

Abstract
The high-throughput expression analysis technologies available today give scientists an overflow of expression profiles but their resolution in terms of tissue specific expression is limited because of problems in dissecting individual tissues. Expression data needs to be confirmed and complemented with expression patterns using e.g. in situ hybridization, a technique used to localize cell specific mRNA expression. The in situ hybridization method is laborious, time-consuming and often requires extensive optimization depending on species and tissue. In situ experiments are relatively more difficult to perform in woody species such as the conifer Norway spruce (Picea abies). Here we present a modified DIG in situ hybridization protocol, which is fast and applicable on a wide range of plant species including P. abies. With just a few adjustments, including altered RNase treatment and proteinase K concentration, we could use the protocol to study tissue specific expression of homologous genes in male reproductive organs of one gymnosperm and two angiosperm species; P. abies, Arabidopsis thaliana and Brassica napus. The protocol worked equally well for the species and genes studied. AtAP3 and BnAP3 were observed in second and third whorl floral organs in A. thaliana and B. napus and DAL13 in microsporophylls of male cones from P. abies. For P. abies the proteinase K concentration, used to permeablize the tissues, had to be increased to 3 g/ml instead of 1 g/ml, possibly due to more compact tissues and higher levels of phenolics and polysaccharides. For all species the RNase treatment was removed due to reduced signal strength without a corresponding increase in specificity. By comparing tissue specific expression patterns of homologous genes from both flowering plants and a coniferous tree we demonstrate that the DIG in situ protocol presented here, with only minute adjustments, can be applied to a wide range of plant species. Hence, the protocol avoids both extensive species specific optimization and the laborious use of radioactively labeled probes in favor of DIG labeled probes. We have chosen to illustrate the technically demanding steps of the protocol in our film.
Anna Karlgren and Jenny Carlsson contributed equally to this study.
Corresponding authors: Anna Karlgren at Anna.Karlgren@ebc.uu.se and Jens F. Sundström at Jens.Sundstrom@vbsg.slu.se
Keywords: Plant Biology, Issue 26, RNA, expression analysis, Norway spruce, Arabidopsis, rapeseed, conifers
Protocol
This non-radioactive mRNA in situ hybridization protocol is optimized for Norway spruce tissues and described in detail. It is based on an Arabidopsis/rapeseed in situ hybridization protocol optimized in our groups, which in turn is based on protocols from the Meyerowitz and Irish labs and optimized by Vivian Irish, Cindy Lincoln and Jeff Long among others 1-4.
PROCEDURE
1. FIXATION AND EMBEDDING
To provide RNA retention tissues need to be fixated and embedded in wax before an in situ experiment is performed. The fixation and embedding is a critical step since poorly fixed materials might give a low or undetectable signal even though the mRNA is highly abundant. The tissues are dehydrated, cleared and embedded in wax in gradual changes to avoid tissue damage. To further avoid shrinkage and swelling of the cells 0.85% NaCl is added to the first ethanol steps in the dehydration of Norway spruce tissues. It is possible to leave out the NaCl in the ethanol (e.g. it is left out in the Arabidopsis/rapeseed protocol). The dehydration step during embedding may be performed on ice to keep RNase activity at minimum or at +4°C. In this protocol the 4% paraformaldehyde fix solution contains 0.25% glutaraldehyde, which is supposed to give a better RNA retention even though that some argue that it probably makes no difference (e.g. it is left out in the Arabidopsis/rapeseed protocol). Most likely any standard fixation and embedding protocol can be used. As seen below the Norway spruce embedding differs slightly from the Arabidopsis/rapeseed protocol.
1.1 Day 1 Collecting plant material and paraformaldehyde fixation
Collect the plant material of interest and dissect it if necessary. Keep the tissues on ice and place them in ice-cold fix solution (see recipes below) directly or as soon as possible. N.B.! Keep on ice all the time!
Apply vacuum to the samples until the paraformaldehyde starts to bubble. Hold the vacuum for up to three hours (e.g. 2 x 15 min for Arabidopsis and rapeseed floral tissues or one hour for Norway spruce male cones) and release the vacuum slowly. The tissues are supposed to sink after the vacuum infiltration of the fixative, but for certain tissues containing a lot of air it is not possible (e.g. Arabidopsis flowers). A piece of gauze can be used to make the tissues stay in the fixative.
Replace the fixative and incubate at +4°C overnight (~12-16 hours) gently shaking.
Option 1
N.B.! Norway spruce embedding version below (steps 4-8). Make sure scintillation tubes or glass vials are used, at least in step 5.
1.2 Day 2 Dehydration N.B.! Norway spruce embedding version.
0.85% NaCl 30 min on ice 50% EtOH + 0.85% NaCl 90 min on ice 70% EtOH + 0.85% NaCl 90 min on ice 85% EtOH + 0.85% NaCl 90 min +4°C 95% EtOH (+ eosin) 90 min +4°C 100% EtOH (+ eosin) 90 min +4°C 100% EtOH (+ eosin) overnight +4°C
1.3 Day 3 Dehydration and embedding N.B.! Norway spruce embedding version.
100 % EtOH (+ eosin) 2 h room temperature 50% EtOH + 50% Histoclear II 1 h room temperature 100% Histoclear II 1 h room temperature 100% Histoclear II 1 h room temperature 50% Histoclear II + 50% Histowax overnight +40-50°C
1.4 Day 4 Embedding N.B.! Norway spruce embedding version.
100 % melted Histowax +60°C (Change morning and evening)
1.5 Day 5 Embedding N.B.! Norway spruce embedding version.
100 % melted Histowax +60°C (Change morning and evening)
1.6 Day 6 Embedding N.B.! Norway spruce embedding version.
100 % melted Histowax +60°C (Change morning and evening)
Option 2
N.B.! Arabidopsis/rapeseed embedding version below (steps 4-8).
1.2 Day 2 Dehydration N.B.! Arabidopsis/rapeseed embedding version.
- N.B.! All steps at +4°C and gently shaking, if not else is stated.
1x PBS 30 min 1x PBS 30 min 30% EtOH 60 min 40% EtOH 60 min 50% EtOH 60 min 60% EtOH 60 min 70% EtOH 60 min 85% EtOH 60 min 95% EtOH (+ eosin) overnight
1.3 Day 3 Dehydration and embedding N.B.! Arabidopsis/rapeseed embedding version.
- N.B.! All steps at room temperature and gently shaking, if not else is stated.
