

Abstract
When injured, tendons tend to heal but with poor structure and compromised function. Tissue engineering is a promising approach to enhancing the quality of healing tendons. Our group and others have identified tendon stem cells (TSCs), a type of tendon-specific stem cells which may be optimal for cellular interventions seeking to restore normal structure and function to injured tendons. However, in vitro expanding of TSCs on regular plastic cell culture dishes only yields a limited number of TSCs before they lose the stemness, i.e., the self-renewal capability and multipotency. In this study, we developed a substrate material for TSCs, engineered tendon matrix (ETM) from decellularized tendon tissues. We showed that ETM in vitro was able to stimulate TSC proliferation and better preserve the stemness of TSCs than plastic culture surfaces. In vivo, implantation of ETM-TSC composite promoted tendon-like tissue formation whereas implantation of TSCs alone led to little such tissue formation. Together, the findings of this study indicate that ETM may be used to effectively expand TSCs in vitro and with TSCs, to enhance repair of injured tendons in vivo.
Keywords: Tendon matrix, stemness, tendon stem cells, differentiation, regeneration
1. Introduction
Tendon injuries are a common problem in both occupational and athletic settings. However, injured tendons typically heal slowly, especially when the tendon injury is substantial or when rupture with tendon retraction occurs. Natural tendon healing also results in the formation of scar tissue, which has inferior mechanical properties making the healed tendons susceptible to re-injury [1]. In recent years, tissue engineering has emerged as a promising approach toward tendon repair or regeneration [2–7]. As a major component in tendons, autologous tenocytes have been used in repair of injured tendons [8]. However, removal of tendon sections for deriving tenocytes leads to formation of a secondary lesion at the donor site. In addition, tenocytes have a limited proliferative potential and quickly lose their phenotype in culture [9]. As a result, tenocytes may not be an ideal cell source for repair of injured tendons. Also, while implantation of bone marrow mesenchymal stem cells (BMSCs) induced a short term benefit in terms of improved mechanical properties at 4 weeks, it produced no visible improvement in the structure of healing tendons afterwards [10]. Moreover, the use of BMSCs even resulted in ectopic bone formation in tendons in a rabbit tendon injury model [11]. Embryonic stem cells (ESCs) can be a potential source for repair of injured tendons [12], but controlling ESC differentiation fates in vivo is more difficult than mesenchymal stem cells (MSCs) due to the pluripotency of ESCs; as a result, implantation of ESCs may risk formation of teratoma [13, 14]. Together, these studies indicate that non-tendon derived stem cells may not be optimal for cell therapies seeking to restore the normal structure and function of injured tendons.
Traditionally, tendons are thought to contain mainly tenocytes, the resident cells responsible for tendon maintenance and repair. In recent years, tendon stem/progenitor cells (TSCs) has been identified in humans, mice, rabbits, and rats [15–18]. TSCs differ from tenocytes in that they possess clonogenicity, self-renewal, and multi-differentiation potential, the three universal criteria of stem cells. Also, as tendon-specific stem cells, TSCs by default differentiate into tenocytes [16]. Moreover, when implanted in vivo, TSCs were able to form tendon-like tissues [15, 16]. Therefore, TSCs may be an ideal cell source for effective repair of injured tendons. However, TSCs in vivo are rare and amounts to less than 5% of total tendon cells [15]. Therefore, it is necessary to expand TSC populations in order to obtain sufficient number of these cells for cell therapy. However, when TSCs are cultured on plastic dishes, a common practice nowadays, they typically lose stemness after only several passages. A major reason for this problem is that plastic surfaces are foreign to TSCs and do not provide an appropriate environment for maintaining their stemness. Indeed, years of intensive stem cell research has uncovered that stem cells need appropriate “niches” in order to keep their stemness [19]. One of the most important niche factors for stem cells is the extracellular matrix (ECM), which is known to be crucial to the normal growth and function of stem cells [20–22]. Undoubtedly, an ideal ECM for TSCs would be a matrix from tendon tissues, which may retain the crucial niche factors including matrix components (e.g. collagen) and growth factors that are able to regulate TSC function. In a recent study, for example, when BMSCs were seeded on decellularized multilayer sliced tendon tissues, they expressed tenomodulin, a specific marker for tenocytes, suggesting that tendon matrix can prime non-tissue-specific BMSCs to differentiate towards tenocytes [23]. As a further advancement, we have developed an engineered tendon matrix (ETM) using decellularized tendon tissues, which can exist in the form of either film or gel for facilitating in vivo implantation. The purpose of this study was to characterize the behavior of TSCs on ETM in terms of self-renewal and multi-potency and to determine their ability to form tendon tissue in vivo. We found that TSCs on ETM exhibited enhanced self-renewal capability and multi-differentiation potential in vitro, and they were able to form tendon-like tissues in vivo.
2. Materials and Methods
2.1. Preparation of ETM
Twelve female New Zealand white rabbits (8–10 weeks old, 3.0 – 4.0 kg weight) were used for both ETM preparation and TSC derivation. The protocol for the use of the rabbits was approved by the University of Pittsburgh IACUC. All rabbits were fully sedated using intra-muscular Ketamine (10 mg/kg) and Xylazine (3 mg/kg) injections and were then sacrificed. After sacrifice, rabbit patellar tendons were obtained by dissection.
For preparation of ETM, patellar tendons were immersed into liquid nitrogen for 5 min and grounded into powder. The powder was then treated with 0.5% trypsin/PBS solution under vigorous agitation at 37°C. The treatment lasted for 24 hrs, and trypsin was changed every 4 hrs. Then the powder was washed three times with PBS, 30 min each time, and treated with a nuclease solution (50 U/ml DNAse and 1 U/ml RNAse in 10 mM Tris-HCl, pH 7.5) at 37°C for 12 hrs. After nuclease digestion, the powder was treated with 1% Triton X-100 for 24 hrs, washed with PBS 6 times with 8 hrs each time, and stored at −80°C for subsequent cell culture or implantation experiments.
2.2. Fabrication of ETM film and gel
ETM powder was dissolved with 3% acetic acid (HAc, ~0.5 M) to make a 5% solution. An ETM film was formed by adding 1 ml ETM-HAc solution in a 35 mm Petri dish and letting it dry overnight under UV light. ETM was also made in a gel form by the addition of sodium hydroxide to ETM-HAc solution.
2.3. Scanning electron microscopy (SEM) of ETM
ETM was sputter coated with gold/palladium and examined under a JEOL (Tokyo, Japan) SEM with an accelerating voltage of 3.0 kV.
2.4. Isolation of rabbit and human TSCs
TSCs were isolated from rabbit patellar tendons. The procedures for isolation of TSCs were similar to our previously published protocol [24]. Using the same protocol, human patellar TSCs were derived from human tendon samples. The protocol for obtaining human tendons and the subsequent culture study was approved by the Institutional Review Board (IRB) of University of Pittsburgh.
