

Abstract
There have been numerous investigations targeted at identifying whether a drug lag exists in the mature markets of the US, EU and Japan. This work focuses on the emerging markets because of the potential they hold for the future of the pharmaceutical industry as a consequence of rapid economic and political development.
The aims of this work are to ascertain whether a drug lag exists in the emerging markets and how it has changed over time from the 1960s to the 2000s. It will also highlight key regulatory barriers which may contribute to drug lag.
The date of the marketing authorisation (MA) approval by the US Food and Drug Administration (FDA) was used as a reference point. A comparison against the company database regarding emerging market specific approval enabled the difference in time and thus the drug lag for that particular market to be calculated.
This work concludes that the overall relative drug lag in the emerging markets has decreased over time and that there are seven key regulatory barriers which need to be targeted in order to make further improvements; ‘Western Approval’, local clinical development (LCD), Certificate of Pharmaceutical Product (CPP), Good Manufacturing Practice (GMP), pricing approval, document authentication and harmonisation.
Keywords: Drug lag, BRIC countries, N-11 countries, Emerging Markets, Marketing authorisation, Regulatory barriers
Introduction
The expression ‘drug lag’ was first forged following the US FDA amendments of 1962 which were a consequence of the thalidomide tragedy of the same year. After the tragedy, the FDA was granted the authority to judge drug efficacy, as well as safety. However, these assessment procedures proved to be time consuming1 and thus the time taken to approve a drug for MA appeared to increase.2 A drug lag is any delay in making a drug available in a particular market for the patient and there are two variations: relative drug lag is a measure of when a drug becomes available in a country i.e. the delay in time between a drug being introduced in one country to another and absolute drug lag is a measure of availability i.e. a comparison of the quantity of drugs available in different countries.2
The first empirical study of drug lag was carried out by Wardell in the 1970s3 revealing a drug lag between the US and UK, whereby the US was introducing fewer new drugs than the UK and the drugs that the US did introduce were done so at a later date. Wardell's first paper was published in 19733 with an updated publication in 1978.4 The FDA rejected all claims of a drug lag following the 1978 publication but this was later retracted. A consequence of Wardell's work was the initiation of further studies from the 1980s. Primarily these were US focused, but with time they became more comprehensive with an international scope. Andersson produced a report in 19922 combining his own research along with that of Wardell and other scientists. By taking all of the studies into consideration, a generalised summary of the findings was derived. The UK and West Germany were found to have the shortest delay in approving new drugs with the longest delay being found in the US, Sweden and Norway. In terms of quantity, the US and Norway introduced the least number of new drugs whereas West Germany, France, Italy and the UK introduced the most.2
It is interesting to note the variation in drug lags between the European countries. Prior to the establishment of the European Medicines Agency (EMA) in 1995, all of the European countries had separate regulatory agencies which led to the duplication of MA application (MAA) reviews throughout the EU. The EMA now presides as the common regulatory authority for all EU member states, which according to Business Monitor International, has ‘significantly improved transparency for regulatory requirements and [has resulted in] much swifter approval’.5
More recently the focus of drug lag has been aimed at Japan where a drug lag still exists today, with new drugs being granted MA approval several years after availability in Western countries.6 Japan has begun to reduce its drug lag by recognising global development whereas previously they required full LCD. However, the requirement for some LCD does still exist which is why a drug lag remains.
The foundation of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) in 1990 has enabled the regulatory authorities of Japan, the US and the EU to discuss scientific and technical aspects related to product registration with industry experts.7 ICH also standardised that the regulatory authorities of these three regions mutually recognised and accepted clinical data.8 ICH implementation has lessened superfluous duplication of efforts and helped to harmonise regulations,5 which in turn has helped to reduce drug lag.