100% EtOH (+ eosin) 30 min 100% EtOH (+ eosin) 30 min 100% EtOH (+ eosin) 60 min 100% EtOH (+ eosin) 60 min 75% EtOH + 25% Histoclear II 30 min 50% EtOH + 50% Histoclear II 30 min 25% EtOH + 75% Histoclear II 30 min 100% Histoclear II 60 min 100% Histoclear II 60 min 100% Histoclear II + ¼ volumes Histowax chip overnight (no shaking)
1.4 Day 4 Embedding N.B.! Arabidopsis/rapeseed embedding version.
Place the vials with tissues at +42°C until the wax chips melt completely. Next add ¼ volume of wax chips and let them melt completely. Move to +60°C and leave the vials for several hours. Finally, replace wax/Histoclear II with freshly melted wax and incubate over night at +60°C.
1.5 Day 5 Embedding N.B.! Arabidopsis/rapeseed embedding version.
100 % melted Histowax +60°C (Change morning and evening)
1.6 Day 6 Embedding N.B.! Arabidopsis/rapeseed embedding version.
100 % melted Histowax +60°C (Change morning and evening)
1.7 Day 7 Embedding (Day 8 in the Arabidopsis/rapeseed protocol) (Film! Technical details are shown in the film)
Preheat petri dishes and/or molds at +60°C. Turn on a hot plate (+60°C) and make sure melted Histowax is available.
Pour the tissues and melted Histowax into a petri dish (or a mold) placed on the hot plate. Add more Histowax so that the tissues are covered. Heat a pair of tweezers and distribute the tissues evenly in the dish. Fill the petri dish (or mold). If the wax hardens before the tissues are in the right position, heat it up again or remove the hardened wax with the heated pair of tweezers.
Let the wax block harden at room temperature before placing it at +4°C. It is better to make several wax blocks instead of embedding the tissues too tightly since the wax block might crack when pieces are cut out during sectioning. Pause! Store at +4°C. The tissues are stable for years. N.B.! Work RNase free from this step and onwards. Use gloves, DEPC-treat MilliQ-water, buffers, glassware etc., autoclave buffers and bake glassware etc. when appropriate.
2. SECTIONING ( Film! Technical details are shown in the film)
Turn on two hot plates (+60°C and +42°C) and make sure melted Histowax is available.
Heat a scalpel and carefully cut out a wax block of tissue from the petri dish (it might crack if one is too violent), or remove it from the mold. When a single wax block has been separated, shape the piece into the right size and shape to facilitate the slicing in the microtome.
The wax piece should have an extra wax “heel” underneath the tissue so it can be attached to the block holder. Heat the block holder and the heel on the hot plate (+60°C) and fuse them together. It might be necessary to put on a drop of Histowax II to make the fusion stable. Let it harden at +4°C.
Cut the wax block into 6-8 μm thick sections connected in a long ribbon using a microtome. N.B.! This will be easier if the wax block is cold, i.e. store the wax block at +4°C and bring it from the fridge when starting the cutting. It can also be beneficial to keep the knife cold.
Study the ribbons of sections in a magnifying glass and mark the ones to be used.
Put RNase-free MilliQ-water on the slides (Probe-On Plus from Fisher Biotechnology, which are pre-cleaned and charged). Cut the ribbon into smaller pieces and put them (“back” or “shiny” side down) on the slides. The slides with ribbons should be placed on a +42°C plate. The ribbon should flatten out (this is important!) in the water. Let the sections sit for a few minutes and then use a Kimwipe/tissue paper to drain of the water.
Leave the slides on the plate for 1-2 hours.
Incubate the slides at +42°C overnight so that the tissues adhere to slides. Pause! It is recommended to use the sections as soon as possible, but they can be stored dry in closed boxes with desiccant up to several days at +4°C.
3. THE MAKING OF PROBES
In situ hybridization detects expression using a labeled probe specific for a certain mRNA. The probe, which is most often a piece of complementary RNA, can be tagged with digoxigenin, DIG. DIG is a small molecule, which can be attached to uridine and thereby incorporated in the RNA probe and then detected using DIG-specific antibodies.
The designing and making of the probe is the most critical step in a RNAin situ hybridization experiment. Care should be taken to make sure the probe has a high specificity and is labeled with high quality UTPs. Sometimes the hybridization temperature and/or reaction length have to be adjusted for specific probes. As always, care should be taken to avoid RNase contamination.
Before labeling, isolate a template according your standard protocols, use e.g. cDNA clones, PCR-fragments, and linearize the template. Sense and antisense fragments are isolated and amplified from the template using gene specific primers. For PCR amplified fragments do not use enzymes that produce 3' overhangs. To all fragments a T7 (or SP6 or T3) overhang is added using primers carrying the T7-sequence or using a vector with a T7-promoter.
The sense (mRNA or (-) strand) probe have the same sequence as the target mRNA and will not hybridize to the target mRNA, while the antisense (anti-mRNA or (+) strand) probe have the complementary sequence and thus hybridize to the target giving the signal of interest. The sense probe is used as a negative control and no signal will be obtained if the experiment worked properly. A positive control is also needed, e.g. a probe that detects a house-keeping gene. The majority of the slides should be hybridized with antisense probes, while only a few slides should be hybridized to the different control probes.
3.1 Probe labeling using In vitro transcription
Reagents from the DIG RNA Labeling Kit (SP6/T7; 11175025910, Roche Applied Science, Mannheim, Germany) or similar are used when labeling the probes. The 20 μl reaction described here should yield ~10 μg of DIG-labeled RNA.
- Make an in vitro transcription to incorporate labeled nucleotides. Add the following reagents to a tube (keep on ice):
Ingredient volume/amount 10 x NTP labeling mixture 2 μl 10 x Transcription buffer 2 μl Protector RNase inhibitor 1 μl (20U) RNA T7 polymerase (SP6 or T3) 2 μl Template DNA (500-1000 ng) x μl RNase- free MilliQ-water y μl, to a final volume of 18 μl Mix the NTP labeling mixture, transcription buffer, RNase inhibitor ( N.B.! if the probe is short, e.g. <500bp, you may increase the RNase inhibitor concentration to 40U), polymerase, template DNA and water and then briefly centrifuge.
Incubate the reaction mix at +37°C for 2 hours ( N.B.! if the probe is short, e.g. <500bp, increase the reaction time to 4-6 hours).
Add 2 μl DNase I and incubate at +37°C for 15 min.
Stop the reaction by adding 2 μl 0.2 M EDTA (pH 8.0). Check the product on gel.
Precipitate the probe by adding 2.5 μl of 4 M LiCl and 75 μl of >99.5% ethanol. Mix well and incubate at -20°C overnight ( N.B.! Do not use tRNA as carrier. Instead use glycogen or acrylamide according to the manufacturers).
Centrifuge (13 000 rpm) for 30 minutes at +4°C and remove the supernatant. Wash the pellet (at least 10 min 13 000 rpm) two times in 70% ethanol (~150-300 μl).