2.5. Cell culture
The ETM film was washed 3 times with PBS, 3 times with 70% ethanol, and finally 5 times with PBS. Rabbit patellar TSCs (rPTSCs) at passage 1 were seeded either on ETM film or in ETM gel in 6-well plates at a density of 4.5 × 104 cells/well and cultured for up to 7 days. The morphology of TSCs grown on ETM film or in ETM gel was either examined directly by using an inverted microscope or histochemically stained with H&E. Population doubling time (PDT) was determined to assess the proliferative capacity of these cells on ETM films according to the method previously published [16].
For H & E staining, TSCs grown on/in ETM were fixed with 4% paraformaldehyde for 30 min at room temperature. Subsequently, the ETM with TSCs was placed in pre-labeled base molds filled with frozen section medium (Neg 50; Richard-Allan Scientific; Kalamazoo, MI). The base mold with ETM and TSCs was quickly immersed in liquid nitrogen cooled 2-methylbutane and allowed to solidify completely. The ETM-TSCs block was cut into 10 μm thick sections, and the sections were placed on glass slides and allowed to dry overnight at room temperature. The sections were rinsed three times with PBS and stained with H&E.
2.6. Characterization of TSCs
TSCs were characterized by immunostaining the following stem cell markers: octamer-binding transcription factor 4 (Oct-4), stage-specific embryonic antigen-1 and -4 (SSEA-1 and SSEA-4), and nucleostemin. The TSCs grown on ETM film or the plastic surfaces of Petri dishes were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature and treated with 0.1% Triton X-100 for 30 min for Oct-4 and nucleostemin staining. After washing the cells with PBS, either mouse anti-Oct-4 (1:350) or goat anti-nucleostemin (1:400) was applied for 2 hrs at room temperature. The cells were washed with PBS for three times, and either Cy-3-conjugated goat anti-mouse IgG antibodies (1:500 for Oct-4) or Cy3-conjugated donkey anti-goat IgG antibodies (1:500 for nucleostemin) was applied for 1 hr at room temperature. In order to stain for SSEA-1 and SSEA-4, fixed cells were blocked with 2% mouse serum for 1 hr and incubated with mouse anti-human SSEA-1 or SSEA-4 antibodies (1:500) for another hour at room temperature. After washing the cells with PBS, TSCs were treated with either fluorescein isothiocyana (FITC)-conjugated goat anti-mouse secondary antibodies (1:500 for SSEA-1) or Cy3-conjugated goat anti-mouse IgG antibodies (1:500 for SSEA-4) for 1 hr at room temperature. The stained cells were examined using fluorescence microscopy. All antibodies used were from Chemicon International (Temecula, CA), BD Biosciences (Franklin Lakes, NJ), or Neuromics (Edina, MN). To quantify the expression of stem cell markers, the stained samples were examined using an inverted fluorescence microscope and images were taken with a 20× objective using a CCD camera. A total of 60 views from 3 wells of a 6-well plate were randomly chosen for each stem cell marker and the number of positively stained cells was manually counted. The percentage of each stem cell marker expression was determined by dividing the number of positively stained cells by the total number of cells stained by H33342.
2.7. Characterization of multi-differentiation potential of TSCs
Multi-differentiation potential of TSCs on ETM or plastic surfaces was tested in vitro for adipogenesis, chondrogenesis, and osteogenesis. TSCs at passage 1 were seeded either on ETM or plastic surfaces in 6-well plates at a density of 2.4 × 105 cells/well in basic growth medium (DMEM plus 10% FBS). To test adipogenic potential, TSCs were cultured in adipogenic induction medium (Millipore, Billerica, MA) consisting of basic growth medium added with dexamethasone (1 μM), insulin (10 μg/ml), indomethacin (100 μM), and isobutylmethylxanthine (0.5 mM). As a test of chondrogenic potential, TSCs were cultured in basic growth medium supplemented with prolin (40 μg/ml), dexamethasone (39 ng/ml), TGF-β3 (10 ng/ml), ascorbate 2-phosphate (50 μg/ml), sodium pyruvate (100 μg/ml), and insulin-transferrin-selenious acid mix (50 mg/ml) (BD Bioscience, Bedford, MA). Finally, the osteogenic potential of TSCs was tested by culturing TSCs in osteogenic induction medium (Millipore, Billerica, MA) consisting of basic growth medium supplemented with dexamethasone (0.1 μM), ascorbic 2-phosphate (0.2 mM), and glycerol 2-phosphate (10 mM).
After 21 days culture, TSCs grown on ETM or plastic surfaces with various differentiation media were stained using Oil Red O for adipogenesis, Safranin O for chondrogenesis, or Alizarin Red S for osteogenesis, respectively (detailed protocols are shown below). The stained samples were examined using an inverted microscope and images were taken with a 20× objective using a CCD camera. A total number of 12 views from each well were randomly chosen. The areas of positive staining were identified manually and computed by a SPOT IMAGING software. The ratio of positive staining was calculated by dividing the stained area by the view area. The values of all views from three duplicate wells (36 views in total) were averaged to obtain the percentage of positive staining, which represents the extent of cell differentiation in the respective induction medium.
2.8. Oil Red O assay for adipogenesis
After removing culture medium, cells were washed with PBS 3 times, for 5 min each time. The cells were then fixed by 4% paraformaldehyde for 30 min at room temperature. Subsequently, the cells were washed with PBS 3 times each for 5 min and then with distilled water 2 times each for 5 min. Finally, the cells were incubated with 0.36% Oil Red O solution (Millipore, Billerica, MA) for 50 min and then washed 3 times with water. Stained samples were examined on an inverted microscope (Nikon eclipse, TE2000-U); images were obtained by a CCD camera on the microscope and analyzed by SPOT™ imaging software (Diagnostic Instruments, Inc., Sterling Heights, MI). Stained lipid droplets of the adipocytes appeared red.
2.9. Safranin O assay for chondrogenesis
Cells were fixed in ice cold ethanol for 1 hr, rinsed with distilled water 2 times each for 5 min, and stained at room temperature for 30 min with Safranin O solution (Sigma, St. Louis, MO). The cells were then rinsed 5 times with distilled water. Chondrocytes producing glycosaminoglycans (GAG)-rich matrix were stained red.
2.10. Alizarin Red S assay for osteogenesis
Using chilled 70% ethanol, cultured cells were fixed for 1 hr. Then cells were rinsed with distilled water twice, each for 5 min, and stained with Alizarin Red S (Millipore, Billerica, MA) at room temperature for 30 min. The osteocytes containing mineral deposits were stained orange-red.