The pharmaceutical industry's largest key markets are currently the US, EU and Japan.9 In general, these mature markets have well-established healthcare systems which are government funded, a high level of integration between healthcare and research and development (R&D) and are prescriber driven. All of these factors combined have helped to minimise drug lag. Healthcare in the emerging markets is considerably different; it is usually self-paid, highly fragmented with limited regulations, experiences minimal integration of R&D and the drive has been mainly originated from the pharmaceutical industry. However, these markets are rapidly growing in terms of their economies and changes to their politics also mean that the regulatory environment is improving. Pricewaterhouse Coopers noted that as countries get richer they tend to spend more on healthcare.10 The emerging markets want greater and timelier access to drugs for their populations and to integrate R&D further in order for them to improve their healthcare environment.
BRIC and N-11 countries
This paper focuses on the BRIC and N-11 countries, looking into the drug lags and key regulatory barriers seen there.
The BRIC countries (Brazil, Russia, India and China) were identified by Goldman Sachs in 2001 with the premise that by 2050, the combined BRIC economies could eclipse the combined economies of the world's current richest countries.11 The BRICS have a present combined economy of $15.435 trillion12 and are essentially the fastest growing emerging markets. By 2025, they are estimated to account for over half the size of the G6 countries (US, UK, France, Germany, Italy and Japan) in economic terms, whereas in 2003 they were worth less than 15%. Also, by 2025 the annual increase in US dollar expenditure by the BRICs could possibly be double that of the G6 and even quadruple by 2050.12
The next set of countries investigated, the N-11, were also identified by Goldman Sachs. The countries (where ‘N’ stands for ‘Next’) are Bangladesh, Egypt, Indonesia, Iran, Mexico, Nigeria, Pakistan, the Philippines, South Korea, Turkey and Vietnam. They were identified four years later than the BRICs, as having the capacity to impact the global economy in a BRIC-like fashion in terms of matching and possibly overtaking the G7 (G6 plus Canada) by 2025. The N-11 are fundamentally the next largest developing economy countries by population after the BRICs. This group of countries is very diverse in terms of development and their economies. South Korea, for example, is essentially a developed country whereas Pakistan is one of the world's poorest countries. It is perhaps due to this diversity that makes the N-11 unlikely to be able to rival the BRICs in terms of their impact on the world's economy. They do however have the capability to rival the G7 in terms of growth. The annual economic expansion of the N-11 is envisioned to overtake the G7 in 2033 and be twice it in 2050.13
Previously the emerging markets have had little commercial interest or impact on the pharmaceutical industry with their limited gross domestic product (GDP). Comparing the E7 (Brazil, China, India, Indonesia, Mexico, Russia and Turkey)10 with the G7 in terms of pharmaceutical expenditure helps to highlight this. In 2004 the E7 spent 0.94% of their GDP on prescription medications compared to 1.31% by the G7 and for the same year the E7 accounted for only 8% of the global market versus 79% by the G7. If these countries manage to reach their predicted GDP growth targets and continue to spend the same proportion of their GDP on prescription medications, then by 2020 the E7 is estimated to account for 14% of a global pharmaceutical market worth $800 billion.10 This demonstrates just how big of a potential the emerging markets hold for the future of the pharmaceutical industry in terms of trade and research.
Drug Lag
This work was carried out in order to study the drug lag seen in the BRIC and N-11 countries as earlier investigations have mainly been focused on the mature markets of the US, EU and Japan. It is important to complete this work in order to produce quantitative data which can be used to build our knowledge and understanding of key regulatory issues in the BRIC and N-11 countries. The aims of this work are to give an industry wide, general overview, of how drug lag has changed from the 1960s until the 2000s. It will identify key regulatory barriers, whether they have contributed to drug lag and pinpoint any areas where improvements could be made.
