

Abstract
The use of infrared laser sources for creation of localized temperature fields has opened new possibilities for basic research and drug discovery. A recently developed technology, Microscale Thermophoresis (MST), uses this temperature field to perform biomolecular interaction studies. Thermophoresis, the motion of molecules in temperature fields, is very sensitive to changes in size, charge, and solvation shell of a molecule and thus suited for bioanalytics. This review focuses on the theoretical background of MST and gives a detailed overview on various applications to demonstrate the broad applicability. Experiments range from the quantification of the affinity of low-molecular-weight binders using fluorescently labeled proteins, to interactions between macromolecules and multi-component complexes like receptor containing liposomes. Information regarding experiment and experimental setup is based on the Monolith NT.115 instrument (NanoTemper Technologies GmbH).
Introduction
We present here a biophysical technology for the analysis of molecular interactions. The technology is named Microscale Thermophoresis (MST), a term that refers to the motion of molecules in microscopic temperature gradients. The thermophoretic effect, although not fully understood on a microscopic level, is very sensitive to the molecule–solvent interface. It allows the quantification of biomolecule interactions by the thermophoretic detection of even minute changes in conformation, charge, and size of a molecule as they are induced by a binding event. Using a titration approach, MST enables one to measure the affinity constants of a wide variety of interactions in the binding equilibrium. In addition, the approach can also be utilized to measure enzymatic activities and modifications of proteins and nucleic acids.1–4
As will be shown later, MST is easy to handle, has a low sample consumption, and measures interactions with essentially no limitation on molecule size or molecular weight. Measurements are performed within minutes in free solution, be it in a buffer of choice or complex biological context like cell extract or blood serum. These properties and the flexible assay design make MST a powerful tool for basic and translational research as well as drug discovery. The technology is complementary to surface-based sensors like surface plasmon resonance (SPR) that measure binding kinetics or calorimetric approaches that directly access the thermodynamic properties of an interaction but require a significantly higher amount of sample material. The scope of this article is to discuss technical details of the MST approach to biomolecular interaction studies, and to highlight different applications and approaches for basic research and drug discovery. It also provides a detailed description of different kinds of molecular information that can be obtained by analyzing the MST signal.
Interaction Analysis
Today there are various methods that measure the affinity of interacting molecules and that provide information on the binding mechanism and binding kinetics. Approaches range from semi-quantitative assays like electrophoretic mobility shift assay (EMSA) and pull-down assays to SPR-based techniques that measure physical constants of binding kinetics and binding affinities. In addition to this surface-based method, truly label-free calorimetric approaches are very widely used, which measure the enthalpy of a reaction (i.e., heat of reaction). Another class of methods is based on changes in diffusion times induced by a binding event. Fluorescence anisotropy (FA) is based on measurement of changes in the rotational diffusion while fluorescence correlation spectroscopy (FCS) basically measures changes in translational diffusion upon complex formation. Both methods are successfully used to address questions of biomolecular interactions, but are best used for studies of interactions that come along with a pronounced increase of the size of the complex compared to the nonbound fluorescently labeled binding partner. Preceding a description of MST, the advantages of some of these methods for a respective field of application are highlighted.
SPR, in general, is a very sensitive method that makes use of electromagnetic surface waves on a thin metal film.5–7 These fields are strongly enhanced in resonance and are very sensitive to the dielectrical properties of the surface and adjacent layers of surface-coupled molecules and solvent. SPR instrumentation allows one to measure time-resolved binding events to a surface immobilized biomolecule. Thus, SPR provides information on binding kinetics and affinity and can, in principle, be used for measuring dissociation constants (KD) from sub nM to low mM.8 Measurements are typically performed in buffers with an intermediate throughput. Drawbacks of the method are that establishing a new assay is quite labor intensive and that the covalent coupling of a molecule to a surface might interfere with the binding event. As with most surface-based kinetic sensors, SPR also suffers from artifacts stemming from mass transport limitations close to an interface (i.e., rebinding effects and concentration depletion).9 Despite these drawbacks, especially when on- and off-rates of a reaction are of interest, the SPR system is the method of choice.
Isothermal titration calorimetry (ITC), the only truly label-free method discussed here, measures the dissipated or absorbed heat of a reaction, allows direct access to the thermodynamics of an interaction and makes the indirect calculation from van't Hoff approaches obsolete. This allows differentiating between enthalpic and entropic binders in a single experiment. In addition, the dissociation constant (KD) and stoichiometry (i.e., number of binding sites on each molecule in the calorimeter cell) are obtained.10–12 To gain a measurable amount of heat, a remarkably high concentration on the order of milligrams per milliliter is required for at least one of the binding partners. Depending on the degree of automatization, the method has a low to intermediate throughput and allows one to measure dissociation constants in the range of nanomolar to submillimolar, in completely identical buffers and with most additives. Since measured directly, calorimetry is the method of choice to measure thermodynamic parameters of an interaction. Drawbacks are preparation time, material consumption, buffer limitations, and that interactions with no or only a small change in enthalpy are not measurable with ITC.