2.11. Quantitative real-time PCR (qRT-PCR)
The specific gene expression of differentiated TSCs grown on ETM and plastic surfaces was determined using qRT-PCR. Total RNA was extracted using an RNeasy Mini Kit with an on-column DNase I digest (Qiagen). First-strand cDNA was synthesized in a 20 μl reaction of 1 μg total RNA through reverse transcription with SuperScript II (Invitrogen). The conditions for the cDNA synthesis were: 65ºC for 5 min and cooling for 1 min at 4ºC, then 42ºC for 50 min, and finally 72ºC for 15 min. The qRT-PCR was carried out using QIAGEN QuantiTect SYBR Green PCR Kit (Qiagen). In a 50 μl PCR reaction mixture, 2 μl cDNA (total 100 ng RNA) were amplified in a Chromo 4 Detector (MJ Research). Rabbit-specific primers were used for stem cell gene expression, including Oct-4 and Nanog, and tenocyte related genes, including collagen types I and III, tenascin C, and tenomodulin. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. The forward and reverse primer sequences and the resultant products were designed according to published methods {Fan, 2008 #5320; Yukata, 2010 #5321; Intawicha, 2009 #5319}. All primers were synthesized by Invitrogen (Carlsbad, CA).
The relative gene expression levels were calculated from 2−ΔCT, where ΔCT was determined by the formula: ΔCT = (CTtarget − CTGAPDH)differentiation − (CTtarget − CTGAPDH)control. In the formula, CTtarget and CTGAPDH are the cycle thresholds of target gene and GAPDH gene, respectively, for each RNA sample. The standard deviation (SD) of the ΔCT was determined from at least three parallel tests.
2.12. Implantation experiments
Six female nude rats (10 weeks old; 200–250g) were used to test the effects of ETM on tendon regeneration in vivo. Rats were housed individually on a 12 hrs:12 hrs light–dark cycle and were cared for in accordance with the Guide to the Care and Use of Experimental Animals. Human patellar TSCs (hPTSCs; 5 × 107 cells) at passage 2 were incubated with 1 ml of 10 μM of CFDA SE (a cell green tracer, Invitrogen, Carlsbad, CA) in PBS at 37°C for 15 min, followed by centrifugation at 3,000g for 10 min. After removing CFDA SE, the labeled cells were cultured with 0.5 ml of PBS at 37°C for 30 min. Then each 10 μl of labeled hPTSCs (106 cells) were mixed with ETM gel, which was made by adding 0.25 ml of 5% ETM-HAC solution to a 24-well plate and adjusting its pH to 7.0 with 10 μl of 4M NaOH.
For implantation experiments, surgeries were performed on the nude rats under general anesthesia using ketamine hydrochloride (75 mg/kg body weight) and xylazine hydrochloride (5 mg/kg body weight), administrated by intramuscular injection. hPTSC-ETM gels were placed subcutaneously into the abdominal areas of the rats. Two pieces of hPTSC-ETM gels were positioned in two distinct places on each rat. In addition, a circular defect with a diameter of 2 mm was created in the rat patellar tendon and filled with hPTSC-ETM gel by injection. In control group, the rats were treated with the same procedures, but they were only implanted with labeled hPTSCs without ETM gel. Tissue samples were harvested at 8 weeks and placed in pre-labeled base molds filled with frozen section medium (Neg 50; Richard-Allan Scientific; Kalamazoo, MI). The base mold with tissue samples was quickly immersed in liquid nitrogen cold 2-methylbutane and allowed to solidify completely. The tissue blocks were then placed on dry ice and subsequently stored in the −80°C freezer until time for histological analysis.
2.13. Histochemical and immunohistochemical analyses of tissue sections
The tissue block was cut into 10 μm thick sections, and they were then placed on glass slides and allowed to dry overnight at room temperature. The sections were rinsed 3 times with PBS, fixed with 4% paraformaldehyde for 30 min, and further washed with PBS for 3 times. The sections were stained with H&E. For immunohistochemical staining, the sections were coated with 5% goat serum and incubated for 30 min at room temperature in a humid chamber. The serum was carefully removed by aspiration, and mouse anti-human collagen type I antibody (1:350; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was applied to the sections, which were then incubated at room temperature for 2 hrs. The tissue section was washed 3 times with PBS, reacted with Cy3-conjugated goat anti-mouse IgG (1:500; Santa Cruz Biotechnology) at room temperature for 1 hr, again washed 3 times with PBS, and then reacted with Hoechst fluorochrome 33342 (1μg/ml; Sigma, St. Louis, MO) at room temperature for 5 min. Finally, the sections were washed with PBS twice.
2.14. Statistical analysis
Data are presented as mean plus and minus standard deviation (SD). At least three replicates for each experimental condition were performed, and the presented results are representative of these replications. One-way analysis of variance (ANOVA), followed by either Fisher's predicted least-square difference (PLSD) for multiple comparisons or two tailed student t-test wherever applicable, was used for statistical analysis. Differences between two groups were considered significant when the p-value was less than 0.05.
3. Results
We successfully fabricated ETM in the form of film and gel out of rabbit patellar tendons. SEM imaging showed that physical mincing and subsequent chemical/biochemical treatments with trypsin, nuclease, and Triton X-100 removed original tendon cells and cellular components (Fig. 1A). Moreover, while the highly organized microstructure of tendon was lost in ETM, collagen microfibrils seemed to be well preserved (Fig. 1B).
Fig. 1.

SEM images of ETM film and gel. The ETM was fabricated from rabbit patellar tendons. Note that ETM film (A) contains rough textures of matrix components on its surface and that ETM gel (B) consists of collagen fibrils (arrows) and interconnected pores (triangle).
To examine the ability of ETM to support cell growth in vitro, we seeded rabbit patellar TSCs (rPTSCs) either on ETM films or in ETM gels. The cells attached well to ETM, and formed numerous colonies (Fig. 2A). After 7 days in culture, a monolayer of cells was formed on the ETM film, whereas cells resided in the pores of ETM gels (Fig. 2B). Frozen section staining with H&E showed that ETM provided a 3-D culture environment where rPTSCs not only grew on ETM surface, but also migrated into the inside of ETM (Fig. 2C). In addition, rPTSCs grew faster in ETM than on plastic surfaces, as indicated by the population doubling times (PDT) of cells on these two substrates (Fig. 2D). The PDT of rPTSCs on ETM films was about 15% lower than that on the plastic surfaces. As a result, rPTSCs formed larger colonies on ETM compared to cells grown on plastic surfaces within the same culture time.
Fig. 2.
