.[19] An aliquot from a 24-hour urine collection was sterile-filtered and stored at −20°C for several days before being ultra-filtered using the 3KMWCO VIVASPIN 500. The ultra-filtrate was used for both creatinine and HGA assays. For HGA determinations, urine samples from untreated patients were then diluted 1:100 using 0.5% acetic acid. Urine from patients being treated with nitisinone was not diluted. Creatinine was measured using the Creatinine Reagent Set from TECO Diagnostics. This assay uses picric acid and an alkaline buffer reagent similar to the Jaffe method. For patients not receiving nitisinone, a correction was made to compensate for the interference of HGA in the assay.
Plasma amino acids were quantitated in our lab using an ion-exchange analyzer with post-column Ninhydrin derivatization using the Biochrom 30. Additional samples of plasma amino acids were also quantified using ion-exchange chromatography by Mayo Laboratory in Rochester, MN. Plasma HGA was measured by AAI Pharma in Germany using HPLC-UV. Twenty microliters of phosphoric acid was added to 190 μl of plasma, and samples were stored at −20°C until shipment to AAI Pharma.
Plasma nitisinone was measured by Children’s Hospital and Regional Medical Center in Seattle, Washington using LC/MS/MS.
2. Results
3.1 Baseline data
At baseline, the untreated and nitisinone groups were well matched for age, functional reach and laboratory parameters (Table 1). Blood and urine testing was performed in duplicate on the initial visit. Averages for each parameter were used for Table 1. P-values were calculated using Wilcoxon rank sums testing. Normal values listed in Table 1 for primary and secondary parameters are taken from referenced literature.[20-26] While not statistically significant, the nitisinone group had worse hip range of motion. Additionally, the nitisinone group had significantly worse 6-minute walk times, get up and go times, and Schober scores.
Table 1. Baseline parameters for control and nitisinone-treated alkaptonuria patients.
| Control | Nitisinone-treated | P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Normal Values | N | Mean | SD | (Range) | N | Mean | SD | (Range) | ||
| Male/Female | 15/5 | 12/8 | ||||||||
| Age | 20 | 51.3 | 6.5 | (40-63) | 20 | 52.2 | 7.9 | (38-68) | 0.72 | |
| ROM-worse hip (°)a | 90 | 20 | 46.4 | 16.1 | (19-90) | 20 | 39.0 | 12.7 | (18-72) | 0.13 |
| ROM-better hip (°) | 90 | 19 | 56.1 | 12.6 | (40-90) | 19 | 51.0 | 11.4 | (33-77) | 0.23 |
| 6MW (ft)b | 1621 (F) 1890 (M) |
20 | 1578 | 205 | (1255-1999) | 20 | 1336 | 369 | (642-1929) | 0.04 |
| Get up & Go (s)b | <10 freely mobile <20 mostly independent |
20 | 7.6 | 1.4 | (5.1-10.7) | 20 | 9.7 | 4.1 | (5.5-20.6) | 0.06 |
| Functional Reach (in)b | <6 predictive of falls >10 inches okay |
20 | 10.2 | 2.9 | (5-15) | 20 | 9.0 | 3.1 | (0.6-12.7) | 0.39 |
| Schober (cm)b | ≥15 | 20 | 11.4 | 1.04 | (10.2-13.5) | 20 | 10.9 | 0.73 | (10-13) | 0.08 |
| Abnormal Aortic Valvec | 17/20 | 14/20 | ||||||||
| Abnormal MRId | 8/20 | 7/18 | ||||||||
| Hgb (g/dl) | 11.1-15.0 (F) 12.7-16.7 (M) |
20 | 14.4 | 1.1 | (11.9-16.1) | 20 | 14.0 | 1.1 | (11.6-15.9) | 0.29 |
| WBC (k/μL) | 3.30-9.60 | 20 | 5.8 | 1.2 | (4.3-8.8) | 20 | 5.8 | 1.4 | (3.0-8.6) | 0.90 |
| ALT (U/L) | 6-41 | 20 | 26.8 | 10.1 | (13-52) | 20 | 24.3 | 13.0 | (10-70.5) | 0.23 |
| Plasma Tyr (μM) | 34-112 | 19 | 58.2 | 8.9 | (46.5-85.5) | 19 | 61.4 | 16.3 | (41-108) | 0.83 |
| Urine HGA (mg/d) | 20-30 | 20 | 5979 | 1840 | (2250-9294) | 20 | 5383 | 2128 | (835-9600) | 0.36 |
Primary outcome parameter
Secondary outcome parameter
Valvular changes including calcification, thickening of valve leaflets, sclerosis or stenosis
Nonspecific white matter changes; not performed on all individuals
3.2 Dropouts
Seven individuals failed to complete the study (Table 2). One patient in the treatment group developed atrial fibrillation within two weeks of enrolling in the study. The study medication was discontinued; the patient died one month later due to myocardial infarction. Two individuals in the control group discontinued participation in the study for personal reasons. Two individuals, one in the control and one in the treatment group, had their second hip replaced during the course of the study. Both individuals returned every four months for the duration of the study. One patient in the treatment group developed corneal irritation with branching subepithelial opacities 6 weeks after enrollment in the study. Upon stopping the nitisinone, the patient’s symptoms improved and the opacities resolved. The study medication was ultimately discontinued, but the patient continued to return every four months under an intent-to-treat design. Another patient in the treatment group had recurrent elevations of liver transaminases, initially related to gall stones, then due to multiple medications. Ultimately, the etiology could not be determined and the nitisinone was stopped. The patient also continued to return every four months under an intent-to-treat design.