FCS and FA are fluorescence-based methods that rely on a comparably pronounced change in diffusion time of the fluorescently labeled species. FCS measures the translational diffusion of molecules in free solution by detecting fluorescence fluctuations of a small number of molecules diffusing into and out of the focus volume of an excitation laser. The data yield the fraction of bound and unbound molecules in a concentration range of μM down to 0.1 nM, and FCS thus reaches high affine binding constants. Assuming that a change in hydrodynamic radius by a factor two is detectable by FCS, a globular protein must increase by a factor of 8 in mass upon binding due to the third root scaling of radius with mass.13 An advantage of FCS is that it provides access to single molecule properties, which remain obscure in all bulk methods. FA in comparison measures changes in the rotational diffusion time of a fluorescently labeled molecule with low molecular weight, typically on the order of a few thousand daltons. Polarized light is used to excite the fluorescent dye. The nonbound fluorescently labeled binding partner diffuses so fast, that the emitted light is essentially not polarized. When a binding event occurs, the tumbling of the fluorescently labeled molecule is slowed down and a higher fraction of the emitted light remains polarized. The interplay between lifetime of the dye and the rotational diffusion of the binding molecule defines the sensitivity of this method. As FCS, FA relies on a comparatively large change in size upon binding, which is why the smaller binding partner has to be labeled fluorescently, bearing a risk of changing the binding properties.
MST in comparison utilizes a very different approach to measure the equilibrium affinity constants of interactions. It is exceptionally sensitive, since it detects changes in size and charge as well as changes in the hydration shell of a molecule, thus combining various aspects of the afore mentioned methods. This makes the method suited to measure a wide range of interactions, from small molecule that bind to proteins, to quantification of affinities of multi-subunit complexes and liposomes. The fluorescence approach of MST in combination with low-volume fluidics allows reducing the amount of sample material to a few microliter at nanomolar concentrations. An equilibrium, binding affinity is determined in about 10 min in free solution, in any buffer and without the need for surface coupling of molecules.
Theoretical Background of Thermophoresis
A temperature gradient in an aqueous solution of (bio-)molecules induces not only a flow of heat but also a flow of molecules. This coupling between a heat-flow and mass flow is known as Ludwig-Soret effect or thermophoresis. It was first observed by Carl Ludwig in 1856.14 While thermophoresis in gases was theoretically predicted before it was experimentally observed, the origin of thermophoresis in liquids remained obscure. The first systematic studies of thermophoresis of salts in aqueous solution were carried out in 1879. Today, it is commonly argued that thermophoresis is a local nonequilibrium effect that requires fluid dynamics, force fields, or particle–solvent potentials.15–19 Other approaches assume molecules in a local thermodynamic equilibrium and apply equilibrium thermodynamics to describe thermophoresis in liquids.20,21 Most approaches have in common that the solvent particle interface and particle charge are of great importance to explain the thermophoretic effect. Notably, the temperature dependence of the diffusion coefficient through the Einstein relation does not suffice to describe thermophoresis as it is typically 10-fold smaller than the measured thermophoresis.
In general, the movement of molecules in a temperature gradient is described phenomenologically with a linear drift response.22 The molecule velocity v=−DTigradT is linearly dependent on the temperature gradient (gradT) with a proportionality constant DTi, termed thermal diffusion coefficient or thermophoretic mobility. We have indexed the different states and types of the solved molecules with index i. In steady state, this drift is counterbalanced by mass diffusion. For low molecule concentrations—here on the nanomolar scale—the balance of molecule flow densities becomes
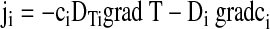 |
(1) |
with diffusion coefficient Di and concentration ci. Full integration of Equation (1) with temperature independent DTi and Di would yield a steady state concentration of
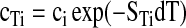 |
(2) |
at the position where the temperature is increased by dT above ambient temperature level. The Soret coefficient STi is defined by the ratio STi=DTi/Di of mobility over the diffusion coefficient Di. Typically, the concentration cTi is depleted and lower than the initial concentration ci. In other words, the thermophoretic mobility DTi and the Soret coefficient are typically positive. However, depending on the properties of a molecule, also an increase of concentration with respect to the initial concentration is observed for biomolecules. In general, the Soret coefficient depends on the size of a molecule, its charge, but also on its interaction with the solvent (i.e., conformation).1 Since at least one of these parameters is changed by a binding event, a wide range of molecular interactions can be analyzed.
Experimental Implementation
The experimental setup consists of an infrared (IR)-Laser coupled into the path of fluorescence excitation/emission using an IR dichroic mirror (Fig. 1A). The IR-Laser is focused into the sample through the same optics that is used for fluorescence detection. This setup allows for the mechanically robust and thus very precise observation of thermophoresis, since IR and visible optics are well aligned. IR-Laser-induced heating is one of the prerequisites for a successful MST analysis, since it has various advantages over conventional contact heating. The IR-Laser allows heating a solution with very high precision and reproducibility in the mK range. This is very important if we consider that for a typical interaction analysis where a serial dilution that contains 10 to 16 different samples is analyzed and compared. Another advantage of the IR-Laser is that it can be focused into the sample. This allows for a localized temperature increase on the order of a few hundred micrometers. The microscopic scale of the temperature distribution is essential for fast MST analysis. The motion of molecules is essentially diffusion limited and restricting the temperature distribution to the μm scale enables one to quantify changes in thermophoretic properties in <30 s. For this work, the spatial temperature distribution and the timescales of sample heating and thermophoresis were simulated by a three-dimensional finite element calculation using Comsol Multiphysics (Fig. 2). It shows the dominant physical effects in an MST experiment for a molecule with diffusion coefficient of D=150 μm2/s and a thermophoretic mobility of DT=5.6 μm2/(sK). The IR-Laser creates a spatial temperature distribution on a length scale of 200 μm. Less than a second after the IR-Laser is turned on, the energy is absorbed and heat dissipation reaches equilibrium, and a steady-state temperature increase of 1 to 10 K (typically 2–6 K) is obtained. After this fast increase in temperature (MST temperature jump or MST T-Jump), thermophoresis sets in and builds up a concentration gradient in the solution. In the following section the evaluation of an MST signal and the information content of the different phases are discussed.