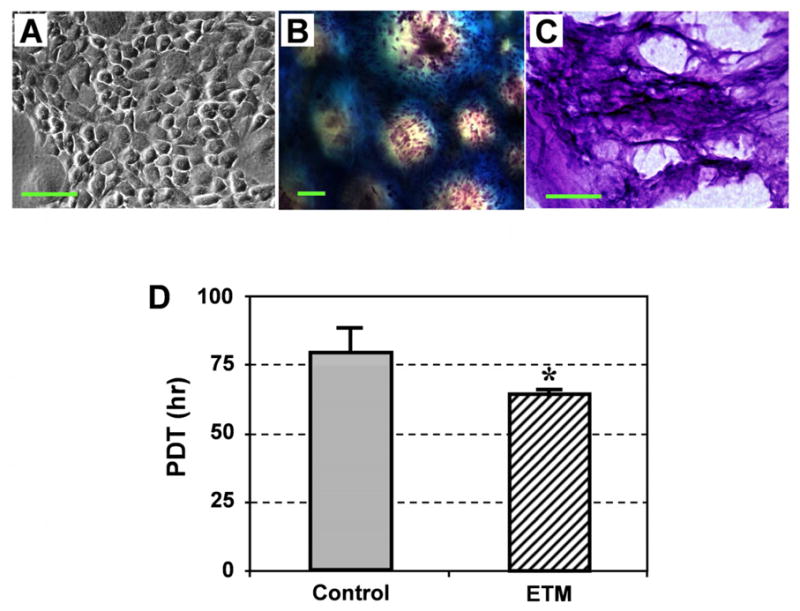
The effects of ETM film and ETM gel on rPTSCs. Colonies of rPTSCs were formed on the ETM film (A), and rPTSCs were also highly proliferated in the ETM gel, as shown by numerous cells identified by H&E staining (B). Frozen section results showed that rPTSCs have migrated into the inside of ETM (C). The population doubling time (PDT) of rPTSCs on ETM films was significantly lower than that of the same cells on control plastic surfaces, indicating that ETM enhanced rPTSC proliferation (D, * p < 0.05). Bar: 100 μm.
Moreover, immunofluorescence assays for stem cell markers revealed that rPTSCs expressed more extensively Oct-4, SSEA-1, SSEA-4, and nucleostemin when they were grown on ETM films (Fig. 3A, C, E, and G), compared to the same cells grown on control plastic surfaces (Fig. 3B, D, F, and H). A quantitative comparison between these two groups showed that the extent of gene expression in all four stem cell markers was signficantly higher on ETM than on control plastic surfaces (Fig. 3I).
Fig. 3.
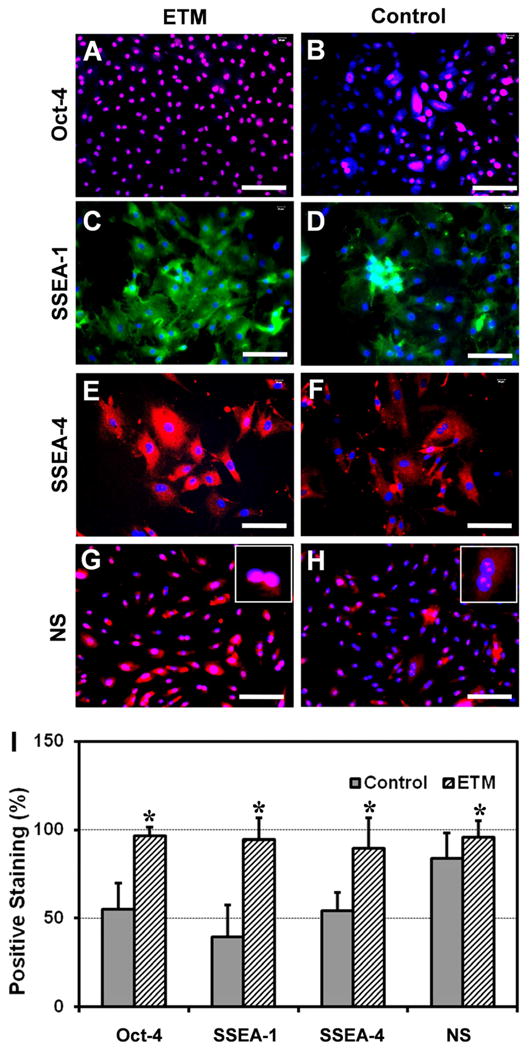
The expression of stem cell markers for rPTSCs grown on ETM and control plastic surfaces. All four stem cell markers, i.e. Oct-4, SSEA-1, SSEA-4, and nucleostemin (NS), were more extensively expressed in rPTSCs grown on ETM films (A, C, E, and G) than those grown on plastic surfaces (B, D, F, and H). The semi-quantitative results of stem cell marker expression are given (I, * p < 0.05 with respect to each own control). Bar: 100 μm.
As a further characterization of the stemness of TSCs on both ETM films and plastic surfaces, we examined the gene expression profiles of rPTSCs grown on ETM and plastic surfaces using qRT-PCR. Three sets of genes were examined, including embryonic stem cell genes Oct-4 and Nanog, tenocyte-related genes collagen types I and III, tenascin C, and tenomodulin, and non-tenocyte-related genes PPARγ, collagen type II, and Runx-2. We found that both stem cell genes and tenocyte-related genes, except tenascin C, were significantly up-regulated in rPTSCs cultured on ETM compared to those on plastic surfaces (Fig. 4). Nevertheless, rPTSCs on both ETM and plastic surfaces expressed minimal non-tenocyte related genes PPARγ, collagen type II, and Runx-2 (data not shown).
Fig. 4.