A reference point in terms of the first MA approval of a drug was essential to base countries′ drug approvals on and this resulted in an assumption being made, which was that the first approval of a drug was granted by the FDA. This date was considered as the initial drug approval date. All of the drugs first granted approval by the FDA between the 1960s and the 2000s were researched using the FDA website;14 their submission and approval dates in all fifteen BRIC and N-11 countries were then collected from the company database. This enabled the drug lag in each country to be calculated if applicable (i.e. if the drug was submitted there). The overall drug lag is the relative lag of that drug – from the time it was approved by the FDA to the time it was approved by a specific country. There are two types of lag which make up the relative drug lag: the first is the submission time lag - the time taken from FDA approval to country specific submission for MA; the second is the review time lag - the time taken from country submission to country approval. The medians for both submission and review time lags were calculated to reduce the influence of any outliers of the data set – the median taking into account the position of the data in numerical order rather than the actual numerical value which the mean utilises.
In total there were 132 different drugs from 9 therapeutic areas. In order to remain in scope with this work and considering the time constraints, the data from all BRIC and N-11 countries was consolidated enabling the results to be plotted graphically, firstly by decade (see Figure 1) and secondly by therapeutic area for all decades (see Figure 2).
Figure 1.
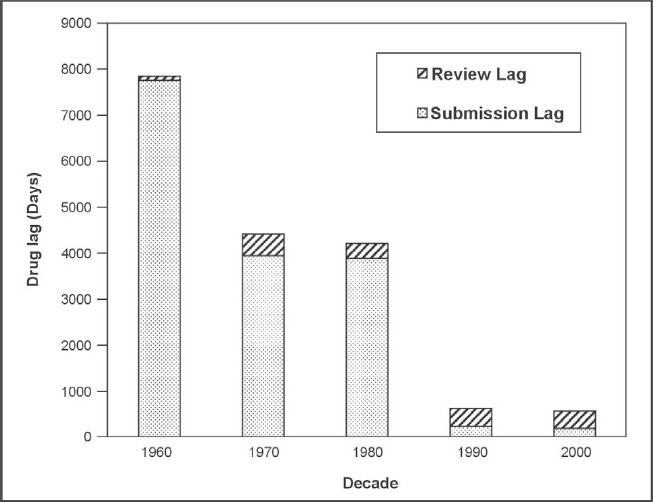
Median lag time from FDA approval to country approval for drugs in BRIC and N-11 countries
Figure 2.
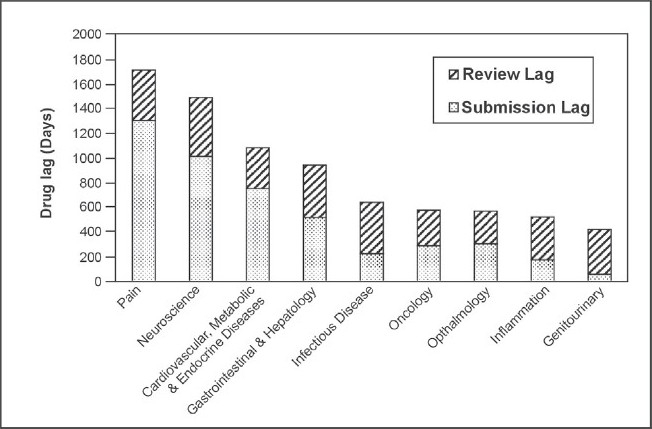
Median lag from FDA approval to country approval for drugs in BRIC and N-11 countries from 1960s-2000s
Future studies could be aimed at analysing the fifteen countries individually in order to get a more specific picture of their personal regulatory strengths and weaknesses, progress they have made so far and future direction.
Figure 1 shows a decrease in the relative drug lag, from 7843 days in the 1960s to 560 days in the 2000s for the BRIC and N-11 countries. Submission lag has continuously decreased throughout the decades (7754 days in the 1960s in comparison to only 190 days in 2000) whereas the MAA review time lag has fluctuated and did increase from 327 days in the 1980s to 388 days in the 1990s. The review lag for the 2000s (until November 2009 when the data was collected) was slightly less than that for the previous decade at 370 days.