Remove the supernatant and let the pellet dry. Resuspend the probe in 30 μl RNase-free water for 1-2 hours on ice. Pause! The probe can be stored at -20°C for more than a year. N.B.! Save 0.5 μl samples from step 21 (before the in vitro transcription), step 23 (before the DNase treatment), step 24 (after the DNase treatment, but before the precipitation) and 1 μl sample from step 27 (when the probe has been resuspended). Run the samples on a nondenaturating 1% TBE gel. If the DNase reaction has worked properly the template-band will disappear. The in vitro transcription should generate a thick RNA band of approximately the correct size. If you see multiple bands, denaturate the samples at +80°C for 5 minutes followed by quenching on ice before loading. If the recovery from step 27 looks bad it might be so because the probe was not resuspended well. Incubate the RNA at +55°C 5-10 minutes to get it into solution.
3.2 Hydrolysis of long probes
N.B.! If your probe is larger than 150 bp it should be reduced to 50-150 bp to give a high signal, due to better penetration. The probe may be chemically degraded in a carbonate buffer (pH 10.2). If the probe is of the correct size skip the hydrolysis step, but remember to add one volume formamide, i.e. 30 μl, to the probe.
Prepare 0.2 M sodium bicarbonate (NaHCO3) and 0.2 M sodium carbonate (Na2CO3). Both should be fresh.
Test the two buffers by mixing 5 ml H2O, 2 ml 0.2 M NaHCO3 and 3 ml 0.2 M Na2CO3. The pH should be ~10.2.
- Calculate the incubation time:
Incubation time (in minutes) = (L0-Lf) / (K*L0*Lf) L0= starting length of the probe in kb Lf = final length of the probe in kb = e.g. 0.100 kb K= rate constant for hydrolysis = 0.11 kb-1min-1 Hydrolyze half of your probe, save the rest for later. Dilute probe to 50 μl with RNase free water and add 20 μl 0.2 M NaHCO3 (add first) and 30 μl 0.2 M Na2CO3. Mix and incubate at +60°C for the calculated incubation time.
Stop the reaction by adding 10 μl of 1 M NaOAc pH 4.7.
Precipitate the probe by adding 10 μl 4 M LiCl2 and 300 μl ethanol ( N.B.! Use glycogen or acrylamide as carrier, if needed). Incubate at -20°C overnight.
Centrifuge (13 000 rpm) for 30 minutes at +4°C and remove the supernatant. Wash the pellet two times in 70% ethanol (~300 μl). Centrifuge (13 000 rpm) at least 10 min in each wash at +4°C.
Remove the supernatant and let the pellet dry. Resuspend the probe in 30 μl RNase-free MilliQ-water. Add one volume formamide, i.e. 30 μl, to the probe. Pause! The probe can be stored at -20°C for more than a year. N.B.! Take 2 μl of the unhydrolyzed probe and 2 μl of the hydrolyzed probe (before the formamide is added) and check on a nondenaturating 1% TBE gel. There should be a difference in size between the two samples. The probes might need to be denaturated at +80°C for 5 minutes followed by quenching on ice before loading.
4. IN SITU HYBRIDIZATION ( Film! Technical details are shown in the film)
Make sure that all solutions are RNase free! It is not necessary to DEPC-treat everything, but use at least autoclaved MilliQ-water. Make sure that all glassware are heated at +200°C overnight or at least for 5 hours. Treat plastic containers and stir bars with 0.1 M NaOH with agitation overnight. Rinse the plastic containers several times with autoclaved MilliQ-water (~500 ml/container). It is crucial to rinse the containers carefully to avoid traces of NaOH that might disturb the hybridization. DEPC-treat all stock solutions, except the Tris-buffers. Check all solutions etc. before starting the experiment to ensure that everything is available. Until step 61 (except steps 51-52) we keep the slides in a rack, which is easily moved between containers.
4.1 In situ section pretreatment
- Deparaffinize/dehydrate sections:
2x 10 min Histoclear II (use glass containers) 2x 1-2 min 100% EtOH 1-2 min 95% EtOH 1-2 min 90% EtOH 1-2 min 80% EtOH 1-2 min 60% EtOH 1-2 min 30% EtOH 1-2 min H2O Wash the slides for 15-20 min in 2x SSC at room temp ( N.B.! Prepare the paraformaldehyde used in step 42 during the incubation time).
Treat the tissues for 30 min with proteinase K at +37°C with gentle agitation (e.g. 1 μg/ml for Arabidopsis/rapeseed and 3 μg/ml for Norway spruce). Mix and preheat to +37°C 100 mM Tris pH 8 and 50 mM EDTA. Add proteinase K immediately before treating the slides. N.B.! The proteinase K treatment needs to be optimized depending on tissue, plant species and age. ( N.B.! Prepare the triethanolamine used in step 45 during the incubation time).
Treat the slides for 2 min with 2mg/ml glycine in 1x PBS at room temperature. ( N.B.! Mix the glycine with PBS the day before using it to ensure that the glycine is properly dissolved).
Wash the slides for 2 minutes in 1x PBS at room temperature.
Wash the slides for 2 minutes in 1x PBS at room temperature.
Incubate the slides for 10 minutes with 4% (w/v) paraformaldehyde (made fresh) in 1x PBS pH 7 at room temperature. Use glass container.
Wash the slides for 5 minutes in 1x PBS at room temperature.
Wash the slides for 5 minutes in 1x PBS at room temperature. (Dispense acetic anhydride into the solution in step 45).
Incubate the slides for 10 minutes in 0.1M triethanolamine (made fresh, pH 8) and acetic anhydride at room temperature.
Wash the slides for 5 minutes in 1x PBS at room temperature.
Wash the slides for 5 minutes in 1x PBS at room temperature.