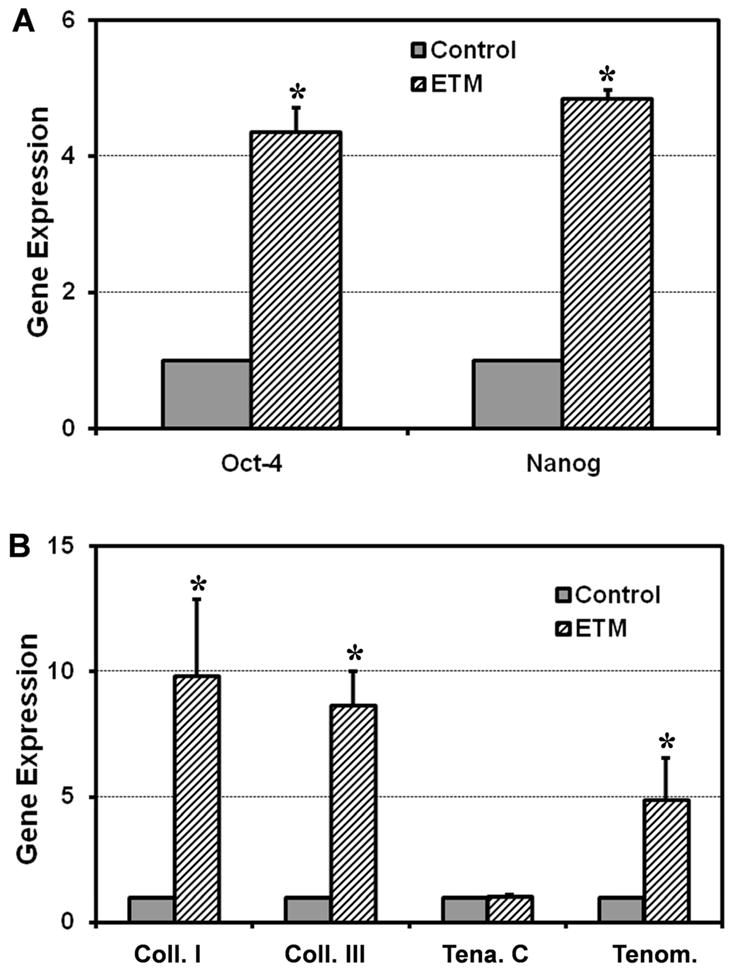
The expression of stem cell and tenocyte related genes in rPTSCs on ETM films and control plastic surfaces. The cells on ETM films expressed markedly higher levels of both stem cell genes (Oct-4 and Nanog) (A). Except tenascin C, tenocyte-related genes (collagen types I and III, and tenomodulin) in rPTSCs on ETM films were also highly expressed than those on control plastic surfaces (B). (* p < 0.05 with respect to each own control. Note that the gene expression levels were normalized with respect to own controls).
We next examined the multi-differentiation potential of TSCs by culturing them in adipogenic, chondrogenic, and osteogenic induction media, respectively. After 3 weeks, more than 41.6% of rPTSCs on ETM were positively stained by Oil Red O (Fig. 5A), and 78.1% of cells were stained by Safranin O, and 54.8% of the cells were stained by Alizarin Red S, respectively (Fig. 5C, E). However, on control plastic surfaces, the percentages of cells stained by Oil Red O, Safranin O, and Alizarin Red S were about 14.5%, 27.7%, and 33.5%, respectively (Fig. 5B, D, and F). A comparison of the percentages of differentiated cells on ETM and control plastic surfaces showed that there were singificant differences between the two groups in terms of the extent of respective adipogenesis, chondrogenesis, and osteogenesis (Fig. 5G).
Fig. 5.
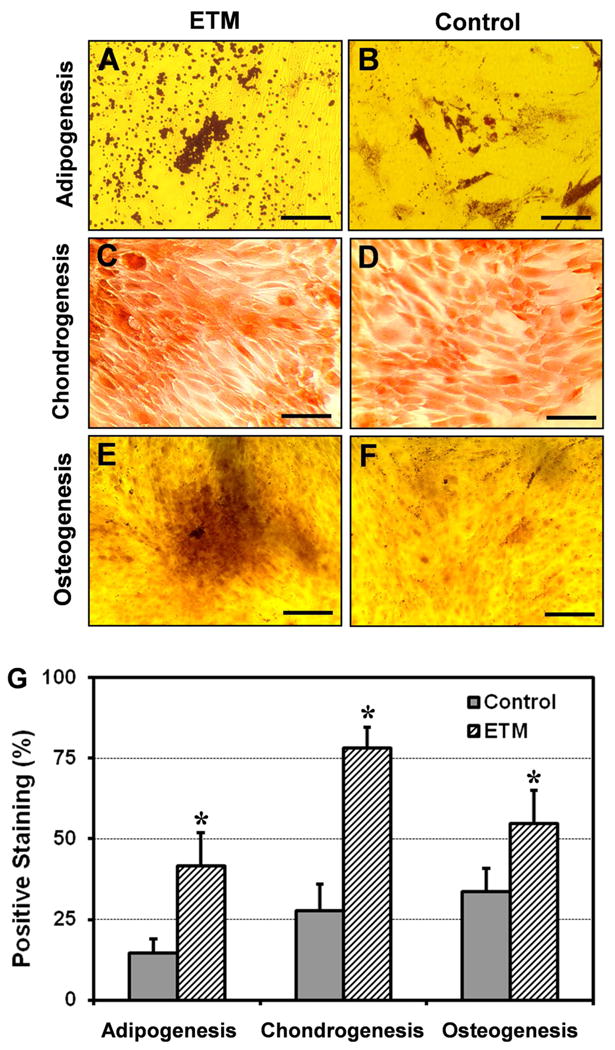
The multi-differentiation potential of rPTSCs on ETM films and control plastic surfaces. When cultured in respective induction media, more extensive adipocytes, chondrocytes, and osteocytes of RPTSCs were found on ETM films (A, C, and E) than on plastic surfaces (B, D, and F). (G) The semi-quantitative analysis of staining results; * p < 0.05 with respect to each own control). Bar: 100 μm.
We further performed in vivo experiments to examine the differentiation capability and fate of TSCs by implanting human patellar TSCs (hPTSCs) into nude rats. We found that when hPTSCs were subcutaneously implanted with ETM into the back of nude rats, tendon-like tissues were formed at 8 weeks after implantation (Fig. 6A–C). However, implantation of hPTSCs alone did not lead to formation of tendon-like tissue (Fig. 6D–F). Furthermore, by immunostaining of human collagen type I, we found that the human collagen type I protein was extensively produced in tendon-like tissues formed by implantation with hPTSCs embedded in ETM gels (Fig. 6A). However, only weak staining was found in the samples with implantation of hPTSC alone (Fig. 6D), or samples without cell implantation at all (control sample, Fig. 6G). Such immunostaining results were also confirmed by H&E staining, which showed tendon-like collagen fiber bundles in ETM+hPTSC samples (Fig. 6C) but not in samples with hPTSC alone (Fig. 6F) and control samples (Fig. 6I).
Fig. 6.