Table 2. Subjects who did not complete the study.
| Groups | ||||
|---|---|---|---|---|
| Reason | Control |
Months on Study |
Nitisinone |
Months on Study |
| Atrial Fibrillation | 1 | 0.5 | ||
| Second hip replacement |
1 | 3 | 1 | 12 |
| Corneal damage | 1 | 2 | ||
| Elevated transaminases |
1 | 28 | ||
| Personal | 2 | 16, 24 | ||
3.3 Compliance
There were 6 missed visits during the course of 36 months. Four patients in the untreated group missed a total of 5 visits and 1 patient from the treatment group missed one visit. For the untreated group, one visit was missed due to recovery from a joint replacement; the other missed visits were because of inability to secure time off work. The one individual with two missed visits withdrew from the study for personal reasons. The missed visit from the treatment cohort was due to a death in the family.
At each visit, patients were queried about compliance with taking the drug. Patient-reported compliance was exceptional; decreased urinary HGA and elevated plasma tyrosine levels, documented on all visits, confirmed compliance. The only exceptions were the two patients removed from nitisinone due to serious adverse events and one patient who electively stopped the medication while taking prednisone and then resumed taking nitisinone on subsequent visits.
Nitisinone concentrations were measured on two occasions for each patient taking nitisinone. Levels were remarkably consistent, suggesting that steady state levels were achieved (Table 3). The mean ± SEM nitisinone concentration resulting from a 2 mg/day dose was 2.22 ± 0.09 μM. In comparison, individuals with hereditary tyrosinemia on nitisinone have plasma nitisinone levels of 40-60 μM.[27]
Table 3. Plasma nitisinone levels (μmol/L).
| Patient Number | Sample 1 | Sample 2 |
|---|---|---|
| 2 | 2.07 | 2.08 |
| 101 | 1.73 | 1.55 |
| 103 | 2.18 | 2.91 |
| 106 | 2.10 | 2.23 |
| 107 | 2.70 | 3.02 |
| 110 | 2.93 | 2.53 |
| 112 | 2.57 | 2.50 |
| 113 | 2.43 | 1.97 |
| 118 | 1.41 | 1.34 |
| 120 | 1.62 | 1.72 |
| 122 | 2.70 | 2.30 |
| 123 | 2.14 | 1.47 |
| 126 | 2.15 | 1.99 |
| 128 | 3.57 | 3.05 |
| 129 | 1.32 | <0.07 |
| 132 | 2.21 | 2.52 |
| 135 | 2.03 | 1.87 |
| 136 | 2.51 | 2.74 |
| 137 | <0.07 | 1.60 |
| 124 | <0.07 | |
| 102 | <0.07 |
Plasma nitisinone concentrations were measured on two separate admissions in 19 nitisinone-treated patients and in 2 untreated control patients (#124 and #102). Patient #129 had the second sample measured when removed from nitisinone; patient #137 had the first sample assayed prior to starting nitisinone.
3.4 Biochemical efficacy
Urine HGA excretion (Fig. 2)
Figure 2. Daily urinary HGA excretion in control and nitisinone-treated alkaptonuria patients over the course of the trial.
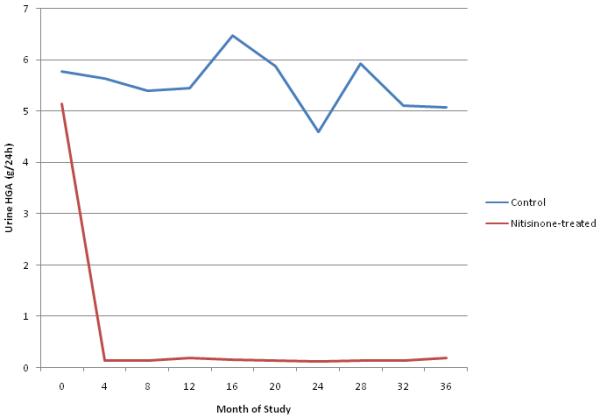
Mean urinary excretion of HGA measured in grams over a 24 hour period throughout the 36 month period. The red line gives the mean for the cohort treated with nitisinone. The blue line represents the mean for the control group.
Mean HGA excretion for the control group was 5.80 grams per day at baseline and persisted at approximately that level for the duration of the study (range, 4.60-6.47 grams per day). Mean urinary HGA excretion in the nitisinone group decreased from 5.1 grams per day at the start of the trial to 125 mg per day by the four-month visit, and ranged from 113 mg to 203 mg per day for the remainder of the trial. Overall, nitisinone reduced urinary HGA excretion by >95%.
Plasma HGA levels
The mean HGA concentration in 38 plasma samples from 33 alkaptonuria patients when they were not receiving nitisinone was 5.74 μg/ml (range 3.15 – 10.5 μg/ml; normal, <0.25). Each patient treated with nitisinone had plasma HGA measured on three separate occasions while receiving nitisinone therapy. Thirty-four of the 55 samples were below the level of quantitation (0.250 μg/ml); a value of 0.250 was assigned to these samples. The mean HGA concentration in plasmas of nitisinone-treated patients was 0.306 μg/ml (range <0.250 – 0.815 μg/ml), meaning that nitisinone had decreased the plasma HGA by 95%, on average. Plasma HGA levels correlated with urinary HGA excretion.