The decrease in relative lag is a consequence of the rapid reduction in submission lag over the decades despite an increase in the MAA review lag. The reduction in the submission lag is a key result of the increase in the commercial interest towards the BRIC and N-11 countries from pharmaceutical companies. It was determined by Cullen that pre 1969, the barriers to drugs going for MA approval were mainly commercial and that post 1969 they were more varied.15 This would support the notion that because these countries were much less commercially appealing to the companies due to their lack of GDP in earlier decades, the submission lag for that time was much longer. The submission lag rapidly decreased from 3885 days in the 1980s to 230 days in the 1990s demonstrating a positive relationship between rapid country GDP growth and a reduction in submission lag. It has been seen that regulatory authorities in the emerging markets are evolving16 and the fluctuation in review lag and particularly the increase from the 1980s versus the 1990s and 2000s could be due to the regulatory authorities of the BRIC and N-11 countries becoming more stringent in their MAA review process. There was less data available for the earlier decades and the 1960s in particular which may explain the large difference between the data collected for the 1960s and 1970s.
Figure 2 shows a general overview of drug lag times for these nine therapeutic areas for the past four decades combined. A trend is seen which indicates that life saving drugs (such as those for the therapeutic areas of oncology and infectious disease) are approved faster than those prescribed for non-life threatening illnesses (i.e. pain drugs which have the longest drug lag time in this work), which is logical. If the data was split by decade, since the 1990s in particular, it is likely that oncology and infectious disease drugs would have the lowest lag times overall due to the ‘fast track’ approval process (essentially a priority review) for certain drugs. This was the case for the HIV/AIDS drug PRO 140 by the FDA in February 200617 and the first oncology drug to receive fast-track status was liposomal doxorubicin in 1995.18 Roberts and Chabner concluded that the MAA review time of drug can be reduced by up to 4 months due to the fast-track process.17 Infectious disease drugs are shown to have the third longest MAA review time lag of 415 days which is longer than would be expected, but this may be due to the potential toxicity of these drugs. However, anticancer drugs are also one of the most toxic classes of prescribed medication19 but the MAA review time lag of the oncology drugs in this work was the lowest of all therapeutic areas at 289 days. This disparity may be due a greater prevalence of infectious disease drugs in older decades compared to oncology drugs and thus the shorter, more modern lag times could have helped to decrease the lag seen for oncology.
Comparing Figure 1 and 2, there is a pronounced difference in the maximum relative lag times – 7843 days and 1717 days respectively. The later decades had much shorter drug lags which is illustrated in Figure 1 and this may have counterbalanced the lengthier older lag times when the data from all decades was combined in order to produce Figure 2, resulting in a much shorter maximum relative lag time.
Regulatory barriers
By researching the company database, this work has identified that there are seven key regulatory barriers affecting the drug lag witnessed in the BRIC and N-11 countries. These barriers are Western approval, LCD, CPP, GMP, pricing approval, document authentication and harmonisation. These barriers need to be overcome in order to reduce drug lag further in the future.
Western approval
There have been few instances to date where a drug has been granted its first worldwide approval in a BRIC or N-11 country. This could be due to the additional testing requirements that these countries have in order to comply with their local guidelines.20 In order to submit an MAA in the BRIC and N-11 countries, companies usually have to wait for an established regulatory authority such as the FDA or EMA to grant approval of that drug. The reliance on these Western countries has led to this barrier being labelled as ‘Western approval’.
LCD
LCD is currently required by China and India at Phase III, compared to Korea who primarily require bridging studies which are not as extensive and are used to ‘bridge the gap’ between the requirements and the data Korea receives from the pharmaceutical company. Mexico and Vietnam are currently considering the requirement of LCD. The BRIC and N-11 countries (and emerging markets in general) have sometimes not been highly regarded enough to be included in global clinical development (GCD) due to lack of infrastructure and resources. Different ethnicities have different metabolisms which can sometimes mean variations in the actions of drugs on patients. Some of the most developed BRIC and N-11 countries such as China, India and Korea have thus decided that they need LCD in order to protect their populations. To carry out LCD, an investigational new drug (IND) application has to be approved as a consequence of positive results, a new drug application (NDA) is submitted in that country and a full MAA review can then be undertaken. LCD was formerly carried out sequentially to GCD, which led to very extreme drug lags. It is preferable for it to be in parallel with, or part of, GCD which has helped to reduce the lag to some extent. GCD has expanded widely in recent years and so LCD may become irrelevant and costly in terms of extra resources. This irrelevance will only increase, as the BRIC and N-11 countries are likely to participate much more in GCD in the near future.