- Dehydrate:
30 sec 30% EtOH 30 sec 60% EtOH 30 sec 80% EtOH 30 sec 90% EtOH 30 sec 95% EtOH 2x 30 sec 100% EtOH
4.2 In situ hybridization ( Film! Technical details are shown in the film)
Decide which slides to use with which probes, do not forget to use both sense and antisense probes as well as a positive control. (N.B.! ProbeOn Plus slides from Fisher Biotechnology have a white frosting that allows them to be sandwiched into pairs during the hybridization/detection steps of the protocol.) The same probe with the same concentration should be used on a specific slide pair. Determine how much hybridization solution to make based on the total number of slide pairs. Preheat the Dextran sulfate in +60°C. The hybridization solution is very viscous from the Dextran sulfate. Put the hybridization solution in +60°C and it will be easier to handle. Air-dry the slides on clean paper towels or Kimwipe/tissue papers before the probe is applied. The slides must be completely dry. For each pair of slides add the probe. It is important to test different probe concentrations (e.g. 0.5, 2 and 4 μl) to find the optimal concentration before the actual experiment is preformed.
| Hybridization solution | 5 slide pairs | 10 slide pairs | 15 slide pairs | 20 slide pairs |
| 10xin situ salts | 100 μl | 200 μl | 300 μl | 400 μl |
| deionized formamide | 400 μl | 800 μl | 1200 μl | 1600 μl |
| 50 % Dextran sulfate (+60 °C) | 200 μl | 400 μl | 600 μl | 800 μl |
| 50x Denhardt's solution | 20 μl | 40 μl | 60 μl | 80 μl |
| tRNA (100mg/ml) | 10 μl | 20 μl | 30 μl | 40 μl |
| H2O (DEPC treated) | 70 μl | 140 μl | 210 μl | 280 μl |
| Total volume | 800 μl | 1600 μl | 2400 μl | 3200 μl |
Dilute the probe in RNase-free MilliQ-water to a final volume of 20 μl. Add one volume (i.e. 20 μl) formamide to the probe giving a total volume of 40 μl. Heat the probe to 80°C for 2 minutes. Place on ice for 2 min. Spin down. Keep the probe on ice. This probe-mixture is enough for one pair of slides. Multiply according the total number of slide pairs for the specific probe.
Add 160 μl of hybridization solution for each pair of slides giving a final volume of 200 μl (i.e. 160 μl hybridization solution + 40 μl probe) for each slide pair. Mix without generating bubbles.
Apply the probe to one slide and slowly fuse the two slides together. (N.B.! Sandwiching can only be done with Probe-On Plus slides, or equivalent slides. If slides without frosting are used use cover slips instead of sandwiching.)
Elevate slides above wet paper towels in a plastic container (which seals tightly) using plastic pipettes. Hybridize +50-55°C overnight (12-16 hours).
4.3 In situ post hybridization (Film! Technical details are shown in the film)
Prepare the in situ post hybridization by warming ~2 L 0.2x SSC (+55°C) and ~2 L 1x NTE (+37°C) overnight. More SSC and NTE are needed if the RNase treatment (steps 57-59 below) is performed.
Dip each slide pair into warm 0.2x SSC (see above) to separate and rinse them. Then place them in a rack in a container with warm 0.2x SSC.
Wash the slides for 60 min in 0.2x SSC with gentle agitation at +55°C. ( N.B.! Prepare the Boehringer block solution used in step 63 during the incubation time.)
Repeat the wash in 0.2x SSC for 60 min. ( N.B.! If the RNase step is performed let the RNase thaw during this incubation, if not continue to step 60.)
Optional: Wash the slides for 5 min in warm 1x NTE (see above) at +37°C with gentle agitation.
Optional: Repeat the wash in warm 1x NTE for 5 min at +37°C with gentle agtation.
Optional: Treat the slides with RNase (20 μg/ml RNase A in 1x NTE) for 30 min at +37°C with gentle agitation. Then wash for 5 min in 1x NTE at +37°C with gentle agitation. Repeat the 1x NTE wash.
Wash the slides for 60 min in 0.2x SSC at +55°C with gentle agitation. ( N.B.! Prepare the block solution used in steps 64-65 and 67 during the incubation time.)
Wash the slides for 5 min in 1x PBS at room temp (Pause! You may store slides in 1XPBS at +4°C overnight if you want to continue the next day.)
Place the slides on the bottom of a large plastic container with the sample side up.
Incubate the slides with 1% Boehringer block in 100 mM Tris pH 7.5, 150 mM NaCl for 45 min at room temperature with gentle agitation. Just cover the slides with the blocking solution. Pour off the blocking solution while the slides still are in the plastic container or place the slides in a new container. When pouring off the solution the slides usually adhere to the plastic container, but make sure that they do not fall out off the container.
Replace the Boehringer block solution with 1.0% BSA in 100 mM Tris pH 7.5, 150 mM NaCl, 0.3% Triton X-100. Perform the incubation as in step 63.
Dilute the anti-dig antibodies (1:1250, i.e. 8 μl antibodies in 10 ml BSA) in the BSA/Tris/NaCl/Triton solution from step 64. Make a puddle of antibody solution in a plastic weigh dish. Sandwich slides together to allow capillary forces to pull up the solution. Drain on Kimwipe/tissue paper and repeat, try to avoid bubbles.
Elevate slides above wet paper towels in plastic container and allow the slides to sit at room temp for 2 hours.
Drain slides on Kimwipe/tissue paper and separate. Place on bottom of plastic container as in step 63. Wash 4x in the BSA/Tris/NaCl/Triton solution. 15 min each time, gentle agitation at room temperature.
10 min 100 mM Tris pH 9.5, 100 mM NaCl, 50 mM MgCl2 . Place on bottom of plastic container as in step 63.
Dip slide in Tris pH 9.5/NaCl/MgCl2 solution to ensure all of detergent is washed off.
Make sandwiches and draw up Western blue solution as in 65. Repeat once.
Place slides in plastic container above wet paper towels in total darkness (wrap in tin foil) for 1-5 days at room temperature. Check after 6, 12 and 24 hours then once every day.
Drain slides, rinse in 1xTE to stop reaction. Put on cover slips before the slides are examined in microscope. The slides can be saved in 1x TE for several months but take photos as soon as possible.
RECIPES/STOCK SOLUTIONS
N.B.! Use RNase-free MilliQ-water as much as possible, e.g. DEPC-treat MilliQ-water (Add 0.1% DEPC. Leave overnight. Autoclave (20 minutes at 15 psi, i.e. 1.05 kg/cm2, on liquid cycle) or use at least autoclaved MilliQ-water. When preparing buffers and stock solutions it is also possible to dissolve RNase-free salts etc. in DEPC-treated water, instead of DEPC-treating the buffers and stock solutions themselves. If you do not DEPC-treat water, buffers and stock-solutions, at least autoclave or filter sterilize them. Pause! Store buffers and stock-solutions at room temperature, if not else is stated. N.B.! You can find many of the buffers as well as hints on how to make them in Molecular Cloning (Sambrook, J., and D.W. Russell. 2001. Molecular Cloning A Laboratory Manual. 3 ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA).
Acetic anhydride in 0.1M triethanolamine (pH 8, volume: 800 ml).
| Ingredient | volume/amount | final concentration |
| Triethanolamine | 10.4 ml | 0.1 M triethanolamine |
| MilliQ-water | 786.4 ml | final volume 800 ml |
| HCl | 3.2 ml | giving pH 8.0 |
| Acetic anhydride | 4.8 ml | 0.6% |
To make 0.1 M triethanolamine buffer, pH 8.0, dilute 10.4 ml of triethanolamine in 786.4 ml of H2O and add 3.2 ml of HCl to bring the pH to 8.0 (check with pH indicator).