3.5 Clinical efficacy
Primary outcome measure
The primary outcome parameter, chosen a priori, was change in total range of motion in the worse hip, evaluated by repeated measures analysis with variable coefficients. By this analysis, individuals in the control group had an average decrease 0.22°/year greater than that of the nitisinone group. This difference was not statistically significant. The analysis was repeated after removing data from the individual who died and the two individuals who had their second hip replaced during the trial. In this case, the worse hip rotation decreased by 0.31°/year more in the control group compared with the nitisinone group. This was also not statistically significant.
Total hip range of motion improved, worsened, or remained the same for some individuals in both the control and nitisinone groups over the 3-year trial period (Figs. 3 & 4). In comparing only the baseline and 3-year range of motion values for patients with at least one native hip remaining, the control group had an average decline of 0.37°/year. (Two of the control patients had the same total range of motion in their two hips at the start of the trial; using the hip with the greatest decline, the average decline for the control group was 1.23°/year.) The nitisinone group had an average gain of 2.0°/year. However, range of motion in each individual varied greatly from visit to visit.
Figure 3. Worse hip range of motion over time in untreated alkaptonuria patients.
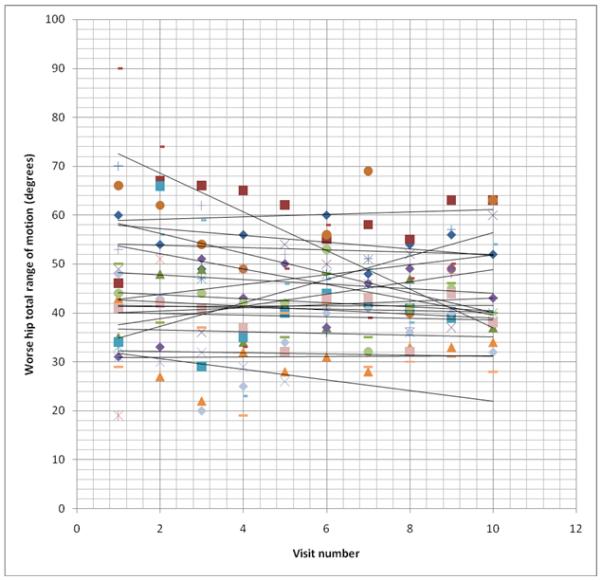
Each set of symbols represents one patient’s total hip range of motion at each visit over the 36 month study period. Best fit lines are drawn depicting the trend of the total range of motion over time. For the purpose of this figure, individuals who died or had their second hip replaced have their data truncated at the time of the event, rather than going to zero as used in the statistical analysis.
Figure 4. Worse hip range of motion over time in nitisinone-treated alkaptonuria patients.
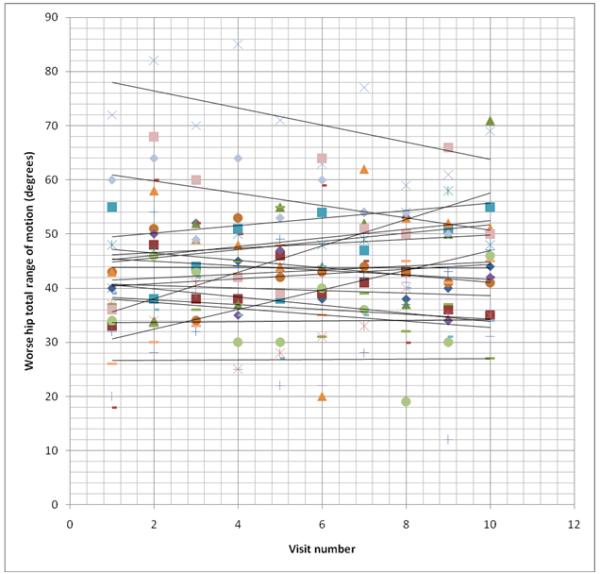
Each set of symbols represents one patient’s total hip range of motion at each visit over the 36 month study period. Best fit lines are drawn depicting the trend of the total range of motion over time. For the purpose of this figure, individuals who died or had their second hip replaced have their data truncated at the time of the event, rather than going to zero as used in the statistical analysis.
Secondary outcome measures
Comparison of the baseline and final values for Schober’s measurement of spinal flexion, 6-minute walk times, timed get up and go, and functional reach yielded no significant differences between the control and the nitisinone groups (Table 4). There were also no significant differences between the groups when repeated measures analyses with random coefficients were employed (data not shown).