Fig. 1.

Experimental setup and MST signal. (A) MST is measured in a capillary with a total volume of 4 μL. The fluorescence within the capillary is excited and measured through the same optical element. An IR-Laser is used to locally heat the sample volume that is observed by fluorescence. T-Jump and thermophoresis are directly observed as a change in fluorescence at different time scales. (B) A typical MST signal for a given capillary is shown on the right. Initially, the molecules are homogeneously distributed and a constant “initial fluorescence” is measured. As soon as the IR-Laser is turned on, a fast T-Jump is observed, followed by thermophoretic molecule motion. The fluorescence decrease is measured for about 30 s. When the IR-Laser is turned off, an inverse T-Jump is observed, followed by the “backdiffusion” of molecules, which is purely driven by mass diffusion, allowing to deduce information on the molecule size. MST, Microscale Thermophoresis; IR, infrared; T-Jump, temperature jump.
Fig. 2.

Simulation of MST. (A) The heating kinetics in a capillary induced by IR-Laser absorption shows that the temperature is equilibrated in <1 s. (B) The temperature depends on the distance from the IR-Laser focus. Within 200 μm distance, a drop of temperature of >70% is observed. (C) Time course of the concentration in the area of the IR-Laser focus. Compared to the MST T-Jump, thermophoresis is a slow process on the order of 1–30 s. (D) Beside thermophoresis and diffusion, a third transport process is induced by local heating. A convective flow of maximally 3 μm/s is observed in the center of the heat spot. This convective transport allows detection of aggregates of fluorescently labeled molecules (e.g., aggregated proteins) in the solution, since convection transports these particles in and out of the detection volume.
Evaluation of MST Signals
The signal obtained by an MST instrument consists of several subsequent processes that can be separated by their respective timescales and dependence on the IR-Laser heat source (Fig. 1B). These events are defined as “Initial Fluorescence” (fluorescence of sample at ambient temperature without laser heating), “MST T-Jump” (fluorescence change induced by sample heating before the thermophoretic molecule transport sets in), “thermophoresis” (fluorescence change induced by thermophoretic motion), “inverse T-Jump” (induced by sample cooling after turning of IR-Laser), and “Backdiffusion” (fluorescence recovery triggered by pure mass diffusion of molecules after heating laser is turned off). Each individual phase of the thermophoretic analysis contains information about the affinity and mechanism of binding. In the following we dissect a typical signal into its component phases and explain the theoretical background and the molecular information it provides.
As can be seen in Equation (2), the Soret coefficient ST determines the steady-state concentration ratio chot/ccold=exp(−ST ΔT)≈1−ST ΔT at a temperature increase ΔT. This linearization is valid, since the depleted concentrations and applied temperature differences (typically 2–6 K) are small compared to the initial concentration and the ∼300 K absolute temperature. The MST experiments discussed here use fluorescence to detect and quantify changes in the concentration of molecules. In principle, fluorescence is measured before IR-Laser heating (Finitial) and after a defined time of IR-Laser heating (Fhot). However, the normalized fluorescence Fnorm=Fhot/Finitial reflects only in part this concentration ratio. Also, the MST T-Jump ∂F/∂T is part of the signal measured by the MST instrument. This T-Jump is a change of fluorescence yield of the dye due to the temperature increase. A temperature dependence of fluorescent dyes is commonly observed and is not necessarily influenced by the binding event. But as shown later, in some cases, binding is not only observed in the thermophoresis signal, but also in the T-Jump. In the linear approximation for times exceeding the time needed for thermophoresis to reach steady state,2 we find: Fnorm=1+(∂F/∂T-ST)ΔT. When analyzing an MST signal, MST T-Jump and thermophoresis can be separated by their respective time scales: fast T-Jump (<1s) and slower thermophoresis (>1s). Thus, the choice of times to calculate Fnorm distinguishes between both processes. As a rule of thumb, when the binding changes the internal state of the labeled molecule, the binding is likely to affect the faster T-Jump signal, whereas size, charge, and hydration shell effects from binding are rather found in the thermophoresis signals. Often, both signals combined enhance the signal-to-noise ratio.