CPP
The World Health Organisation (WHO) primarily devised CPPs as a way of enabling regulatory authorities to ascertain the GMP and quality status of the drug product20 which has been submitted for MAA review. It helps to establish that the drug product in the approval market is the same quality as that of the product which is being imported. A CPP also provides information on the product itself and its regulatory status in the issuing market. A CPP can be issued by an authority when a drug receives approval in that country and may be required for product submission or approval in another country. However, the FDA will not issue a CPP unless the product will be exported from within the US. For authorities requiring a CPP at the time of submission, the applicant company has to wait for the issuing country to have approved the drug which can take up to 18 months, thus significantly delaying registration in the new market.20 The time taken to issue a CPP varies between authorities but may take from 2 weeks up to 9 months. Regulatory authorities can require a CPP to be issued from the ‘source country’ i.e. the manufacturer. However, in today's global environment, pharmaceutical companies can have manufacturing split over separate specialised sites in order to increase efficiency and reduce costs and therefore the source country may not be the same as that where the drug has firstly been granted MA approval. This may further delay submission or approval of the drug. The pharmaceutical industry is proposing that authorities should accept a single CPP from recognised, competent agencies as long as the GMP status of the manufacturing site is included.20 Furthermore, regulatory authorities sometimes require more than one CPP or that a CPP is issued with a marketed statement, which adds further to drug lag, because despite a drug being approved, the company has to wait for product launch in the issuing market before it can request a CPP and thus submit for MAA review.
GMP
Most WHO CPPs carry a GMP statement but some recipient authorities continue to request a separate GMP certificate which is an unnecessary duplication and waste of resources. Some agencies, including the UK's Medicines and Healthcare products Regulatory Agency (MHRA) and Ireland's Irish Medicines Board (IMB) have stopped issuing GMP statements.
Pricing approval
Price certificate requirement is another important barrier Brazil, Egypt and Indonesia. Price certificates are an agreement between the pharmaceutical company and health authority as to the price at which the drug will be sold when MA is granted. The review and approval process for pricing is sometimes integrated into that of the MAA review, delaying the latter further. In order to reduce drug lag, it would be preferable to carry out these processes separately, particularly as they have different focuses – the drug review process being scientific and the pricing review process being policy based. It is thus the pricing approval and negotiation that is the source of the time delay with the price certificate just being a tool used within that process.
Document authentication
Authorities can request that CPPs, GMP and price certificates are legalised and notarised. Notarisation can take only a few weeks to be carried out – this is done by a notary, of which most companies now have their own. Legalisation however is a lengthy process. It is granted by the embassy of the importing country based in the exporting country and can take many months, but it is dependent on the legislation of the exporting country. In 1961, the Hague convention brought the legalisation issue to a solution for many countries in the form of apostille.21 Apostille is a standardised and faster process than legalisation and is accepted by the health authorities of the signatory countries in principal. The apostille certificate ensures that documents require no further legalisation in order to be recognised by the member states. Of the BRIC and N-11 countries, six are signatories of the Hague convention; China, India, Korea, Mexico, Russia and Turkey.21 In practice, the health authorities have not translated this into the local regulation and therefore legalisation is still required by many health authorities of the signatory countries. It would reduce the submission lag further if regulatory authorities agreed to accept authentication by the notary of the submitting company, the regulatory agency who was actually issuing the certificate or by not requiring this additional step.