Elevate slide rack with broken 10 ml plastic pipettes or similar in a container of triethanolamine with a stir bar in the bottom.
Dispense 4.8 ml acetic anhydride into the triethanolamine a few minutes before putting slides in so that it is mixed well.
N.B.! Make the 0.1M triethanolamine fresh and add acetic anhydride just before incubation. N.B.! Use glass container. Triethanolamine buffer has to be used because acetic anhydride is unstable in water.
Agarose (1%) gel in 1 x TBE (volume: 100 ml)
| Ingredient | volume/amount | final concentration |
| Agarose | 1 g | 1% |
| 1 x TBE | up to 100 ml |
Mix agarose with 1 x TBE. Heat/boil until the agarose has melted. Cast a gel. Run the gel in a 1 x TBE buffer.
Dextran sulfate
Dilute 5 g Dextran sulfate to 10 ml with DEPC-treated MilliQ-water (i.e. 50% (w/v)) and dissolve in 60°C. Pause! Store at -20°C.
0.5 M EDTA pH8.0 (titriplex III, volume 500 ml)
| Ingredient | volume/amount | final concentration |
| EDTA (372.24 g/mol) | 93.06 g | 0.5 M EDTA |
| MilliQ-water | up to 500 ml | |
| NaOH | ~ 10 g pellets | giving pH 8.0 |
| DEPC | 0.5 ml | 0.1% DEPC |
Dissolve EDTA in water (happens at pH 8.0), adjust to a final volume of 500 ml. Add 0.1% DEPC. Leave over night. Autoclave.
Fix solution/fixative (steps 1-3, 42) - 4% (w/v) paraformaldehyde in 1x PBS, pH 7.0 (650 ml)
| Ingredient | volume/amount | final concentration |
| MilliQ-water | 585 ml | |
| 10x PBS | 65 ml | 1x PBS |
| 10M NaOH | 520 μl | giving pH ~11 |
| Paraformaldehyde | 26 g | 4% |
| Concentrated HCl | 449 μl | giving pH ~7 |
Mix water, PBS and NaOH and heat to +60-70°C (the temperature and the high pH will make it easier to dissolve the paraformaldehyde.
Add (in fume hood) paraformaldehyde.
When the solution has cleared place it on ice and adjust pH to 7.0 by adding 449 μl concentrated HCl.
N.B.! Make fresh. N.B.! In the spruce protocol glutaraldehyde was added to a final concentration of 0.25%, i.e. 6.5 ml 25% glutaraldehyde to the total volume of 650 ml or 10 ml 25% glutaraldehyde to the total volume of 1000 ml (remember to reduce the water with an equal volume). N.B.! Add a few drops of Tween 20 and/or Triton X-100, after the pH is adjusted, to reduce surface tension to facilitate the fixation in steps 1-3. DO NOT USE IN STEP 42! N.B.! When fixing plant tissues a smaller volume (e.g. 100 - 200 ml) of fixative is generally needed.
10x in situ salts (volume 100 ml)
| Ingredient | volume/amount | final concentration |
| 5 M NaCl | 60 ml | 3M NaCl |
| 1 M Tris-Cl pH 8.0 | 10 ml | 0.100 M Tris |
| 1 M Na Phosphate pH 6.8 | 10 ml | 0.100 M Na Phosphate |
| 0.5 M EDTA | 10 ml | 0.050 M EDTA |
| MilliQ-water | up to 100 ml |
Mix a Na Phosphate buffer with pH 6.8 (46.3 ml 1 M Na2HPO4 + 53.7 ml 1 M NaH2PO4). Mix NaCl, Tris, Na phosphate buffer, EDTA and water. Autoclave. Pause! Store at -20°C.
4 M LiCl2 (lithium chloride, volume: 100 ml)
| Ingredient | volume/amount | final concentration |
| LiCl2 (42.39 g/mol) | 16.956 g | 4M LiCl2 |
| MilliQ-water | up to 100 ml | |
| DEPC | 0.1 ml | 0.1% DEPC |
Dissolve LiCl2 in water, adjust to a final volume of 100 ml. Add 0.1% DEPC. Leave over night. Autoclave. Pause! Store at +4°C.
1 M MgCl2 · H2O (magnesium chloride, volume: 100 ml)
| Ingredient | volume/amount | final concentration |
| MgCl2 · H2O (203.30 g/mol) | 20.33 g | 1 M MgCl2 · H2O |
| MilliQ-water | up to 100 ml | |
| DEPC | 0.1 ml | 0.1% DEPC |
Dissolve MgCl2 in water, adjust to a final volume of 100 ml. Add 0.1% DEPC. Leave over night. Autoclave. N.B.! MgCl2 is very hygroscopic. Do not store opened bottles for long periods of time.
5 M NaCl (sodium chloride, volume: 1 l)
| Ingredient | volume/amount | final concentration |
| NaCl (58.44 g/mol) | 292.2 g | 5 M NaCl |
| MilliQ-water | up to 1l | |
| DEPC | 1 ml | 0.1% DEPC |
Dissolve NaCl in water, adjust to a final volume of 1 l. Add 0.1% DEPC. Leave over night. Autoclave.
1 M NaOAc pH 4.7 (sodium acetate, volume: 100 ml)
| Ingredient | volume/amount | final concentration |
| NaOAc (82.03 g/mol) | 0.8203 g | 1 M NaOAc |
| MilliQ-water | up to 100 ml | |
| Acetic acid | giving pH 4.7 | |
| DEPC | 0.1 ml | 0.1% DEPC |
Dissolve NaOAc in water. Adjust the pH to 4.7. Adjust to a final volume of 100 ml. Add 0.1% DEPC. Leave over night. Autoclave.
0.2 M Na2CO3 pH11.4 (sodium carbonate, volume: 10 ml)
| Ingredient | volume/amount | final concentration |
| Na2CO3 | 0.21198 g | 0.2 M Na2CO3 pH=11.4 |
| MilliQ-water | up to 10 ml |
Dissolve Na2CO3 in water; adjust to a final volume of 10 ml. The pH should be 11.4. N.B.! Make fresh.
0.2 M NaHCO3 pH8.2 (sodium bicarbonate, volume: 10 ml)
| Ingredient | volume/amount | final concentration |
| NaHCO3 | 0.16802 g | 0.2 M NaHCO3: pH=8.2 |
| MilliQ-water | up to 10 ml |
Dissolve NaHCO3 in water; adjust to a final volume of 10 ml. The pH should be 8.2. N.B.! Make fresh.