A typical MST equilibrium experiment for quantification of an interaction is set up as a titration experiment. The concentration of a fluorescently labeled molecule is kept constant as low as 1 nM, whereas the concentration of an unlabeled binding partner is varied. This means that the target concentration is typically varied starting at a concentration of at least about 10 times above the expected dissociation constant down to sub-stoichiometric concentration with respect to the labeled molecule. The MST signal will detect the binding by a quantification of the change in the normalized fluorescence (i.e., the amplitude of the MST signal). By denoting x the fraction of labeled molecules bound to their targets, the changing fluorescence signal depending on the concentration of target is given by:2
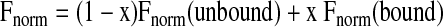 |
(3) |
Here, Fnorm (unbound) is the normalized fluorescence of unbound labeled molecules, and Fnorm (bound) is the normalized fluorescence of complexes (i.e., of labeled molecules bound to their targets in saturation). The unbound state acts as a reference state. As can be seen from Equation (3), differences in normalized fluorescence of the bound and unbound state will allow determination of the fraction bound and thus the dissociation constant. In the results shown in this work, Fnorm is plotted directly or after subtraction of the baseline value (i.e., ΔFnorm). All values are multiplied by a factor of 1,000, yielding a relative fluorescence change in per thousand [‰]. Figure 1 shows a typical thermophoretic time trace and gives an overview of the processes that lead to a change in normalized fluorescence.
Chemical Equilibrium
The temperature enhancement by MST is chosen to be small in order not to yield a significant change of the binding probability. This should be the case for most binding events. As a result, the binding is reported by the change of the molecule thermophoresis upon binding and this isothermal chemical equilibrium is settled long before the actual heating event of the thermophoresis experiment. For fast chemical equilibria with a relaxation time t<tthermophoresis, MST measures the KD effectively at the slightly higher temperature. If the chemical equilibrium is much slower, the effects of temperature could be recorded by analyzing thermophoresis over a longer time.
Even if the temperature applied by MST is large (>5 – 10 K), the change in binding concentration induced by the temperature dependent change in the chemical equilibrium will not significantly perturb the MST measurement. The reason is that the concentration change in an MST experiment is small (at most a factor of two) and highly comparable along the binding titration curve, which changes the concentrations of binder over several orders of magnitude. We thus expect for most interactions that even for an exaggerated temperature increase, no significant shift in the binding behavior is to be expected. This can be experimentally confirmed by performing experiments increasing MST temperatures and comparing the resulting dissociation constants.
MST T-Jump
As noted in the beginning of this review, heating of a solution by an IR-Laser is a fast process that takes place on the order of less than a second. In general, the corresponding MST T-Jump allows quantification of the affinity of an interaction by changes in the intrinsic temperature dependence of the fluorescence. Typically, the fluorescence intensity of a dissolved fluorescent dye changes with temperature.23 This change in yield of fluorescence is an inherent property of most fluorescent dyes. A change in temperature of the sample may lead to a change in intensity that is due to a change in absorption, fluorescence lifetime, quantum yield, and a spectral shift of the emitted fluorescence.23 These properties are also dependent on the local viscosity and environment of the dye, namely, the local conformation and local amino acid composition of the protein the dye is coupled to as well as the surrounding aqueous solution.24,25 Thus, the intrinsic temperature dependence of a dye is different, when the local environment changes. If a molecule binds to a labeled protein in close proximity to the fluorescent dye or induces a conformational change of the protein at a position close to the dye, this binding event also changes the temperature dependence of the fluorescence and will be detected in MST T-Jump. As can be seen from the above discussion, the T-Jump signal is generated by local effects. A crude upper limit of the range of this effect can be estimated to 1–2 nm by considering the reported interactions between fluorescent dyes and the protein's tryptophans.26 This provides further information on the binding mode/binding site of a molecule. The sensitivity of the MST T-Jump can be modulated by the dye chosen for the MST experiment. The quantum yield of dyes, especially of the structurally more flexible dyes, are very strongly influenced by the polarity of the medium24 and thus dependent on the composition of the local biomolecule scaffold. Besides probing changes in the static local surrounding of a dye, the fast IR-Laser-induced MST T-Jump also probes the rigidity of this scaffold of dipoles. This rigidity of the protein's scaffold/conformation is often changed by a binding event (e.g., structure of the protein gets more rigid or even more flexible) and thus also the fluorescence response to the MST T-Jump is changed and reports the binding and allows its quantification. The MST T-Jump and the thermophoretic signal can be easily separated, since the effects that lead to a temperature-induced change in fluorescence as well as the heating itself are fast processes that happen almost instantaneously after start of IR-Laser heating. The timescale of the MST T-Jump is very sensitive to the inner diameter of the capillaries and care has to be taken that a suited capillary geometry is chosen.