Harmonisation
It is evident that a lack of harmonisation between countries can lead to ‘unnecessary duplication of effort’ and a ‘waste of valuable resources’.20 In short, this can increase drug lag. The first harmonisation initiative to be implemented was the Association of South East Asian Nations (ASEAN) in 1967. Three out of the fifteen countries studied in this work are ASEAN members; Indonesia, the Philippines and Vietnam. Collaborations are underway to instil the Sectoral Mutual Recognition Arrangement for GMP Inspection of Manufacturers of Medicinal Products which would harmonise quality standards and is expected to be in place by January 2011.20 The GMP harmonisation will help to improve pharmaceutical trade between ASEAN member states by removing impeding barriers. It is the feeling of some industry experts that the countries of Latin America (Brazil and Mexico in this work) seem to be diverging from regulatory harmonisation. However it should be noted that in 2008 Brazil's regulatory authority Agencia Nacional de Vigilancia Sanitaria (ANVISA) announced that they were going to harmonise with Argentina's Administracion Nacional de Medicamentos, Alimentos y Tecnologia Medica (ANMAT) with the intention of also sharing analytical expertise.20 This is a positive move by these two Latin America countries which may hopefully lead the way for other countries in the region. The EU has seen a large reduction in drug lag, not just due to the establishment of the EMA but due to there being clear expectations of the agency and set approval timelines. The EMA has to commit to meet its 200 day approval timeline and it is upon this commitment which the agency is assessed. Both the EMA and ASEAN initiatives have been established for many years now and based upon the progress seen recently in the BRIC and N-11 countries, it would be possible to suggest further harmonisation for them as it is clear that harmonisation does work in bringing countries together and can be highly effective in reducing drug lag provided that clear guidelines and targets are set.
Conclusion
A drug lag is any delay in making a drug available in a particular market for the patient which can have very serious consequences. Mature markets which once had very lengthy drug lags, such as the US and EU, have seen dramatic improvements over time due to the evolvement of their regulatory environments. The BRIC and N-11 countries are the fastest growing emerging markets in terms of their GDP and this work has revealed a large reduction in the relative drug lag of these countries between the 1960s and 2000s, possibly due to the increased commercial interest from the pharmaceutical industry and changes in the political climate of the countries. Further reduction of the drug lags in these 15 countries is possible if the key regulatory barriers of ‘Western Approval’, LCD, CPP, GMP, pricing approval, document authentication and harmonisation are targeted.
Future studies in this field could be focused on the BRIC and N-11 ‘Key Markets’ – namely Brazil, China, India, Korea, Mexico, Turkey and Russia. By examining these markets individually, it would be easier to target the areas where they can specifically improve their regulatory barriers, thus leading the way for the rest of the BRIC and N-11 countries.
Declaration
The authors declare that the work enclosed is purely their own and has not been taken from the work of others; save the extent that such work has been cited and acknowledged within the text of this work.
At the time of this work both authors were employees of Pfizer Ltd, Kent, United Kingdom. As of 1st March 2010, Arun Mishra will be an employee of GlaxoSmithKline, Brentford, Middlesex, United Kingdom. There are no competing interests.
Acknowledgments
The authors would like to thank Pfizer Ltd for allowing them to take the time to carry out this work and to their fellow colleagues, Julie Dennis and Fraser Stodart for their advice and support.
References
- 1.Henninger Daniel. Drug Lag. The Concise Encyclopedia of Economics, Library of Economics and Liberty. 1993. [Accessed 24 September 2009]. Available at: http://www.econlib.org/library/Enc1/DrugLag.html .