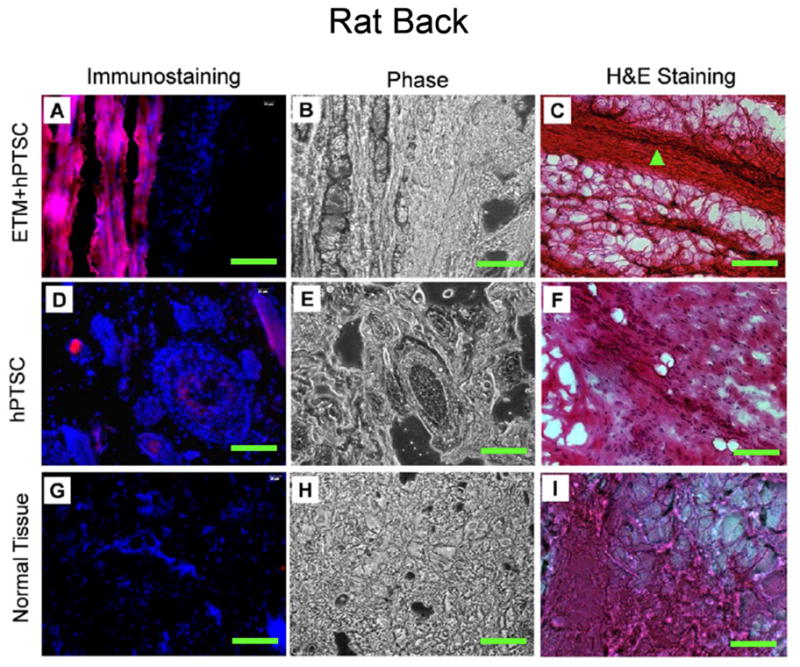
Tissue formation after implantation of hPTSCs with and without ETM into the back of nude rats. The cells were implanted with ETM (A–C) or without ETM (D–F). Normal tendon tissues of the same rats were used as control (G–I). It is seen that the implantation of hPTSCs with ETM resulted in newly formed, tendon-like tissues, as shown by immunostaining for human collagen type I (A, red bands) and H&E staining for collagen bundles (C, triangle). In contrast, implantation of hPTSCs alone did not result in the formation of tendon-like tissues (D). Bar: 100 μm.
Similar results were found when hPTSCs were implanted into the patellar tendons of nude rats instead of their backs (Fig. 7). Compared to the hPTSC alone group (Fig. 7D–F), the collagen fibrils were thicker and more organized in the tendon tissue sections of rats in the ETM+hPTSC group (Fig. 7A–C) at 8 weeks after implantation. Moreover, injured tendons in the ETM+hPTSC group almost restored their organized, parallel structure (Fig. 7C), but those in the hPTSC alone group appeared to retain their injury gaps (Fig. 7F). Overall, the quality of tendon healing was much better when hPTSCs were transplanted in conjunction with ETM, resulting in healed tendons almost comparable to normal tendon tissue (Fig. 7I).
Fig. 7.
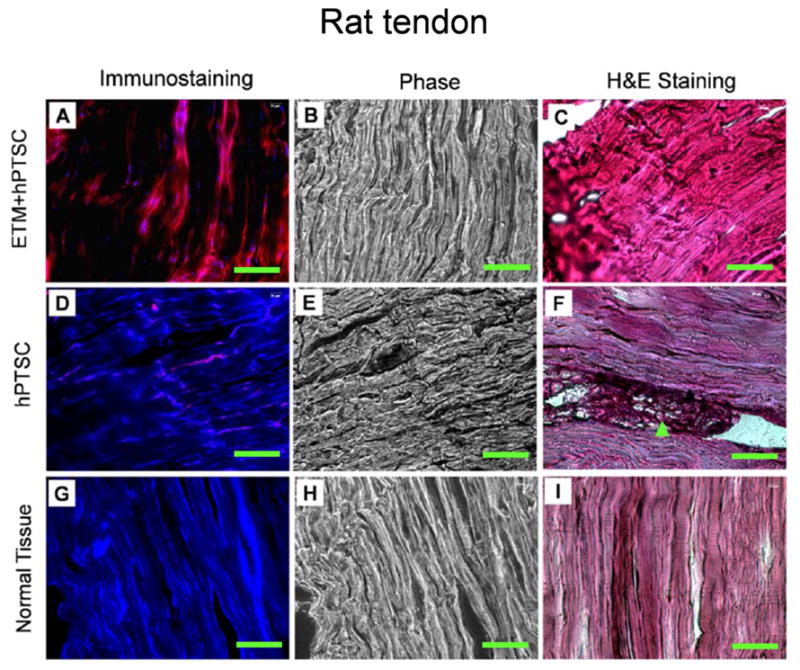
Tissue formation after implantation of hPTSCs with and without ETM into rat patellar tendons. The cells were implanted with ETM (A–C), without ETM (D–F). Normal tendon tissues of the same rats were used as control (G–I). Similar to the implantation into the back of rats, implantation of hPTSCs together with ETM resulted in newly formed, human tendon-like tissues (A, red bands). However, implantation of hPTSCs without ETM resulted in poor tissue formation (F, arrow). Bar: 100 μm.
4. Discussion
In this study, we developed a soluble decellularized tendon matrix, or ETM, by simply mincing tendon tissues, then biochemically digesting them using trypsin and nuclease treatments, and eventually chemically treating them using acetic acid (HAc). Our processing procedures were similar to those protocols that had already been used in collecting collagen proteins from tissues such as skins, corneas, and tendons [25–27]. It is likely that the intermolecular links were largely lost as a result of the relatively harsh physical and biochemical treatments in preparation of ETM. Previous studies showed that treatment with 0.05 M HAc for 1 hr could cause apparent intermolecular bond rearrangement in collagens [27] and that treatment with 0.5 M HAc could result in loss of cross-linking between collagen helices and fibril ultrastructure [25]. Nonetheless, ETMs retained considerable bioactivity in terms of promoting TSC growth based on the findings of our in vitro study. Specifically, in culture, the ETMs, either in the form of thin films (2D culture) or as a gel (3D culture), proved effective in stimulating TSC proliferation (Fig. 2). In fact, TSCs on ETM grew more quickly than on plastic culture substrate, which serves as a “gold standard” in cell culture nowadays.
In addition to stimulating TSC proliferation, ETM was able to better preserve the stemness of TSCs than plastic surfaces. This is evidenced by the more extensive expression of stem cell markers, Oct-4, SSEA-1, SSEA-4, and nucleostemin in TSCs grown on ETM compared to those grown on plastic surfaces (Fig. 3). Among these stem cell markers, Oct-4 is a transcription factor which is typically expressed in embryonic stem cells (ESCs) during development and mediates pluripotency in ESCs [28]. Oct-4 is essential for establishing and maintaining undifferentiated pluripotent stem cells; it is not expressed in differentiated cells [29]. SSEA-1 and SSEA-4 are two molecules characteristic of undifferentiated pluripotent human or mouse stem cells [30–32].
SSEA-1 may be used as a marker of cell status to test the long-term phenotypic stability of ESC cultures [32]. Being exclusively expressed in the nucleoli of stem cells and cancer cells but not in committed and terminally differentiated cells, nucleostemin controls the cell-cycle progression [33]. Together, the substrate-dependent expression of the above stem cell marker proteins in TSCs reveals that more TSCs were kept in an undifferentiated state when they were cultured on ETM than on plastic surfaces.