Thermophoresis
The analysis of thermophoresis is the standard procedure used for evaluating MST data. Thermophoresis takes place on a different timescale than the T-Jump and is essentially a diffusion limited transport process, which in general becomes significant more than one second after start of IR-Laser heating. Changes in thermophoresis of the fluorescently labeled species reflect any changes that have an influence on thermophoretic mobility (DT) and diffusion coefficient (D). This means size, charge, and changes in the solvation shell of a molecule. It is not a localized effect as MST T-Jump, but instead indicates a global change of the fluorescently labeled molecule. One of the main advantages of MST is that it is not only dependent on a change in size of a complex with respect to the binding partners involved but also on charge and changes in the hydration shell. Thus, thermophoretic properties are altered even when a low-molecular-weight compound, peptide, or even ion interacts with a much larger fluorescently labeled protein.3
For an analysis, the ratio of fluorescence values after 30 s of laser heating compared to the fluorescence after ∼1 s of IR-Laser heating is plotted versus the concentration of the unlabeled binding partner. This way, the normalized fluorescence does not include the MST T-Jump, but instead only the fluorescence change induced by thermophoretic motion (i.e., concentration change). The T-Jump can be included in the analysis by plotting the fluorescence ratio of initial fluorescence to fluorescence after 30 s of laser heating. In most cases this is possible, but care has to be taken that when the MST T-Jump shows a concentration dependence of its own whose amplitude is not of the same direction as thermophoresis (i.e., increasing vs. decreasing amplitude). In this case the two signals of opposite sign may cancel out, when analyzed in conjunction. Notably, it is not necessary that thermophoresis reaches a steady state to obtain a dissociation constant.
Backdiffusion
The molecule flow induced by MST leads to a concentration inhomogeneity in the solution. After the heating IR-Laser is turned off, the temperature gradient (i.e., temperature increase) vanishes on approximately the same fast time scales that it takes to establish the gradient. As soon as the temperature gradient as driving force for thermophoresis is not present any more, the spatial concentration gradient starts to relax as well. The time it takes to reach a homogeneous distribution of molecules depends on the diffusion velocity of molecules and thus on their size. The Backdiffuison is therefore suited to detect changes in the size of molecules. In principle, this process is similar to Fluorescence Recovery After Photobleaching (FRAP) experiments27 and allows one to measure the hydrodynamic radius of molecules.28 Backdiffusion provides also important information on the reliability of an experiment. If no change in size is expected (i.e., in case of small molecule binding), a change in backdiffusion indicates aggregation or oligomerization of molecules.
Initial Fluorescence
Beside the signals described above, which are either directly (T-Jump, Thermophoresis) or indirectly (Backdiffusion) induced by IR-Laser heating, the initial fluorescence provides useful, IR-Laser–independent information for interaction analysis as well. Typically, when analyzing an MST experiment, the initial fluorescence values of different samples vary randomly due to the manual sample preparation. A reproducibility of fluorescence intensities, when preparing each sample manually, is between 5% and 10%. In MST experiments, the relative changes in fluorescence are measured, and thus small changes in initial fluorescence have no influence on the result as long as the fluctuations in concentration do not significantly affect the biochemical reaction itself. In some cases, the initial fluorescence might already report changes of the binding state of a molecule. The MST instrumentation measures thermophoresis in geometrically very well-defined sample container that allows one to measure fluorescence intensities of the individual samples with high precision and thus changes in the intensity are observed with exceptionally high precision. It is frequently encountered, when using an MST instrument, that the interaction of two molecules slightly changes the initial fluorescence of a sample. These changes are likely caused by changes in the electrostatic surrounding of the fluorescent dye when complex formation takes place and, where possible, a dissociation constant may as well be calculated from changes in initial fluorescence. When these changes in the fluorescence are very strong, the contribution of the species with higher fluorescence intensity is overestimated and the results have to be corrected with a correction factor obtained by measuring the fluorescence of the unbound and fully bound state (Baaske et al.2 and supplementary information therein). Exceptionally strong changes in initial fluorescence might also indicate that the bound or unbound state is aggregating and fluorescent material is lost before the MST analysis (e.g., in the micro reaction tube).
Other Transport Processes
The localized heating of the sample induces another transport process, namely, thermal convection.29 On the bottom of the capillary, the convective flow is centered toward the detection volume, whereas on top, the flow is directed toward the periphery. The convective flow leads to a steady in- and out-flow of molecules to the volume that is analyzed with a velocity of several μm/s (Fig. 2). The convective flow can be used to gain additional information if, for example, the fluorescently labeled molecule preparation contains macroscopic aggregates (i.e., greater than several micrometer) or if its aggregation is induced by addition of compounds or other binding partners. Aggregated molecules are efficiently transported by the convective flow toward the heat spot and out of the heated region. A fluorescent particle in the solution is observed as a peak in fluorescence intensity during the IR-Laser heating phase. As a basic rule, homogeneity of the labeled sample is an important prerequisite for a successful MST analysis as it is the main determinant of noise in the system.
The convection flow has an influence on the concentration change induced by thermophoretic motion, and the steady-state concentration profile reflects the effects of convection, thermophoresis, and diffusion.29
Experiments and Experimental Conditions
In the following, various examples are shown, where affinities have been determined by MST. In all experiments, one of the molecules is labeled fluorescently, whereas the unlabeled molecule is titrated from concentration higher than the expected dissociation constant down to sub stoichiometric concentrations with respect to the fluorescently labeled molecule. Typically, the concentration of the labeled molecule should be in the order of the KD or lower. A much too high concentration of the labeled binding partner would lead to a significant shift of the inflection point of the binding curve and an uncertainty in the determination of the dissociation constant. However, using the full quadratic dependency obtained from the law of mass action (Baaske et al.2 and supplementary material therein) a dissociation constant is precisely determined even at concentrations slightly higher than KD, since not only the inflection point but also the shape of the dose response curve is taken into account.