Table 4. Secondary outcome parameters.
| Test | Controla | Nitisinoneb |
|---|---|---|
| Schober’s (cm) | ||
| Initial | 11.42 | 10.85 |
| Final | 11.36 | 10.97 |
| Change | −0.06 | +0.12 |
| 6-Minute Walk (ft) | ||
| Initial | 1578 | 1336 |
| Final | 1600 | 1505 |
| Change | +28 | +169 |
| Timed Get Up & Go (s) | ||
| Initial | 7.55 | 9.7 |
| Final | 7.02 | 8.38 |
| Change | −0.53 | −1.32 |
| Functional Reach (cm) | ||
| Initial | 10.2 | 8.99 |
| Final | 9.0 | 7.12 |
| Change | −1.20 | −1.87 |
Results listed are the mean for each group
Initial N=20, Final N=19
Initial N=20, Final N=18
Exploratory outcome measures
As a measure of aortic valvular disease, peak aortic velocity was recorded in individuals at the start of the study and annually thereafter. Aortic stenosis corresponded to a peak velocity ≥2.5 m/s (normal, <2.0 m/s). Aortic sclerosis was defined as thickened valve leaflets with a peak velocity between 2.0 and 2.5 m/s. At the start of the study, one of 19 individuals evaluated in the nitisinone group had aortic stenosis, with a peak velocity of 3.7 m/s (Table 5). Two additional individuals had aortic sclerosis with peak velocities of 2.2 and 2.4 m/s. By the conclusion of the study, all 3 patients maintained their initial classification; only the patient with established aortic stenosis showed significant progression. In the control group, 3 of 20 subjects at the start of the study had aortic stenosis with peak velocities of 2.7, 2.6 and 2.5 m/s. Two individuals were borderline with peak velocities of 2.1 and 2.0 m/s. At the end of the study, 4 of the 18 patients remaining on study were diagnosed as having aortic stenosis (velocities 3.6, 3.0, 2.8, and 2.6 m/s) and 6 patients had aortic sclerosis (peak velocity 2.0-2.5 m/s).
Table 5. Peak aortic velocity at study initiation and at 36 months.
| Control Group | Nitisinone-Treated Group | ||||
|---|---|---|---|---|---|
| Patient # | Baseline Velocity (m/s) |
Final Velocity (m/s) |
Patient # | Baseline Velocity (m/s) |
Final Velocity (m/s) |
| 102 | 1.6 | 1.4 | 137 | 1.1 | 1.3 |
| 117 | 1.5 | 1.9 | 126 | 1.9 | 1.9 |
| 130 | 2.1 | 2.6 | 107 | 1.8 | 1.7 |
| 133 | 1.8 | 2.1 | 135 | 1.3 | 1.2 |
| 105 | 2.0 | 1.8 | 123 | 1.0 | 1.4 |
| 1 | 1.2 | 1.1 | 110 | 1.0 | 1.2 |
| 119 | 1.7 | 1.3 | 113 | 1.2 | 1.7 |
| 3 | 1.3 | 2.0 | 132 | 1.5 | 1.1 |
| 127 | 1.8 | 2.4 | 128 | 0.9 | 1.1 |
| 121 | 1.8 | 2.0 | 118 | 1.3 | 1.9 |
| 138 | 2.6 | 3.6 | 136 | 1.4 | 1.3 |
| 124 | 2.7 | 3.0 | 2 | 1.4 | 1.1 |
| 104 | 1.2 | Discontinued Study | 101 | 2.4 | 2.4 |
| 109 | 1.1 | Discontinued Study | 120 | 1.1 | 1.3 |
| 116 | 1.3 | 1.5 | 129 | 1.0 | 1.9 |
| 114 | 1.9 | 2.1 | 112 | 2.2 | 2.4 |
| 111 | 1.3 | 1.1 | 106 | 1.3 | 1.6 |
| 134 | 2.5 | 2.8 | 122 | 3.7 | 4.3 |
| 131 | 1.9 | 2.3 | 115 | 1.7 | Deceased |
| 125 | 1.9 | 1.8 | 103 | Replaced | Replaced |
Aortic sclerosis (peak velocity 2.0-2.5 m/s) is italicized. Aortic stenosis (peak velocity ≥2.5 m/s) is in bold.
Bone density
The extensive disc degeneration and calcification of alkaptonuria complicate bone densitometry measurement in this population. Nevertheless, we measured lumbar spine posterior-anterior, lateral vertebral body, and central or mid-vertebral body bone mineral densities, as well as bone mineral content, in all patients at the study initiation and at 36 months. Measurement of mid-vertebral body (central vertebral body) bone density was considered the most accurate representation of vertebral density, since calcification outside of and at the edges of the vertebral bodies creates spuriously elevated results. Both male and female mid-vertebral bone density values were compared to the available Hologic, Inc. reference data that was obtained in adult women. Central or mid-vertebral body Z-scores ranged from −3 to +4.2 at baseline, and did not change significantly at 36 months (data not shown). DXA information was also collected at the femur and radius, but the data were not analyzed because it is unclear how ochronosis affects these measurements.
3.6 Safety
There were 8 serious adverse events (SAEs) involving 6 patients, all receiving nitisinone, in this study.
SAE #1
This event involved a 61 year-old male who developed atrial fibrillation 2 weeks after starting nitisinone. Nitisinone was discontinued. The patient received intravenous diltiazem and returned to a regular rhythm, but suffered a fatal myocardial infarction one month later. This adverse event was considered unrelated to nitisinone. Literature reviews, as well as the package insert from the manufacturer, find no reports of cardiac arrhythmias or adverse cardiac events. The drug manufacturer lists “cyanosis” as the only cardiovascular adverse reaction that occurred in <1% of patients. Our patient has an extensive family history of cardiac disease, as well as a personal history of hypertension and hyperlipidemia. Initial EKG was normal, but chest CT revealed heavy coronary calcification. These risk factors were present prior to the patient receiving nitisinone.