In addition to stem cell marker expression, we also showed that TSCs on ETM expressed higher levels of stem cell-related genes (Oct-4 and Nanog) than cells on plastic surfaces (Fig. 4A). Nanog, a unique homeobox transcription factor, was found to be expressed in pluripotent stem cells, and its expression was down-regulated when the cells were differentiated [34]. Therefore, the expression of both stem cell genes indicates that TSCs on ETM retained better stemness than the same cells on plastic surfaces.
Moreover, while TSCs on both ETM and plastic surfaces exhibited minimal expression of non-tenocyte related genes, the former exhibited more extensive adipogenesis, chondrogenesis, and osteogenesis in the same differentiation induction media than the latter (Fig. 5). Again, these data suggest that more stem cells that retained multi-differentiation potential were present in cultures with ETM as culture substrate; in other words, the stemness of TSCs was better preserved on ETM than on plastic surfaces. Thus, ETM may be used as an ideal culture substrate to effectively expand TSCs for cell therapy of injured tendons.
Interestingly, when TSCs were grown on ETM, tenocyte-related genes, including collagen type I and III, and tenomodulin were highly expressed (Fig. 4B). Therefore, we suspect that there likely existed at least two sub-populations of cells in TSC cultures: a “genuine” stem cell group and a progeny cell group. In the formal group, cells expressed Oct-4, SSEA-1, and SSEA-4; in the latter, cells were differentiating towards tenocytes and as a result, they did not express stem cell genes but express tenocyte related genes (collagen types I and III, and tenomodulin) instead. Thus, ETM not only promotes TSC proliferation, it also promotes the differentiation of TSCs into tenocytes, a default differentiation fate of TSCs in vivo under physiological conditions.
The in vitro findings above may explain why ETM plus TSCs functioned better than TSCs alone in terms of the quality of tissues formed in vivo in this study. In the nude rat model, hPTSCs were able to form good, tendon-like tissues only when they were implanted together with ETM (Fig. 6A–C, and Fig. 7A–C). In contrast, implanting hPTSCs alone formed little tendinous tissues (Fig. 6D–F). Such an effect may partially be expected from the in vitro culture tests of TSCs, as ETM would stimulate TSCs to proliferate and produce more TSCs (or self-renewal) and more tenocytes (default TSC differentiation) that are needed for effective repair of injured tissues.
In previous experiments, Matrigel, a basement membrane product, was used as a scaffold when TSCs were implanted in vivo [15, 16]. While tendon-like tissues were formed, it was found that fatty, cartilage-like, and bone-like tissues were also formed, indicating that Matrigel was not an ideal matrix for tendon-like tissue formation by TSCs. On the contrary, we used ETM in our implantation experiment and found no evidence that those non-tendinous tissues were formed, further confirming that ETM preferentially promoted tenocyte differentiation (Fig. 4B) and as a result, formation of tendon-like tissues in vivo.
The results of this study are in line of the concept that stem cells and appropriate scaffolds are two vital components in tissue engineering of injured tissues [3]. Furthermore, the fact that TSCs alone did not lead to the formation of good tendon-like tissues in vivo emphasizes the critical role of ECM scaffolds in the repair/regeneration process of injured tissues such as tendons. Unlike commonly used foreign scaffolds including collagen gels [10, 11] and polymer fibers [35], ETM, a scaffolding material derived from native tendon tissues, provides an amiable niche for TSCs to maintain optimal self-renewal ability and multi-potency while not activated, but quickly turn into tendon progenitor cells when TSCs are activated by inflammatory factors, cytokines, or growth factors in an injured microenvironment such as injured tendons [15, 36, 37].
While the results from this study have shown that ETM was effective in promoting TSC proliferation in vitro and forming tendon-like tissues by TSCs in vivo, there are a few limitations of this study. First, while it is likely that major matrix components (e.g. collagen types I and III) are kept in ETM during its preparation, the exact biochemical composition of ETM is not known. For example, whether the growth factors bound to native tendon matrix (e.g., TGF-β, and bFGF) are preserved and remain active is still an open question [38]. Second, in light of long healing process of tendons [10], the 8-week implantation test in this study is relatively short. A longer period of in vivo study should be performed in a future study in order to better evaluate whether the use of ETM, along with TSCs, can eventually restore normal structure and function to injured tendons. Functional evaluation of the efficacy of ETM -TSC treatment on injured tendons by mechanical testing should also be performed in future research.
5. Conclusions
A tendon tissue engineering scaffold, ETM, has been developed from tendon tissues in this study. ETM was found to promote TSC proliferation and help preserve the stemness of TSCs during in vitro expansion. Moreover, together with TSCs, ETM promoted the formation of tendon-like tissues in vivo. We therefore suggest that ETM may be used in tissue engineering for effective repair or possible regeneration of injured tendons.