After preparing up to 16 samples of a serial dilution and mixing it with the labeled binding partner, capillary forces are used to fill the samples into MST capillaries. To avoid sample evaporation and allow for storage of samples, a wax is used for sealing the capillaries. The samples are placed on a tray, which is inserted in the instrument. A fluorescence scan is performed across the capillaries to determine the position of the capillaries with micrometer precision. After this scan, up to 16 subsequent thermophoresis measurements are performed to determine the binding affinity.
The main source for noise when performing an MST experiment is a not sufficiently high sample quality, meaning inhomogeneities like aggregates being present in the solution. When setting up an MST assay, typically in a first step the quality of a sample in the assay buffer is tested. If necessary, sample quality can be enhanced by spinning it down, or by adding additives like detergent, dithiothreitol, or bovine serum albumin (BSA). As MST works in basically any buffer, changes in buffer composition, ionic strength, or pH can be tested as well.
Beside sample quality, the quality of the capillary and the stability of sample within the capillaries are important. High reproducibility and low sample consumption are achieved using thin glass capillaries with a total volume of <4 μL. The capillaries containing the sample material provide a fixed geometry, which is very important to obtain a reproducible temperature increase (i.e., constant IR-Laser absorption length) and a constant convection pattern. Thus, capillaries have to comply with certain criteria to allow a successful analysis of thermophoretic properties. The difference of inner diameter between individual capillaries is less than a micrometer and the glass quality is chosen such that artifacts from visible light and especially IR-Laser light diffraction are avoided.
Also, the surface properties of the capillaries are important to grant sample stability. Standard glass that is physically treated to achieve homogeneous surface properties can be used in most cases. However, in some cases an adsorption of sample material to glass surfaces is observed in the scan of capillary positions. Due to this effect, only part of the molecules is mobile in the temperature gradient. Adsorption is readily observed by the experimenter due to high fluorescence values from the walls of the capillaries. Capillaries covalently modified with hydrophilic or hydrophobic polymers are available to suppress the adsorption process and keep biomolecules stable in solution (NanoTemper Technologies). In general, also buffer additives like detergents (e.g., Tween-20), proteins (e.g., BSA), or reductive agents (e.g., dithiothreitol) can be used to stabilize samples in solution.
Protein–Small Molecule Interaction
P38 versus BIRB/SB203580
p38 is a serine/threonine protein kinase in the mitogen-activated protein kinase family. There are four isoforms of p38 (p38α, p38β, p38γ, and p38δ), and p38α is considered as the key isoform involved in modulating inflammatory response in rheumatoid arthritis and inflammatory pain.30 Two well-characterized small molecule antagonists SB 20358031 and the clinical candidate BIRB-79632 were used in this study. Whereas the first compound competes with ATP for the binding site on the kinase, BIRB-796 binds adjacent to the active site and directly inhibits enzymatic activity by affecting the conformation of the ATP site.33 Our experiment describes the direct measurement of the compounds binding to the fluorescently labeled p38α (Fig. 3). The concentration of the protein is kept constant at 25 nM, whereas the compound BIRB-796 is titrated from the micromolar to the sub-nanomolar range. The binding of the low-molecular-weight compound to the proteins is readily observed as a change in the thermophoretic property of the fluorescently labeled protein upon complex formation. The complex shows a stronger decrease of normalized fluorescence than the unbound protein. The dissociation constant is determined to be 6±2 nM in good agreement with literature values.33 Also, the evaluation of the MST T-Jump response is shown in Figure 3, which in case of p38 BIRB-796 interaction yields a binding curve with a KD of 11±6 nM, in good agreement with the corresponding thermophoresis result. This experiment shows that thermophoresis (and in some cases also the MST T-Jump) is sufficiently sensitive to observe interactions that do not considerably alter the size or mass of a protein. The direct binding experiment of BIRB-796 shows that MST is not restricted to assays where size or mass of molecules is changed considerably. The amplitude of the change in normalized fluorescence that is observed for the BIRB binding shown in Figure 3 depends on the chemistry of the compound that is titrated (e.g., Charge), its binding site, and the conformational change induced upon binding. Typically, response units (i.e., amplitude) in the range of 5–100 units ΔFnorm [‰] (1,000×(Fnorm (bound)−Fnorm (unbound))) are observed that can be either positive or negative.
Fig. 3.

Protein–small molecule interactions. Direct interaction study between fluorescently labeled p38 protein kinase and the small molecule inhibitor BIRB-796. The p38 concentration is kept constant at 25 nM and the small molecule is titrated from about 2,500 to about 0.3 nM. The difference in normalized fluorescence [‰] is plotted for analysis of thermophoresis (A) and MST T-Jump (B). The protein showed a stronger decrease in fluorescence in the bound than in the unbound state. The KD is fitted to 6±2 nM (A) and 11±6 nM (B) in good agreement with literature values. In general, the amplitude of MST T-Jump and thermophoresis may differ in magnitude or even direction. Note that the 50% point of the binding curve only coincides with the KD for negligible labeled binder concentration. The law of mass action law yields a shift of the 50% point depending on the concentration of offered binding partner. Error bars represent standard error of n=3 measurements.