SAE #2
Six weeks after initiating nitisinone, a 48 year-old male developed corneal irritation. On slit lamp examination, branching, subepithelial opacities were seen (Figure 5), consistent with the classic pattern previously seen with tyrosine crystal deposition.[16] The patient was placed on a 40 gram/day protein restricted diet but, 6 days later, the examination and symptoms were unchanged. Nitisinone was discontinued and the patient made a complete recovery. Two attempts were made to restart nitisinone; on each subsequent attempt, symptoms recurred. Nitisinone was stopped permanently, but the patient continued to return every 4 months under an intent-to-treat design.
Figure 5. Slit lamp exam of the cornea of a nitisinone-treated patient showing branching, subepithelial opacities.
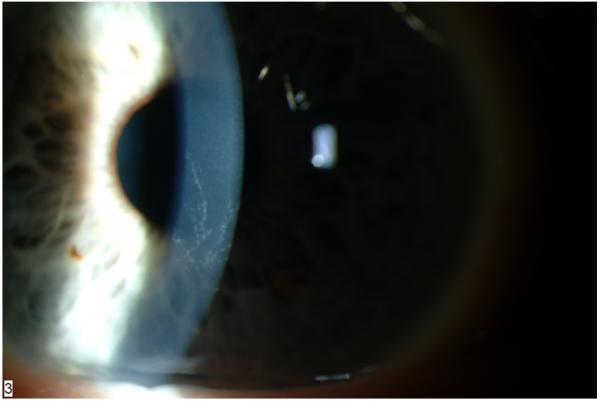
SAE #3-5
These events all involved a 54 year-old female with elevated liver transaminases. Eleven months after starting nitisinone, she developed right upper quadrant pain associated with an ALT of 163 U/L (normal, 6-41 U/L). Nitisinone was stopped. Testing revealed a 10 mm stone obstructing the common bile duct; the patient underwent a cholecystectomy with resolution of all symptoms. A regimen of three weeks on nitisinone, one week off, was initiated. Twelve months after resuming nitisinone, the patient returned for a routine admission and was found to be overmedicated with analgesics. Liver transaminases included an ALT of 225 U/L and AST of 409 U/L (normal, 9-34 U/L). Pain medications were adjusted and liver enzymes returned to normal (ALT 34 U/L; AST 14 U/L) within one week. However, 4 months later, the patient again returned for routine follow up and had an ALT of 166 U/L. No etiology for the elevated enzymes could be determined. Nitisinone was discontinued for the remainder of the study, but the patient continued to return every 4 months for follow up.
SAE #6
This event involved an elevated creatine kinase in a 57 year-old male. During a routine admission, the creatine kinase was 357 U/L (normal, 52-386 U/L), but on repeat testing the next day, it was 678 U/L. The patient had worked strenuously in the days immediately preceding his admission and reported shoulder soreness. The rise in creatine kinase was attributed to this manual labor, but the nitisinone was held until the creatine kinase returned to normal. The patient resumed nitisinone without further elevations of creatine kinase.
SAE #7
This event involved a 48 year-old female with hilar and mediastinal adenopathy noted at her 48-month admission. Tissue biopsy ultimately confirmed the diagnosis of sarcoidosis. The nitisinone was held during the diagnostic evaluation, and was not restarted because the study was terminated in the interim.
SAE #8
The final SAE involved a 56 year-old female with anemia at her 16-month visit. Hemoglobin slowly decreased to a low of 8.6 g/dL, but generally stayed in the 9 g/dL range. Colonoscopy and endoscopy, performed locally, revealed no source of bleeding. The patient was suffering from an open, draining, Achilles tendon wound and was diagnosed with anemia of chronic disease and iron deficiency anemia. Near 28 months into the study, she underwent a series of revisions to close the Achilles tendon wound and, six months later, both knees were replaced. Following this surgery, her hemoglobin levels returned to normal and remained stable. She remained on nitisinone throughout the study.
Plasma tyrosine levels (Fig. 6)
Figure 6. Plasma tyrosine concentrations in control and nitisinone-treated alkaptonuria patients over time.
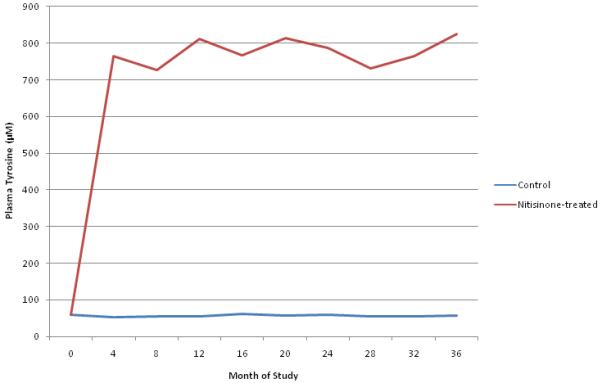
Mean plasma tyrosine levels (μM) measured at each visit over the 36 month study period. The red line is the mean for the nitisinone-treated group. The blue line represents the mean for the control group.