Acknowledgments
The funding support from NIH grants AR049921 and AR049921S2 (JHW) for this work is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butler DL, Juncosa N, Dressler MR. Functional efficacy of tendon repair processes. Annu Rev Biomed Eng. 2004;6:303–29. doi: 10.1146/annurev.bioeng.6.040803.140240. [DOI] [PubMed] [Google Scholar]
- 2.Bagnaninchi PO, Yang Y, El Haj AJ, Maffulli N. Tissue engineering for tendon repair. Br J Sports Med. 2007;41:e10. doi: 10.1136/bjsm.2006.030643. discussion e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler DL, Juncosa-Melvin N, Boivin GP, Galloway MT, Shearn JT, Gooch C, et al. Functional tissue engineering for tendon repair: A multidisciplinary strategy using mesenchymal stem cells, bioscaffolds, and mechanical stimulation. J Orthop Res. 2008;26:1–9. doi: 10.1002/jor.20456. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Zou XH, Yin GL, Ouyang HW. Tendon tissue engineering with mesenchymal stem cells and biografts: an option for large tendon defects? Front Biosci (Schol Ed) 2009;1:23–32. doi: 10.2741/S3. [DOI] [PubMed] [Google Scholar]
- 5.Kuo CK, Marturano JE, Tuan RS. Novel strategies in tendon and ligament tissue engineering: Advanced biomaterials and regeneration motifs. Sports Med Arthrosc Rehabil Ther Technol. 2010;2:20. doi: 10.1186/1758-2555-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo UG, Lamberti A, Maffulli N, Denaro V. Tissue engineered biological augmentation for tendon healing: a systematic review. Br Med Bull. 2010 doi: 10.1093/bmb/ldq030. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Ramanath HS, Wang DA. Tendon tissue engineering using scaffold enhancing strategies. Trends Biotechnol. 2008;26:201–9. doi: 10.1016/j.tibtech.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Cao Y, Liu Y, Liu W, Shan Q, Buonocore SD, Cui L. Bridging tendon defects using autologous tenocyte engineered tendon in a hen model. Plast Reconstr Surg. 2002;110:1280–9. doi: 10.1097/01.PRS.0000025290.49889.4D. [DOI] [PubMed] [Google Scholar]
- 9.Yao L, Bestwick CS, Bestwick LA, Maffulli N, Aspden RM. Phenotypic drift in human tenocyte culture. Tissue Eng. 2006;12:1843–9. doi: 10.1089/ten.2006.12.1843. [DOI] [PubMed] [Google Scholar]
- 10.Awad HA, Boivin GP, Dressler MR, Smith FN, Young RG, Butler DL. Repair of patellar tendon injuries using a cell-collagen composite. J Orthop Res. 2003;21:420–31. doi: 10.1016/S0736-0266(02)00163-8. [DOI] [PubMed] [Google Scholar]
- 11.Harris MT, Butler DL, Boivin GP, Florer JB, Schantz EJ, Wenstrup RJ. Mesenchymal stem cells used for rabbit tendon repair can form ectopic bone and express alkaline phosphatase activity in constructs. J Orthop Res. 2004;22:998–1003. doi: 10.1016/j.orthres.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Song XH, Yin Z, Zou XH, Wang LL, Hu H, et al. Stepwise differentiation of human embryonic stem cells promotes tendon regeneration by secreting fetal tendon matrix and differentiation factors. Stem Cells. 2009;27:1276–87. doi: 10.1002/stem.61. [DOI] [PubMed] [Google Scholar]
- 13.Blum B, Bar-Nur O, Golan-Lev T, Benvenisty N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat Biotechnol. 2009;27:281–7. doi: 10.1038/nbt.1527. [DOI] [PubMed] [Google Scholar]
- 14.Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, et al. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson's disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–40. doi: 10.1634/stemcells.2005-0393. [DOI] [PubMed] [Google Scholar]
- 15.Bi Y, Ehirchiou D, Kilts TM, Inkson CA, Embree MC, Sonoyama W, et al. Identification of tendon stem/progenitor cells and the role of the extracellular matrix in their niche. Nat Med. 2007;13:1219–27. doi: 10.1038/nm1630. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Wang JH. Characterization of differential properties of rabbit tendon stem cells and tenocytes. BMC Musculoskelet Disord. 2010;11:10. doi: 10.1186/1471-2474-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rui YF, Lui PP, Li G, Fu SC, Lee YW, Chan KM. Isolation and characterization of multipotent rat tendon-derived stem cells. Tissue Eng Part A. 2010;16:1549–58. doi: 10.1089/ten.TEA.2009.0529. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Akinbiyi T, Xu L, Ramcharan M, Leong DJ, Ros SJ, et al. Tendon-derived stem/progenitor cell aging: defective self-renewal and altered fate. Aging Cell. 2010;9:911–5. doi: 10.1111/j.1474-9726.2010.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–78. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- 20.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920–6. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 21.Lee ST, Yun JI, Jo YS, Mochizuki M, van der Vlies AJ, Kontos S, et al. Engineering integrin signaling for promoting embryonic stem cell self-renewal in a precisely defined niche. Biomaterials. 2010;31:1219–26. doi: 10.1016/j.biomaterials.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 22.Tanentzapf G, Devenport D, Godt D, Brown NH. Integrin-dependent anchoring of a stem-cell niche. Nat Cell Biol. 2007;9:1413–8. doi: 10.1038/ncb1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omae H, Zhao C, Sun YL, An KN, Amadio PC. Multilayer tendon slices seeded with bone marrow stromal cells: a novel composite for tendon engineering. J Orthop Res. 2009;27:937–42. doi: 10.1002/jor.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Wang JH. Mechanobiological response of tendon stem cells: implications of tendon homeostasis and pathogenesis of tendinopathy. J Orthop Res. 2010;28:639–43. doi: 10.1002/jor.21046. [DOI] [PubMed] [Google Scholar]
- 25.Lian JB, Morris S, Faris B, Albright J, Franzblau C. The effects of acetic acid and pepsin on the crosslinkages and ultrastructure of corneal collagen. Biochim Biophys Acta. 1973;328:193–204. doi: 10.1016/0005-2795(73)90345-0. [DOI] [PubMed] [Google Scholar]
- 26.Shuttleworth CA, Forrest L. Changes in guinea-pig dermal collagen during development. Eur J Biochem. 1975;55:391–5. doi: 10.1111/j.1432-1033.1975.tb02174.x. [DOI] [PubMed] [Google Scholar]
- 27.Davison PF, Cannon DJ, Andersson LP. The effects of acetic acid on collagen cross-links. Connect Tissue Res. 1972;1:205–16. [Google Scholar]
- 28.Greco SJ, Liu K, Rameshwar P. Functional similarities among genes regulated by OCT4 in human mesenchymal and embryonic stem cells. Stem Cells. 2007;25:3143–54. doi: 10.1634/stemcells.2007-0351. [DOI] [PubMed] [Google Scholar]
- 29.Pesce M, Scholer HR. Oct-4: gatekeeper in the beginnings of mammalian development. Stem Cells. 2001;19:271–8. doi: 10.1634/stemcells.19-4-271. [DOI] [PubMed] [Google Scholar]
- 30.Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109:1743–51. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- 31.Henderson JK, Draper JS, Baillie HS, Fishel S, Thomson JA, Moore H, et al. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 2002;20:329–37. doi: 10.1634/stemcells.20-4-329. [DOI] [PubMed] [Google Scholar]
- 32.Cui L, Johkura K, Yue F, Ogiwara N, Okouchi Y, Asanuma K, et al. Spatial distribution and initial changes of SSEA-1 and other cell adhesion-related molecules on mouse embryonic stem cells before and during differentiation. J Histochem Cytochem. 2004;52:1447–57. doi: 10.1369/jhc.3A6241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai RY, McKay RD. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 2002;16:2991–3003. doi: 10.1101/gad.55671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–9. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 35.Cooper JA, Lu HH, Ko FK, Freeman JW, Laurencin CT. Fiber-based tissue-engineered scaffold for ligament replacement: design considerations and in vitro evaluation. Biomaterials. 2005;26:1523–32. doi: 10.1016/j.biomaterials.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Theise ND. The stem cell niche and tissue biology. Stem Cell Rev. 2006;2:169–70. doi: 10.1007/s12015-006-0044-5. [DOI] [PubMed] [Google Scholar]
- 37.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 38.James R, Kesturu G, Balian G, Chhabra AB. Tendon: biology, biomechanics, repair, growth factors, and evolving treatment options. J Hand Surg Am. 2008;33:102–12. doi: 10.1016/j.jhsa.2007.09.007. [DOI] [PubMed] [Google Scholar]