Competitive Experiment
P38-Tracer versus BIRB
Although the amplitude and direction of the signal contains valuable information on the interaction, it is not straight-forward to design a high-throughput screening (HTS) assay with this direct binding approach. In such a screening approach the number of data points per titration is typically reduced to only a few concentrations. By defining an amplitude cut-off value, compounds are identified as hits or as nonbinders. By using the direct binding approach, this cut-off value is difficult to define, since most compounds differ in the strength of their MST response and the direction of the signal. This high information content is advantageous to characterize an interaction in detail, but for an HTS approach it is not desirable and to overcome this drawback, a competitive approach (i.e., reporter assay) can be used. To establish such an assay here, a fluorescently labeled tracer molecule (Tracer 178; Invitrogen) is used at a concentration of 50 nM and mixed with a serial dilution of active p38 starting at 1 μM. For the experiment described here, a 50 mM Tris buffer pH 7.6 containing 150 mM NaCl, 10 mM MgCl2, and 0.05% Tween-20 was used. The samples are incubated for 10 min and are measured at a temperature increase of 3 K for 30 s. A decreasing MST signal with increasing protein concentration (Fnorm [‰] starting at 805 units, decreasing to 738 units) is observed with a sigmoidal behavior that allows deducing a KD of about 80 nM (Fig. 4). This experiment is sufficient to characterize the interaction between Tracer and the p38 kinase. Following this experiment, 150 nM of p38 protein is mixed with 25 nM of Tracer 178. To this stock solution a serial dilution of the compound SB203580 (MW=377.4 Da) starting at 4 μM is added. This molecule is known to have a high affinity to the protein p38 IC50=34 nM in vitro and 600 nM in cells. After incubation of 20 min the MST signal of the samples is measured (Fig. 4B). The signal shown in Figure 4 starts at an Fnorm level of about 760 units. Thus, a significant amount of the tracer is in complex with the protein. When increasing the concentration of SB203580, the MST signal increases to about 805 units, which is exactly the signal level we expect for free Tracer 178 thermophoresis. The signal allows one to determine an IC50 of 80 nM and taking the competition and the protein concentration into account a dissociation constant of 20 nM in good accordance with literature values.30
Fig. 4.

Competitive MST. (A) Binding of kinase p38 to fluorescently labeled compound (Tracer 178; Invitrogen). The protein p38 is titrated against the fluorescently labeled Tracer 178. Upon binding of the protein, the tracer thermophoresis is changed. The affinity is determined to 80 nM. (B) 150 nM of p38 and 25 nM of Tracer 178 are mixed with increasing amounts of the compound SB203580. The replacement of the tracer by the compound is observed with MST. Since the binding is observed in a competition setup with two p38 binding compounds present, the solid line is only a guide to the eye. The amplitude of the MST signal directly reflects the fraction of the tracer bound to p38. Error bars represent standard error of n=3 measurements.
Performing a competition experiment using MST has several advantages. It is essentially label free, since all molecules of interest (i.e., p38 and SB203580) are not labeled. It is site specific, since a very strong MST response with the expected amplitude is only generated when a compound competes with the tracer. This allows defining a cut-off value and to reduce the number of data points to set up a screening project with high amount of compounds. Hits identified with the tracer approach can be verified with a follow-up direct binding study. Since no size changes of molecules are necessary for an MST analysis, also a fluorescently labeled protein-protein complex can be formed and information on the ability of a compound to interrupt this interaction can be obtained. By doing so, the competitive approach is designed as a functional assay instead of detecting only molecule binding.
Protein–DNA and Protein–Protein Interactions
Heat-stable protein kinase inhibitor versus catalytic subunit of protein kinase A
Conformational control of protein kinases is an important way of modulating catalytic activity. Crystal structures of the C (catalytic) subunit of PKA (protein kinase A) in complex with physiological inhibitors and/or nucleotides suggest a highly dynamic switching between open and more closed conformations. The underlying molecular mechanisms have been analyzed in detail using the SPR technique.34 Here we show the detailed binding analysis of the physiological PKA inhibitor heat-stable protein kinase inhibitor (PKI), in the presence and absence of nucleotide cofactors. It could be shown that the affinity of the inhibitor PKI is strongly enhanced in the presence of ATP/Mg2+. This is consistent with a model, where a high-affinity complex consisting of PKI and the C subunit could only be formed in the closed state, which is dependent on the presence of both metal ions and nucleotide. For this experiment, the inhibitor PKI has been labeled fluorescently with the dye NT-647 (NanoTemper Technologies) and the C subunit of PKA is titrated in presence of ATP/Mg2+ and absence of ATP/Mg2+. A concentration of 40 nM of fluorescently labeled PKI is mixed with a serial dilution of the catalytic subunit of PKA (C-subunit) (final PKI concentration: 20 nM). The experiment was performed in a Tris buffer pH 7.6 containing 1 mM ATP and 5 mM MgCl2, as well as in a buffer containing 50 μM EDTA instead of ATP and MgCl2. Figure 5 shows the result of the MST experiments. In the absence of ATP and MgCl2, a strong reduction in the affinity from 2.3±0.8 to 489±105 nM is observed. This is an example of another class of interactions where the MST approach has particular advantages to standard techniques. Since MST measures affinities in low volume capillaries and without surface coupling of molecules, studies of complexes involving multiple components can be performed, whereas only one of these has to be fluorescently labeled. Thus, information on the interdependencies of interactions is obtained by analyzing a set of experiments in the presence of various proteins/compounds of interest. In other assays, such interactions are notoriously difficult to measure because of material consumption and stability issues of complexes.