Mean plasma tyrosine in the untreated group remained approximately 60μM throughout the course of the study. Patients in the nitisinone group exhibited persistent, approximately ten-fold elevations in plasma tyrosine (range for individual values, 332μM to 1528μM; range for patient averages, 670μM to 826μM).
Nutrition
At baseline, there were differences between the two groups in mean total protein, phenylalanine, and tyrosine intake. Mean total protein intake for the control group was 92.4 g/d while mean phenylalanine and tyrosine intakes were 4.0 g/d and 3.1 g/d, respectively. Mean total protein intake for the nitisinone group was 68.6 g/d while phenylalanine and tyrosine intakes were 2.9 g/d and 2.3 g/day, respectively. The differences between these baseline means were statistically significant for all three measurements. However, these measurements remained approximately consistent throughout the study. Protein, phenylalanine, and tyrosine intakes were 89.8, 3.9, and 3.1 g/day, respectively, for the control group and 68.8, 3.0, and 2.3 g/d, respectively, for the nitisinone group.
3. Discussion
Consistent with the initial proof of principle study in 2002,[13] data from this clinical trial demonstrate that 2 mg of nitisinone daily reduces HGA secretion, a reflection of total body HGA production, by 95%. This decrease was rapid and sustained throughout the study and corresponded to a mean 95% reduction of plasma HGA levels. In fact, more than 60% of plasma HGA values were below the level of detection (0.250 μg/ml), similar to individuals without alkaptonuria. The close relationship between urine and plasma HGA reflects the fact that nearly all of the HGA produced in the body is excreted by the kidney through both filtration and active secretion.[28] This emphasizes the critical role of the kidney in removing HGA from the plasma, whose very slightly elevated HGA levels in alkaptonuria patients (5-6 μg/ml) nevertheless destroy connective tissue over the course of decades. Although there was a difference in protein, phenylalanine, and tyrosine consumption between the two groups, this difference was present at baseline when urine and plasma HGA levels were comparable, and persisted throughout the trial; therefore, reductions in urinary and plasma HGA levels cannot be attributed to dietary differences.
It is not surprising that plasma tyrosine levels increased dramatically with nitisinone treatment. Children with hereditary tyrosinemia are treated with nitisinone and tyrosine-restricted diets; in general, treating physicians attempt to limit plasma tyrosine levels to 500 μM.[27, 29] Noncompliance with dietary restriction has been problematic in many children with hereditary tyrosinemia and ocular effects have been extensively investigated. In 1998, Holme and Lindstedt reported ocular symptoms in 13 patients with hereditary tyrosinemia treated with nitisinone.[17] The symptoms were transient, although they did recur several times in a few patients. One patient developed corneal crystals that resolved within a few days without cessation of nitisinone. Holme and Lindstedt concluded that there was no clear correlation between plasma tyrosine levels and the occurrence of eye symptoms. Similarly, Gissen et al. reported ophthalmologic findings in 11 patients with hereditary tyrosinemia treated with nitisinone.[18] Four of the patients were noncompliant with the protein restricted diet. Plasma tyrosine levels reached 1240 and 1410 μmol/L but no patient developed corneal toxicity. Finally, Ahmad et al. reported a case of one patient with hereditary tyrosinemia who was noncompliant with dietary restrictions and who developed corneal opacities with tyrosine levels between 238 and 602 μmol/L.[16] The opacities resolved with dietary adjustment; again demonstrating the reversibility of the corneal lesions.
We did not protein restrict the adults enrolled in this trial, allowing us to determine, for the first time, how high plasma tyrosine levels would rise, and what side effects might occur under those conditions. In fact, plasma tyrosine levels averaged 800 μM, with individual levels reaching as high as 1500 μM; these levels were remarkably well tolerated. A single individual developed a keratopathy classical for tyrosine toxicity approximately 6 weeks following initiation of oral nitisinone. Interestingly, this individual’s plasma tyrosine levels, measured while symptomatic, were approximately 200 μM below the average for the treated cohort. Thus, in our small sample of nitisinone-treated patients, about 5% experienced corneal toxicity regardless of plasma tyrosine level, suggesting that some predisposition to toxicity exists independent of the peak plasma tyrosine concentration. Another patient developed elevated liver transaminases that may have been exacerbated by nitisinone use; however, her medical history was complicated and the patient was already on multiple medications associated with possible hepatotoxicity. Overall, the side effect profile of nitisinone was quite reassuring. The low frequency of toxicity in the absence of dietary protein restriction has implications for hereditary tyrosinemia patients treated with nitisinone.
While biochemical proof of efficacy was straightforward, confirming clinical efficacy has proven much more challenging. Total hip range of motion was selected as the primary outcome parameter based on natural history data collected at the National Institutes of Health. Sixteen patients had been followed over a period of up to 3 years with serial joint examinations as part of detailed physical medicine and rehabilitation evaluations. Of the 16 patients, 11 had decreased external rotation of the hip, 5 patients remained the same, and no patient showed improvement. Internal rotation yielded similar findings, with 14 patients worsening in range of motion, 2 remaining the same, and no patient showing improvement. In fact, 14 of the 16 patients experienced a loss in total hip rotation averaging 9.6 ± 5.9 degrees/year.