1 M Na2HPO4 · 2 H2O (volume 500 ml)
| Ingredient | volume/amount | final concentration |
| Na2HPO4 · 2 H2O (177.99 g/mol) | 88.995 g | 1 M Na2HPO4 |
| MilliQ-water | up to 500 ml | |
| DEPC | 0.5 ml | 0.1% DEPC |
Dissolve Na2HPO4 in water, adjust to a final volume of 500 ml. Add 0.1% DEPC. Leave over night. Autoclave.
1 M NaH2PO4 · H2O (volume 500 ml)
| Ingredient | volume/amount | final concentration |
| NaH2PO4 · H2O (137.99 g/mol) | 68.995 g | 1 M NaH2PO4 |
| MilliQ-water | up to 500 ml | |
| DEPC | 0.5 ml | 0.1% DEPC |
Dissolve NaH2PO4 in water, adjust to a final volume of 500 ml. Add 0.1% DEPC. Leave over night. Autoclave.
5x NTE (total volume: 1l)
| Ingredient | volume/amount | final concentration |
| 5 M NaCl | 500 ml | 2.5 M NaCl |
| 1 M Tris-Cl pH 8 | 25 ml | 50 mM Tris-Cl pH 8 |
| 0.5 M EDTA | 5 ml | 5 mM EDTA |
| MilliQ-water | 470 ml | final volume 1l |
Mix NaCl, Tris, EDTA and water. Do not DEPC-treat. Autoclave.
10 x PBS (phosphate-buffered saline) pH 7.0 (total volume: 1l)
| Ingredient | volume/amount | final concentration |
| 5 M NaCl | 260 ml | 1.3 M NaCl |
| 1 M Na2HPO4 | 70 ml | 70 mM Na2HPO4 |
| 1 M NaH2PO4 | 30 ml | 30 mM NaH2PO4 |
| MilliQ-water | 640 ml | final volume 1l |
| DEPC | 1 ml | 0.1% DEPC |
Mix NaCl, Na2HPO4, NaH2PO4 and water. Adjust the pH to pH 7.0 with HCl. Add 0.1% DEPC. Leave over night. Autoclave.
20x SSC pH 7.0 (total volume: 1l)
| Ingredient | volume/amount | final concentration |
| NaCl (58.44 g/mol) | 175.32 g | 3 M NaCl |
| Na Citrate (294.1 g/mol) | 88.23 g | 0.300 M Na Citrate |
| MilliQ-water | up to 1 l | |
| HCl | giving pH 7.5 | |
| DEPC | 1 ml | 0.1% DEPC |
Dissolve NaCl and Na Citrate in ~ 800 ml water. Adjust the pH to pH 7.0 with HCl. Adjust the volume to 1 l with water. Add 0.1% DEPC. Leave over night. Autoclave.
5 x TBE (volume: 1l)
| Ingredient | volume/amount | final concentration |
| Tris (121.14 g/mol) | 54 g | ~ 0.45 M Tris |
| Boric acid (61.83 g/mol) | 27.5 g | ~ 0.45 M boric acid |
| EDTA (372.24 g/mol) | 3.7 g | ~ 0.01 M EDTA |
| MilliQ-water | up to 1l |
Mix Tris, boric acid, EDTA and water. The pH should be ~ 8.3. Dilute to 1 x TBE before use.
10 x TE pH 8.0 (Tris EDTA, volume: 1l)
| Ingredient | volume/amount | final concentration |
| 1 M Tris-Cl, pH 8.0 | 100 ml | 0.100 M Tris-Cl |
| 0.5 M EDTA pH8.0 | 100 ml | 0.050 M EDTA |
| MilliQ-water | up to 1l |
Mix Tris, EDTA and water. Do not DEPC-treat. Autoclave. N.B.! Tris should not be DEPC-treated! Use RNase-free powder and dilute with DEPC-treated/RNase-free MilliQ- water.
1 M Tris-HCl pH 7.5 - 9.5 (volume 500 ml)
| Ingredient | volume/amount | final concentration |
| Tris (121.14 g/mol) | 60.57 g | 1 M Tris |
| MilliQ-water | up to 500 ml | |
| HCl | giving pH 7.5 | |
| HCl | giving pH 8.0 | |
| NaOH | giving pH 9.5 |
Dissolve Tris in DEPC-treated water. Adjust to the desired pH. Adjust to a final volume of 500 ml. Autoclave. N.B.! Tris should not be DEPC-treated! Use RNase-free powder and dilute with DEPC-treated/RNase-free MilliQ- water. N.B.! In Molecular Cloning (see above) there is a table describing how much concentrate HCl is needed to achieve the correct pH.
MATERIAL AND METHODS
The in situ protocol is presented above and in the film. Here additional information of the plant material and probes used in this specific example are described.
Plant material and experimental design
Male cones from Picea abies [L.] Karst. (Norway spruce) were collected in Uppsala, Sweden, autumn 2007. Eight cones were sectioned and sections from each cone were used in each treatment. Three proteinase K concentrations were tested; 1, 3 and 5 μg/μl and each concentration were treated with or without RNase. Slides not treated with RNase were left in PBS during RNase treatment. Arabidopsis thaliana [L.] Heynh. and Brassica napus L., were grown under controlled conditions in a culture chamber with 22°C/18°C day/night temperatures and a photoperiod of 16 h. Young inflorescences containing floral buds, stages 0-10 (stages according to Smyth 5) were studied.
cRNA probes
Total RNA was isolated from P. abies male cones as previously described 6 and from A. thaliana and B. napus using TRIzol (Gibco BRL, Frederick, Maryland, USA) and the Qiagen RNeasy Plant Mini Kit (Qiagen, Hilden, Germany), respectively, in accordance with manufacturers' recommendations. The cDNA was synthesized from 0.5-1 μg total RNA, depending on species, using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, California, USA) following the manufacturer's instructions. AtAP3 and BnAP3 sense and antisense fragments and P. abies sense fragments were isolated and amplified using gene specific primers (Table 1). DAL13 antisense fragments were isolated and amplified using primers as in 7. A T7 overhang was added to fragments from A. thaliana and P. abies using modified reverse primers carrying the T7-sequence (Table 1). A 500 bp fragment was isolated using BnAP3 specific primers (Table 1) and cloned into a pGEM-T vector with a T7-promotor. For the P. abies fragments all PCR reactions were performed in a final volume of 20 μl containing 0.2 U Phusion High-Fidility DNA Polymerase (Finnzymes, Espoo, Finland), 1x Phusion HF buffer, 200 μM of each dNTP, 0.3 μM of each primer and 25-50 ng cDNA and a standard PCR program was used with annealing temperature 60-55°C (touch-down -1°C/cycle) followed by 55°C. Sense and antisense cRNA probes for all three species were synthesized with in vitro transcription using the DIG RNA Labeling Kit (SP6/T7; Roche Applied Science, Mannheim, Germany) according to the manufacturer's recommendations and long probes were digested to approximately 100-150 bp fragments using a Na2CO3 buffer according to 3.