- 2.Andersson F. The drug lag issue: the debate seen from an international perspective. International Journal of Health Services. 1992;22(1):53–72. doi: 10.2190/9Y32-X86Y-M3F0-JQFC. [DOI] [PubMed] [Google Scholar]
- 3.Wardell W M. Introduction of new therapeutic drugs in the United States and Great Britain: An international comparison. Clin. Pharamacol Ther. 1973;14:773–790. doi: 10.1002/cpt1973145773. [DOI] [PubMed] [Google Scholar]
- 4.Wardell W M. The drug lag revisited: Comparison by therapeutic area of patterns of drugs marketed in the United States and Great Britain from 1972 through 1976. Clin. Pharamacol. Ther. 1978;24:499–524. doi: 10.1002/cpt1978245499. [DOI] [PubMed] [Google Scholar]
- 5.Business Monitor International. Pharmaceutical Regulatory Convergence: The Case For A Global Agency. 2009 [Google Scholar]
- 6.Hashimoto J, Ueda E, Narukawa M. The Current Situation of Oncology Drug Lag in Japan and Strategic Approaches for Pharmaceutical Companies. Drug Information Journal. 2009;43(6):757–765. [Google Scholar]
- 7.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (n.d.) History and Future of ICH. [Accessed 11 January 2010]. Available at: http://www.ich.org/cache/compo/276-254-1.html .
- 8.European Medicines Agency (2006) ICH Topic E6 (R1) Guideline for Good Clinical Practice. [Accessed 25 January 2010]. Available at: http://www.ema.europa.eu/pdfs/human/ich/013595en.pdf .
- 9.European Federation of Pharmaceutical Industries and Associations (2009) The Pharmaceutical Industry in Figures. [Accessed 22 January 2010]. Available at: http://www.efpia.eu/Content/Default.asp?PageID=559&DocID=4883 .
- 10.Pricewaterhouse Coopers (2007) Pharma 2020: The Vision - Which Path will you take? [Accessed 15 December 2009]. Available at: http://www.pwc.com/pharma2020 .
- 11.Wilson D, Purushothaman R. Global Economics Paper No: 99. s.l. The Goldman Sachs Group, Inc; 2009. Dreaming With BRICs: The Path to 2050. [Google Scholar]
- 12.Wikipedia contributors (n.d.) BRIC. Wikipedia, The Free Encyclopedia. [Accessed 29 September 2009]. Available at: http://en.wikipedia.org/wiki/BRICs .
- 13.Wilson D, Stupnytska A. Global Economics Paper No: 153. s.l. The Goldman Sachs Group, Inc; 2007. The N-11: More Than an Acronym. [Google Scholar]
- 14.U.S. Food and Drug Administration (n.d.) Drugs @ FDA. [Accessed 26 October 2009]. Available at: http://www.accessdata.fda.gov/Scripts/cder/DrugsatFDA/index.cfm .
- 15.Cullen R. Pharmaceuticals inter-country diffusion. Managerial and Decision Economics. 1983;4:73–82. [Google Scholar]
- 16.McAuslane N, Cone M, Collins J, Walker S. Emerging Markets and Emerging Agencies: A Comparative Study of How Key Regulatory Agencies in Asia, Latin America, the Middle East and Africa Are Developing Regulatory Processes and Review Models for New Medicinal Products. Drug Information Journal. 2009;43:349–359. [Google Scholar]
- 17.AIDS info (2009) PRO 140. [Accessed 21 December 2009]. Available at: http://www.aidsinfo.nih.gov/DrugsNew Drug Detail T.aspx?
- 18.Medscape Today. Postmarketing Surveillance for Oncology Drugs: Approval of Oncology Medications. 2009. [Accessed 21 December 2009]. Available at: http://www.medscape.com/viewarticle/585694_2 .
- 19.McLeod Howard L. Clinically relevant drug-drug interactions in oncology. [Accessed 21 December 2009];British Journal of Clinical Pharmacology. 1998 45(6):539–544. doi: 10.1046/j.1365-2125.1998.00719.x. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1873644/Courtesy of the British Pharmacological Society . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAuslane N, Cone M, Collins J. A crossregional comparison of the regulatory environment in emerging markets. R&D Briefing 50. 2006. Available on request at: http://www.cmr.org/institute .
- 21.The Official Apostille Service. Hague Convention. 2008. [Accessed 18 December 2009]. Available at: http://www.apostille.org.uk/hague.asp .