

Abstract
Not all Phase IV studies are post-marketing surveillance (PMS) studies but every PMS study is a phase IV study. Phase IV is also an important phase of drug development. In particular, the real world effectiveness of a drug as evaluated in an observational, non-interventional trial in a naturalistic setting which complements the efficacy data that emanates from a pre-marketing randomized controlled trial (RCT). No matter how many patients are studied pre-marketing in a controlled environment, the true safety profile of a drug is characterized only by continuing safety surveillance through a spontaneous adverse event monitoring system and a post-marketing surveillance/non-interventional study. Prevalent practice patterns can generate leads that could result in further evaluation of a new indication via the RCT route or even a signal that may necessitate regulatory action (change in labeling, risk management/minimization action plan). Disease registries are another option as are the large simple hybrid trials. Surveillance of spontaneously reported adverse events continues as long as a product is marketed. And so Phase IV in that sense never ends.
Keywords: Non-interventional/observational, post-marketing safety surveillance, generalizability, effectiveness, real world
JUST as Phase I is sometimes referred to as the acid test of drug development (where the rubber meets the road), since it is the first time that the drug is being tested in humans, Phase IV may be considered as the real test since for the first time that the drug is tested in the real world.
Drug products are launched after regulatory authorities have scrutinized a vast amount of data from animal and clinical studies and found it to show that the drug is sufficiently effective and adequately safe in specified indications. The popular notion is that drugs are thoroughly studied before they are marketed, so that everything about the drug is known the time of launch. Few realize that while enough is known the time of launch to avoid calamities, catastrophes and disasters, a lot of the thorough knowledge that we have about well-established products is obtained after the drug has been marketed and hundreds of thousands of patients have been exposed to the product through commercial sales. This is so because of 3 primary reasons:
It is not possible to study more than a few thousand patients in clinical trials. The economics of the pharmaceutical industry does not allow for more money and time to be spent on pre-launch development than is done currently. Anymore pre-launch spending would make drugs even more expensive than they are today and render them unmarketable and also delay its reach to patients.
A lot of the additional knowledge about drugs comes from scientific, rather than commercial interest, through research done by individual workers in universities and research institutions and by groups of investigators with academic interest in the drug or in therapeutics. Generally, such studies are possible only after the drug receives regulatory approval and becomes commercially available.
Some of the new knowledge about a drug is obtained by serendipity when doctors all over the world use the drug in a wide spectrum of patients, with varied ethnicity, various underlying diseases, and a range of concomitant medication.
RCTs are essential to prove efficacy or the fact that a drug works but are inevitably limited in generalizability as extrapolation of the results from RCTs can only be to patients included in the RCTs under controlled conditions (strict inclusion and exclusion criteria, drug provided free of cost, compliance monitored, etc). In the real world no patient can be excluded; even pregnant and lactating women, those with hepato-renal dysfunction, on multiple concomitant medications for concomitant clinical conditions must be treated. How the drug performs in such real world conditions is a test of its effectiveness. All studies conducted in a phase IV setting, i.e., after marketing authorization approval per label are called phase IV studies. Of these, those mandated by the regulatory authority to be conducted as observational studies in a naturalistic setting per label are called PMS studies.
Non-Interventional Studies (NIS)
By definition, an NIS is a study conducted to assess safety, tolerability and effectiveness of marketed medicines in clinical practice, i.e., in a naturalistic setting where choice of therapy is consistent with approved prescribing information (no study drug to be supplied) and in line with current practice at the study site; other aspects of patient-care, including clinical examinations, laboratory investigations, and the use of instrumentation, other invasive and non-invasive procedures are in consonance with current practice at the study site. The drug is prescribed per routine practice per label, the doctor's decision to prescribe precedes decision to enroll preferably by at least a month, there is no systematic assignment of treatment, there is no protocol (it is called an observational plan), informed consent essentially comprises a data privacy clause, there is no investigator indemnity, and there are no additional diagnostic tests or visits beyond what would anyway be done per usual practice. In the current scenario, NIS do not need regulatory approval but it is a good practice to register it on the Clinical Trials Registry of India (CTRI) website.
There is a mistaken perception that such studies are done only to increase sales. In fact since one is merely capturing actual use of the product there should be no real increase in sales. There is a debate on whether off-label use should be captured. If a company were to do such a study it may be wrongly perceived that company is trying to promote off- label prescribing. However the point is that it is only through knowledge of real world practice that sometimes new ways of using the product are discerned, e.g., a new route (nimesulide per rectal as reported in Nise PMS study), or new indication, e.g., low dose aspirin as an anti-platelet agent. The company should not use this data but instead apply to the regulator to do a formal study in this new indication. Thus medicine advances.