This rate of loss, which was seen in our previous natural history study and served as the basis for power estimates for our clinical trial, was not recapitulated in either the treated or control cohort in this three-year trial. When looking specifically at baseline and final data, the control group lost an average of 0.37°/year, while the treated group gained 2.0°/year. However, repeated measures analysis yielded only a 0.22°/year difference between the two groups. Regardless of the method of analysis, benefits attributable to nitisinone would be muted by the inclusion, under our intent-to-treat design, of 4 individuals who did not receive the drug for 8-35 months (Table 2). All of the hip measurements were performed by a single examiner, and the passive range of motion measurement was employed to reduce error due to patient effort. Nevertheless, there was significant variability in hip ROM from visit to visit, indicating that other parameters, such as pain, contributed to the measurement’s reliability.
Why was the loss of hip range of motion less in our untreated clinical trial group than in our previous untreated natural history group? Our trial population was older and had already experienced substantial hip involvement prior to enrollment in the trial. Once the degenerative cascade is initiated, there may be no way to halt the process. In addition, the rate of loss of ROM may decrease as patients approach immobility. Another reason for the slow rate of loss of ROM in our patients was that they all received the same aggressive musculoskeletal rehabilitation. It was striking that all patients, even in the control group, responded to consistent, yet gentle, strengthening and flexibility exercises. In addition to general exercises designed specifically for patients with alkaptonuria, patients also received instruction on individualized exercises targeting problematic areas. The response to this therapy reflects the critical role of physiatry in the therapy of alkaptonuria.
In addition to joint degeneration, aortic valve disease is a significant concern for individuals with alkaptonuria. While echocardiographic changes were not among our primary or secondary outcome parameters, they were evaluated for all patients at trial initiation and annually thereafter. Patients with alkaptonuria commonly exhibit thickening of the aortic valve by the fifth decade of life, and a fraction of such patients progress to mild or moderate aortic stenosis; some require aortic valve replacement.[13] In the present study, none of the 18 nitisinone-treated patients lacking signs of aortic stenosis at study onset progressed to aortic sclerosis or stenosis. In contrast, 7 of 17 control patients lacking signs of aortic stenosis at study onset developed aortic sclerosis or stenosis by study completion. These limited data suggest that nitisinone may impede progression of aortic valve disease, and warrant further exploration.
Bone density might also be an outcome parameter to gauge the clinical efficacy of nitisinone treatment, but measuring bone density is problematic in patients with alkaptonuria. Extensive intervertebral disc calcification, in conjunction with reactive arthritic changes such as osteophyte formation, substantially alters spinal measurements, giving spuriously elevated results. Although the underlying pathophysiology is not understood, patients with alkaptonuria have significant osteopenia and osteoporosis; in our 3-year study, nitisinone did not appear to influence bone density.
A longer clinical trial might be necessary to demonstrate the clinical efficacy of oral nitisinone for alkaptonuria patients. On first principles, nitisinone treatment early in life should prevent ochronotic vertebral disk and joint destruction, but demonstrating this may require one or two decades. Fortunately, the present study provides incentive for performing such a study. First, it confirms the 95% reduction in plasma and urinary HGA consequent to nitisinone treatment. It is unknown how low HGA needs to be reduced for maximal efficacy, but we believe that a 95% reduction in HGA production should be salutary. In addition, any benefit associated with significantly higher doses of nitisinone to achieve a greater reduction in HGA must be weighed against possible adverse reactions. Unlike in hereditary tyrosinemia where very slight quantities of FAA and MAA are toxic to the liver and kidneys within weeks or months, HGA does its damage over decades. Second, our findings demonstrate the tolerability of a daily dose of 2 mg of nitisinone; in general, the side effects to anticipate include corneal keratopathy and hepatotoxicity. Third, this work shows that nitisinone can be used in adults without protein restriction, and it sets the limits of plasma tyrosine concentration that can be reached with near-total inhibition of para-hydroxyphenylpyruvate oxidase. In addition, this study indicates that, without nitisinone treatment, perhaps the most important intervention for alkaptonuria patients is a regular program of physiatry administered by an experienced practitioner.
Acknowledgements
This study was funded by the intramural program of the National Human Genome Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None of the authors have a conflict of interest to disclose.