RESULTS
Here in the result section we describe three different examples of typical results from in situ experiments.
The genetic mechanism that regulates reproductive development in gymnosperms and angiosperms by specifying male and female organ identity appears to be evolutionary conserved7-9 despite that the two seed plant lineages separated 285 million years ago 10. In A. thaliana the floral homeotic genes APETALA3 (AP3 11) and PISTILLATA (PI 12) are necessary and sufficient to specify male reproductive development within the context of a flower, and this function appears to be conserved within the angiosperm lineage 13. In gymnosperms, which lack flowers and have their reproductive organs arranged in separate male and female cones (Fig 1A-C), genes homologous to AP3 and PI are specifically expressed in the pollen bearing organs of the male cone, the microsporophylls 7,14. Hence, a similar set of homologous genes in both angiosperms and gymnosperms defines the pollen-bearing organs.
Gene specific probes directed against AtAP3 and BnAP3, respectively, gave signals from stage 3 in young floral buds in a region corresponding to whorl two and three in A. thaliana and B. napus. In later staged buds the signals were restricted to developing petals and stamens (Fig. 2A and B). These results are in agreement with previous studies 11,15,16. Probes directed towards the AP3 homologue in P. abies, DAL13, produced signals in the microsporophylls of P. abies male cones both before and after apical meristem termination (Fig. 2C and D), as expected from previous studies 7,14. Sense probes gave no signal above background (Fig. 3A-B and data not shown). Taken together, these results demonstrate expression of A. thaliana AP3 and its orthologs in B. napus and P. abies in developing male reproductive organs.

Figure 1.A.thaliana and B. napus flowers and pollen producing male cones of the conifer P. abies Pictures show inflorescences with floral buds and open flowers of A.thaliana and B.napus in A and B respectively. Note that angiosperm flowers apart from the sterile perianth harbors both male and female reproductive organs; stamens and carpels. Shown in C is a twig of the gymnosperm P. abies with green vegetative needles and red reproductive male cones.
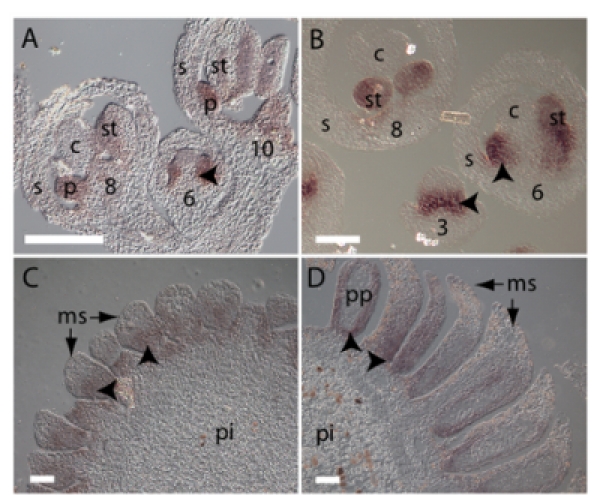
Figure 2. Expression of A. thaliana AP3 and homologous genes in B. napus and P. abies. Micrographs show longitudinal sections of A. thaliana and B. napus inflorescences in A and B, respectively and P. abies male cones in C and D. Sections hybridized with antisense probes directed against AtAP3 in A and BnAP3 in B show signal in second and third whorl floral organs. Numbers indicates floral stages according to Smyth 5. Antisense probe directed towards the DAL13 gene gave signal in the pollen bearing microsporophylls of P. abies male cones, as shown in C-D. Examples of microsporophylls are indicated by arrows. Arrowheads in A-D point to hybridization signal, which appears as purple. In C and D phenolic compounds give brownish unspecific color to individual cells in the central pith. s, sepal; p, petal; st, stamen; c, carpel; ms, microsporophyll; pi, pith; pp; pre-pollen cells. Bar: 100 μm.
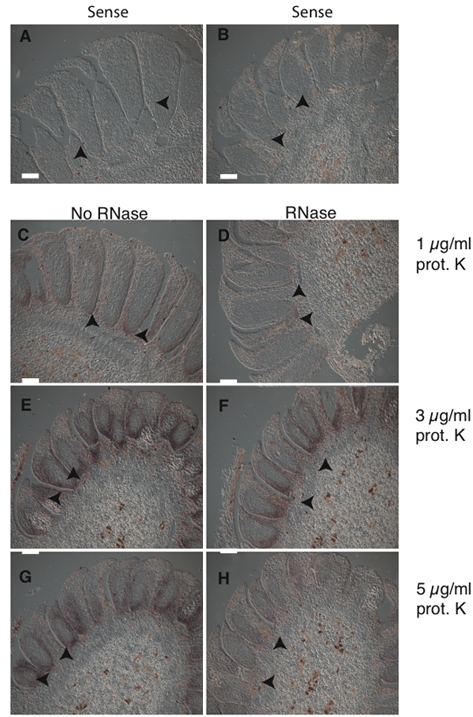
Figure 3.DAL13 expression in male cones from P. abies. All Micrographs (A-H), are showing longitudinal sections of male cones after apical meristem termination. Arrowheads point to cell types in which DAL13 is expressed. Sections in A and B are hybridized with a sense control probe to determine background staining. Micrographs C to H are hybridized with a DAL13 antisense probe. Sections from experiments with and without RNase treatment are shown in D, F, H and C, E, G respectively. Micrographs from experiments with 1 μg/ml proteinase K are shown in C and D, 3 μg/ml in E and F, and 5 μg/ml in G and H. Bar: 100 μm.
Discussion
Two aspects of the P. abies protocol were optimized compared to the A. thaliana/B. napus protocol, RNase treatment and proteinase K concentration. The RNase removes background signals and thus increases the signal specificity 17. The proteinase K treatment is needed to permeabilize the tissues so the probe can enter and hybridize to the RNA pool of interest. The DAL13 antisense probe gave a higher signal without RNase-treatment (Fig. 3C, E, G) compared to sections treated with RNase for 30 min (Figure 2D, F and H). Since the RNase-treatment did not enhance the signal it was removed from the protocol. The proteinase K treatment were tested at three different concentrations; 1, 3 and 5 µg/ml. In the case of P. abies a low or no signal was observed using 1 µg/ml proteinase K (Fig. 3C-D). A strong signal was obtained with both 3 µg/ml (Fig. 3E-F) and 5 µg/ml (Fig. 3G-H) proteinase K. Since no apparent difference in signal strength was found when using 5 µg/ml proteinase K compared to 3 µg/ml, we chose to use 3 µg/ml in our protocol. It is likely that the need of a higher proteinase K concentration in the P. abies male cones compared to the A. thaliana and B. napus floral buds is due to more compact tissue with higher levels of phenolics and polysaccharides.