NIS thus allow information to be collected on the actual use of a particular drug. Usual clinical practice must always be adhered to in these studies and no additional diagnostic or monitoring procedures (i.e., additional visits) can be applied to the patients. Besides providing greater knowledge about drug effects, non-interventional studies can also be a good way of further mapping risks in the real world.
Large simple trial (LST)
It is a hybrid between a randomized clinical trial and an observational study (e.g., cohort study). A large number of participants are randomized to treatment groups, with follow-up per routine practice. Simple refers to the effort of enrolling physicians and participants and the objective is to interfere with routine practice as little as possible (Table 1). One example of such a study was the VOLUME study on Exubera (inhaled insulin) which was a requirement of US FDA as part of a risk management plan. In fact there is a school of thought that going forward regulators may insist on sponsors doing pre-marketing studies (phase III) that closely mimic the real world (instead of the typical RCTs which would be done in phase IIb – robust proof of efficacy) so that there is greater confidence of the drug's effectiveness prior to approval. This could minimize drug withdrawals but has the potential to delay launch of the drug. While an RCT maximizes validity but has limited generalizability, and an observational study has limited validity (depends on appropriateness of design and control for bias) but maximizes generalizability, a large simple trial maximizes validity and generalizability.
Table 1.
Large Simple Trial
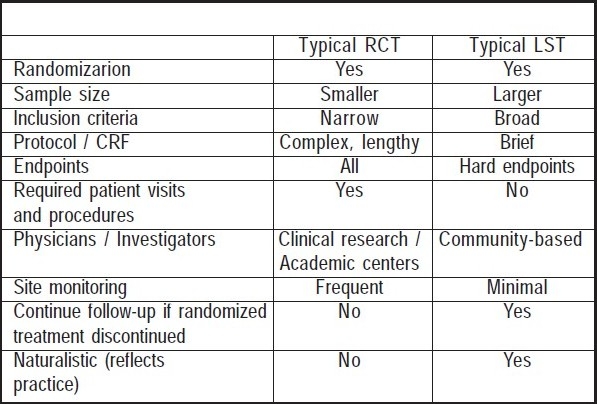
Post-Marketing Surveillance (PMS) Studies
Adverse reactions that occur in fewer than 1 in 3,000 – 5,000 patients are unlikely to be detected in Phase I – III investigational clinical trials, and may be unknown at the time a drug is approved. These rare adverse reactions are more likely to be detected when large numbers of patients are exposed to a drug after it has been approved and marketed.
Safety monitoring, nevertheless, is just one form of Post-PMS. Another is the planned collection of clinical data relating to the use of a drug through the conduct of PMS studies. These could be general, open studies where unlike pre-marketing studies, the selection of patients is not strictly defined by stringent inclusion and exclusion criteria, but governed by the permissible indications and contra-indications of the drug as stated in the text of prescribing information. This ensures that information is collected in a varied spectrum of patients, and makes it likely that the study will yield data that may not have been captured in Phase III studies.
PMS studies exemplify the difference between efficacy and effectiveness. Efficacy is judged within the controlled environment of a clinical trial with strict inclusion and exclusion criteria and close monitoring and ensured compliance. Effectiveness is the real test of a drug when it is used in a much larger population, with varied organ system function, concomitant drugs and where monitoring and compliance are not always ensured. In other words, a PMS study is a non-interventional study requested by regulatory authorities to verify the safety, tolerability and effectiveness of a marketed drug in a particular population per the locally approved label.
Conducting such general, open-label PMS studies is a regulatory requirement in countries such as Japan and the Philippines. In India, PMS data used to be submitted to the Drugs Controller General of India (DCGI) within 2 years of launch. Now Periodic Safety Update Reports (PSURs) are filed at regular intervals as specified in the revised Schedule Y of the Drugs and Cosmetics Act. Most, other regulatory authorities, however, do not insist on PMS studies. Instead, in countries such as Germany, regulators may require a company to conduct controlled clinical studies under precisely defined enrollment criteria, to investigate specific concerns and gather information about the drug under specific conditions of use when there is a suspected problem.
The outcomes of such studies could be signals, pharmacoepidemiological information, need for controlled studies, labeling changes with modified undesirable effects section, indications and dosing schedules, and regulatory action (boxed warning, risk minimization action plan, withdrawal). Other phase IV studies could be RCTs, in vitro studies, outcomes research (burden of illness) and pharmacoeconomic studies, drug utilization studies, practical clinical trials, and investigator-initiated research in practice.