References
- [1].Garrod AE. About alkaptonuria. Lancet. 1901;158:1484–1486. [Google Scholar]
- [2].Garrod AE. The Croonian lectures on inborn errors of metabolism. Lecture II. Alkaptonuria. Lancet. 1908;172:73–79. [Google Scholar]
- [3].Piro A, Tagarelli A, Tagarelli G, Lagonia P, Quattrone A. Archibald Edward Garrod: the physician father of biochemistry Metabolism: clinical and experimental. 2009;58:427–437. doi: 10.1016/j.metabol.2008.12.001. [DOI] [PubMed] [Google Scholar]
- [4].La Du BN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria. The Journal of biological chemistry. 1958;230:251–260. [PubMed] [Google Scholar]
- [5].O’Brien M, La Du BN, Bunim J. Biochemical, pathologic and clinical aspects of alcaptonuria, ochronosis, and ochronotic arthropathy. American Journal of Medicine. 1963;34:813–838. W. [Google Scholar]
- [6].Kayser MA, Introne W, Gahl WA. Alkaptonuria. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Scriver CR, Sly WS, Childs B, editors. Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2007. [Google Scholar]
- [7].Milch RA. Studies of alcaptonuria: mechanisms of swelling of homogentisic acid-collagen preparations. Arthritis and rheumatism. 1961;4:253–267. doi: 10.1002/art.1780040304. [DOI] [PubMed] [Google Scholar]
- [8].Milch RA, Murray RA. Studies of alcaptonuria: adsorption of homogentisic acid solutions on collagen chromatographic columns. Arthritis and rheumatism. 1961;4:268–274. doi: 10.1002/art.1780040305. [DOI] [PubMed] [Google Scholar]
- [9].La Du BN, O’Brien WM, Zannoni VG. Studies on ochronosis. I. The distribution of homogentisic acid in guinea pigs. Arthritis and rheumatism. 1962;5:81–87. doi: 10.1002/art.1780050110. [DOI] [PubMed] [Google Scholar]
- [10].Zannoni VG, Malawista SE, La Du BN. Studies on ochronosis. II. Studies on benzoquinoneacetic acid, a probale intermediate in the connective tissue pigmentation of alcaptonuria. Arthritis and rheumatism. 1962;5:547–556. doi: 10.1002/art.1780050603. [DOI] [PubMed] [Google Scholar]
- [11].Zannoni VG, Lomtevas N, Goldfinger S. Oxidation of homogentisic acid to ochronotic pigment in connective tissue. Biochim Biophys Acta. 1969;177:94–105. doi: 10.1016/0304-4165(69)90068-3. [DOI] [PubMed] [Google Scholar]
- [12].Introne WJ, Kayser MA, Gahl WA. GeneReviews at Gene Tests: Medical Genetics Information Resource. University of Washington; Seattle: Alkaptonuria. database online. Updated [July, 2009] [Google Scholar]
- [13].Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, Gahl WA. Natural history of alkaptonuria. The New England journal of medicine. 2002;347:2111–2121. doi: 10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- [14].Anikster Y, Nyhan WL, Gahl WA. NTBC and alkaptonuria. American journal of human genetics. 1998;63:920–921. doi: 10.1086/302027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Suwannarat P, O’Brien K, Perry MB, Sebring N, Bernardini I, Kaiser-Kupfer MI, Rubin BI, Tsilou E, Gerber LH, Gahl WA. Use of nitisinone in patients with alkaptonuria. Metabolism: clinical and experimental. 2005;54:719–728. doi: 10.1016/j.metabol.2004.12.017. [DOI] [PubMed] [Google Scholar]
- [16].Ahmad S, Teckman JH, Lueder GT. Corneal opacities associated with NTBC treatment. American journal of ophthalmology. 2002;134:266–268. doi: 10.1016/s0002-9394(02)01532-5. [DOI] [PubMed] [Google Scholar]
- [17].Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione) Journal of inherited metabolic disease. 1998;21:507–517. doi: 10.1023/a:1005410820201. [DOI] [PubMed] [Google Scholar]
- [18].Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. Journal of inherited metabolic disease. 2003;26:13–16. doi: 10.1023/a:1024011110116. [DOI] [PubMed] [Google Scholar]
- [19].Lustberg TJ, Schulman JD, Seegmiller JE. The preparation and identification of various adducts of oxidized homogentisic acid and the development of a new sensitive colorimetric assay for homogentisic acid. Clin Chim Acta. 1971;35:325–333. doi: 10.1016/0009-8981(71)90202-6. [DOI] [PubMed] [Google Scholar]
- [20].Moll J, Wright V. Normal range of spinal mobility: an objective clinical study. Ann Rheum Dis. 1971;30:381–386. doi: 10.1136/ard.30.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Enright P, Sherrill D. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med. 1998;158:1384–1387. doi: 10.1164/ajrccm.158.5.9710086. [DOI] [PubMed] [Google Scholar]
- [22].Steffen T, Hacker T, Mollinger L. Age and gender-related test performance in community-dwelling elderly people: six-minute walk test, berg balance scale, timed up and go test, and gait speed. Phys Ther. 2002;82:128–137. doi: 10.1093/ptj/82.2.128. [DOI] [PubMed] [Google Scholar]
- [23].Podsiadlo D, Richardson S. The timed up and go: a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc. 1991;39:142–148. doi: 10.1111/j.1532-5415.1991.tb01616.x. [DOI] [PubMed] [Google Scholar]
- [24].Schober P. Ledenwirbel saoule und kreuzachmerchen. Munchener medizinische Wochenschrift. 1937;84:366. [Google Scholar]
- [25].Macrae I, Wright V. Measurement of back movement. Ann Rheum Dis. 1969;28:584. doi: 10.1136/ard.28.6.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Joint Motion: Method of Measuring and Recording. American Academy of Orthopedic Surgeons; Chicago: 1965. T.C.o.J. Motion. [Google Scholar]
- [27].Sniderman King L, Trahms C, Scott CR. Tyrosinemia Type 1, GeneReviews at Gene Tests: Medical Genetics Information Resource. University of Washington; Seattle: database online. Updated [October, 2008] [Google Scholar]
- [28].Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation. Molecular genetics and metabolism. 2002;77:136–142. doi: 10.1016/s1096-7192(02)00121-x. [DOI] [PubMed] [Google Scholar]
- [29].Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121–126. doi: 10.1002/ajmg.c.30092. [DOI] [PubMed] [Google Scholar]