During the fixation and embedding some differences between the A. thaliana/B. napus protocol and the P. abies protocol are evident. The tissue of interest is fixed to provide RNA retention and this is a critical step since poorly fixed materials might give a low or undetectable signal even though the mRNA is highly abundant. The tissue is dehydrated, cleared and embedded in wax in gradual changes to avoid tissue damage, and in some protocols 0.85% NaCl is added to the ethanol in the dehydration to further avoid shrinkage and swelling of the cells 3. The dehydration step during embedding can be performed on ice to keep RNase activity at minimum. In the P. abies protocol the 4% paraformaldehyde fix solution contains 0.25% glutaraldehyde, which is supposed to give a better RNA retention 17 even though that some argue that it probably make no difference 3. After sectioning the material is fixed onto slides and pretreated (i.e. dewaxed, rehydrated, permeabilized, treated to reduce non-specific binding of the probe and finally dehydrated) before the hybridization.
In the protocol presented here the whole detection procedure can be performed in ordinary laboratories, the signal develops usually within 1-3 days and the ratio between signal over background can continuously be monitored. The DIG-labeled probes usually give a more distinct signal as compared to radioactive labeled probes, are more stable and can be reused in consecutive experiments. On the other hand, the sensitivity is reduced compared to radioactive probes 17. In situ hybridization detects expression using a labeled probe specific for a certain mRNA. Radio-labeling is a robust and sensitive labeling method but it requires handling of radioactivity and it takes several weeks before the results can be detected. Antigen-labeled probes on the other hand, are more stable, less hazardous and require exposure to the detection medium for a few days only 17. Thus, antigen-labeling methods are currently dominating. The most critical step regardless of protocol is the probe design. Care should be taken to make sure the probe has a high specificity and is labeled with high quality UTPs. Sometimes the hybridization temperature and/or reaction length have to be adjusted for specific probes.
Our modified protocol based on DIG-labeled probes will facilitate localization of mRNA expression simultaneously in species ranging from A. thaliana to P. abies, and should with minor adjustments be usable in other plant species. The protocol has, beside male cones, also been tested on different stages of vegetative shoots and in both zygotic and somatic embryos from P. abies with good results (unpublished results; Karlgren et al.).
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are very grateful to Anders Bovin who provided the music for our film and to Michael Elliot Stocks for volunteering to be the speaker voice. Support was provided by Carl Trygger Foundation, the strategic research program Agricultural Functional Genomics (AgriFunGen) at the Swedish University of Agricultural Sciences, the Swedish Research Council (VR), and the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning (FORMAS).
References
- Carr SM, Irish VF. Floral homeotic gene expression defines developmental arrest stages in Brassica oleracea L. vars. botrytis and italica. Planta. 1997;201(2):179–188. doi: 10.1007/BF01007702. [DOI] [PubMed] [Google Scholar]
- Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353(6339):31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- Jackson D. In situ hybridization in plants. In: Gurr SJ, McPherson MJ, Bowles DJ, editors. Molecular Plant Patology: a practical approach. Oxford: University Press; 1991. [Google Scholar]
- Leitch AR, Schwarzacher T, Jackson D, Leitch IJ. In situ hybridization: a practical guide. Oxford: BIOS Scientific Publishers Limited; 1994. [Google Scholar]
- Smyth DR, Bowman JL, Meyerowitz EM. Early flower development in Arabidopsis. Plant Cell. 1990;2(8):755–767. doi: 10.1105/tpc.2.8.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo H, Lino-Neto T, Tavares RM. An improved method for high-quality RNA isolation from needles of adult maritime pine trees. Plant Molecular Biology Reporter. 2003. pp. 333–338.
- Sundstrom J, et al. MADS-box genes active in developing pollen cones of Norway spruce (Picea abies) are homologous to the B-class floral homeotic genes in angiosperms. Dev Genet. 1999;25(3):253–266. doi: 10.1002/(SICI)1520-6408(1999)25:3<253::AID-DVG8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Tandre K, Svenson M, Svensson ME, Engstrom P. Conservation of gene structure and activity in the regulation of reproductive organ development of conifers and angiosperms. Plant J. 1998;15(5):615–623. doi: 10.1046/j.1365-313x.1998.00236.x. [DOI] [PubMed] [Google Scholar]
- Winter KU, et al. MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc Natl Acad Sci U S A. 1999;96(13):7342–7347. doi: 10.1073/pnas.96.13.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard L, et al. Chloroplast and nuclear gene sequences indicate late Pennsylvanian time for the last common ancestor of extant seed plants. Proc Natl Acad Sci U S A. 1994;91(11):5163–5167. doi: 10.1073/pnas.91.11.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack T, Brockman LL, Meyerowitz EM. The homeotic gene APETALA3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens. Cell. 1992;68(4):683–697. doi: 10.1016/0092-8674(92)90144-2. [DOI] [PubMed] [Google Scholar]
- Bowman JL, Smyth DR, Meyerowitz EM. Genes directing flower development in Arabidopsis. Plant Cell. 1989;1(1):37–52. doi: 10.1105/tpc.1.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EM, Irish VF. Evolution of genetic mechanisms controlling petal development. Nature. 1999;399(6732):144–148. doi: 10.1038/20172. [DOI] [PubMed] [Google Scholar]
- Sundstrom J, Engstrom P. Conifer reproductive development involves B-type MADS-box genes with distinct and different activities in male organ primordia. Plant J. 2002;31(2):161–169. doi: 10.1046/j.1365-313x.2002.01343.x. [DOI] [PubMed] [Google Scholar]
- Carlsson J, et al. Microarray analysis reveals altered expression of a large number of nuclear genes in developing cytoplasmic male sterile Brassica napus flowers. Plant J. 2007;49(3):452–462. doi: 10.1111/j.1365-313X.2006.02975.x. [DOI] [PubMed] [Google Scholar]
- Teixeira RT, Farbos I, Glimelius K. Expression levels of meristem identity and homeotic genes are modified by nuclear-mitochondrial interactions in alloplasmic male-sterile lines of Brassica napus. Plant J. 2005;42(5):731–742. doi: 10.1111/j.1365-313X.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- Ying S, Kay AB. The technique of in situ hybridization. Methods in Molecular Medicine. 2001;56:263–283. doi: 10.1385/1-59259-151-5:263. [DOI] [PubMed] [Google Scholar]