Adverse event monitoring
The number of patients one would need to observe to have a 95% chance of detecting 1, 2, or 3 cases of an adverse reaction at a given incidence of the reaction can be gauged from this table:1
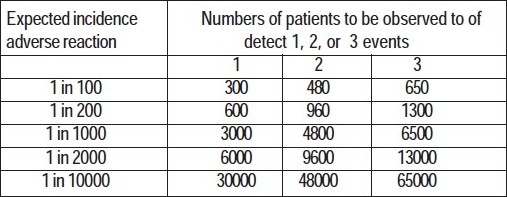
It is evident from this that rare, but fatal side effects such as aplastic anaemia seen with chloramphenicol, or retinal damage with high dose chloroquine therapy can only be detected if there is a system of collecting adverse event information from customers once a drug has been marketed.
Safety monitoring continues for the life of a drug. Pharmaceutical companies with worldwide operations have established large global systems to track, investigate, and evaluate adverse drug events (Aes) for their products on a continuing basis and report them to regulatory authorities around the world. When a doctor fills up adverse event report (AER) form s/he is playing a vital role in this global endeavor. S/he is in effect a continuing surveillance source for products and shoulders responsibility for the safety of patients. The reporting of Aes helps the company evaluate them for relatedness and accordingly this may lead to change in labeling, if required.
Serious and unexpected suspected adverse reactions (SUSARs) are reported to regulatory authorities on a continual basis and non-serious ones are compiled and reported periodically.2 Changes in prescribing information are undertaken by the company on the basis of these reports, either at the behest of regulatory authorities or, often, voluntarily. On occasion the drug may be withdrawn from the market.
Case-Control Studies
PMS studies conducted after the launch of a product are part of Phase IV development of the drug. Some of these studies may be retrospective case-control evaluations. These are done to evaluate rare suspected side effects. For example, when there was a suspicion that use of oral contraceptives may be associated with an increased incidence of thrombophlebitis (clotting of blood in the deep veins) and thromboembolism (blockage of smaller arteries due to detached blood clots) case-control studies were carried out. A group of cases of thromboembolism were compared with age matched controls that were as similar to the cases as possible, but without the disease. The fact that the rate of oral contraceptive consumption among the two groups did show a statistical difference indicated that oral contraceptive use is in fact associated with a 2-4 fold increase in incidence of embolic phenomena. Cohort and cross-sectional studies may also be done as part of comparative observational studies in pharmacovigilance planning.
Drug Utilisation Studies (DUS)2
Such studies describe how a drug is marketed, prescribed, and used in a population, and how these factors influence outcomes, including clinical, social, and economic outcomes.3 These studies provide data on specific populations, such as the elderly, children, or patients with hepatic or renal dysfunction, often stratified by age, gender, concomitant medication, and other characteristics. DUS can be used to determine if a product is being used in these populations. From these studies denominator data can be developed for use in determining rates of adverse drug reactions. DUS have been used to describe the effect of regulatory actions and media attention on the use of drugs, as well as to develop estimates of the economic burden of the cost of drugs. DUS can be used to examine the relationship between recommended and actual clinical practice. These studies can help to determine whether a drug has the potential for drug abuse by examining whether patients are taking escalating dose regimens or whether there is evidence of inappropriate repeat prescribing. Important limitations of these studies can include a lack of clinical outcome data or information of the indication for use of a product.
Registry
A prospective observational study of patients with certain shared characteristics (e.g., particular disease, exposure, or risk factor) that collects ongoing and supporting data over time on well-defined outcomes of interest for analysis and reporting. Properly designed and executed, registries can provide a real-world view of clinical practice, patient outcomes, safety, and comparative effectiveness.
Conclusion
Thus, we find that product launch is merely a milestone in drug development, albeit an important one, rather than a mark of the end of the development process. Inevitably, however, the investments made late in Phase IV, usually a declining phase of the product life-cycle, are much smaller than commitments during the early growth phase. Not only have most of the important questions been answered but also the commercial interest in answering residual or newly emergent questions is low towards the end of the patent period and the potential commercial gains from use of new data are small in the face of emerging new therapies that have been designed to surpass the older agents. Commercial development of a drug in fact only ends with, or close to, the end of patent life. But surveillance of spontaneously reported AE continues as long as a product is marketed. And so Phase IV in that sense never ends.
References
- 1.Grahame-Smith DG, Aronson JK, editors. Oxford Textbook of Clinical Pharmacology. 2nd ed. Oxford University Press; 2004. p. 117. (reprinted), Chapter 9. [Google Scholar]
- 2.International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH) Guideline on Pharmacovigilance Planning, E2E. 2004 Nov 18th; doi: 10.1111/j.1365-2125.1994.tb05705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strom BL, editor. Pharmacoepidemiology. 3rd ed. New York, NY: John Wiley and Sons, Ltd; 2002. [Google Scholar]