Fig. 5.

Protein–protein interactions. The affinity of the catalytic subunit of protein kinase A (C-subunit) to the pseudosubstrate inhibitor heat-stable protein kinase inhibitor is analyzed. (A) In presence of ATP/Mg2+, a high affinity of 2.3±0.8 nM is observed. (B) In absence of the cofactor a dissociation constant of 489±105 nM is obtained.
DNA versus Ku70/80
The DNA repair protein Ku acts as a heterodimer of Ku70 and Ku80 and binds to DNA ends produced during the generation of programmed double-strand breaks (DSBs) induced by V(D)J or class switch recombination, or accidently by a variety of DNA damaging agents. It has been shown that Ku binds much stronger to DNA ends than to internal DNA regions.35 Although there are a number of reports indicating the possible binding of Ku to nicked DNA,36 or to single-to-double-stranded DNA transition,37 indisputable evidence exists that Ku preferentially binds to DNA ends. The DNA end binding activity of Ku highlights its major functions in genome stability and maintenance and in the survival of cells after introduction of DSBs. The loading of Ku onto DNA ends is thought to be one of the first steps in the repair of DSBs by nonhomologous DNA end joining (NHEJ), where the Ku heterodimer recruits the catalytic subunit of DNA-dependent protein kinase, and thus orchestrates the overall DSB repair process.35
The resolved three-dimensional structure of Ku revealed a ring-shaped molecule perfectly evolved to bind DNA termini. Nevertheless, the ring-shaped Ku structure may also increase the complexity of Ku-DNA binding kinetics, since DNA can accommodate multiple Ku subunits, by translocation of the threaded Ku molecule along the double helix. In addition, Ku may exhibit cooperative DNA binding characteristics during the loading process. However, using computer simulation and curve-fitting, several comprehensive mechanistic models and rate constants were provided that closely approximate the experimental data for DNA helices that bind one, two, or three Ku molecules under both kinetic and equilibrium conditions.38
Here we have measured the binding of Ku to fluorescently labeled 50 bp dsDNA using the MST technology. Since more than one Ku molecule (1–3) can bind to 50 bp dsDNA oligos, we expected to distinguish in this analysis between initial binding state and final binding state, which should exhibit different affinities to dsDNA. In the binding reaction, 50 nM AlexaFluor 532–labeled dsDNA was incubated with the indicated amount of unlabeled Ku. The binding was analyzed using MST directly after mixing of protein and DNA, or 30 min later (Fig. 6). As expected, we observed binding of Ku to DNA immediately after assembling the reaction with a calculated KD of about 100 nM. However, a more complicated binding was detected in samples incubated for 30 min. In this case a biphasic binding pattern was detected: first, a high affinity binding with an IC50 of 2 nM (red), which correlates well with previously reported SPR and EMSA data,38 and then the lower affinity binding (KD=100 nM, black). While the high affinity binding is presumably related to the binding of Ku to DNA ends, the second phase (KD=100 nM) might correlate to the translocation or binding of Ku to internal DNA positions. Please note that the 50% point of the binding curve only coincides with the KD for negligible labeled binder concentration. The mass action law yields a shift of the 50% point depending on the concentration of offered binding partner, in our case the dsDNA. Further work is needed to elucidate the binding mode observed in the MST signal. The biphasic DNA binding behavior at equilibrium indicates heterogeneity in the number of binding sites and positions occupied by the proteins. These results demonstrate the advantages of the MST technology for the analysis of molecular interactions in solution. Although the technology relies on equilibrium analysis, the time dependencies of slow binding events can be measured, since a measurement time of 10–30 s/sample is fast enough to allow multiple determinations in a time frame of a few minutes.
Fig. 6.

DNA–protein interactions. Binding of Ku to a fluorescently labeled dsDNA oligomer. The thermophoresis of the fluorescently labeled DNA oligomer shows a biphasic signal after 30 min of incubation. This signal reflects two different binding modes of Ku. A low affinity binding possibly to the center of the DNA molecule (KD=100 nM, black) and a high affinity reflects binding to ends of the DNA molecule (IC50=2 nM, red). The binding curve after 30 min of incubation was fitted to two differing models for [Ku70/80] smaller and larger than 6 nM, respectively.
Interactions of Membrane Proteins
SNARE receptor interactions in liposomes
In eukaryotes, most intracellular membrane fusion reactions are mediated by the interaction of complementary SNARE proteins that are present in both fusing membranes. The following experiment shows the result of two different liposome populations with compatible SNAREs incorporated in their membranes that bind to each other, followed by membrane fusion.39,40 One liposome population contains the neuronal SNARE protein synaptobrevin-2 (syb-2), whereas the other contains a receptor complex consisting of SNAP-25, syntaxin-1A and a fragment of syb-2 (residues 49–96) that is labeled with Alexa Fluor 488. Full length syb-2 binds to the acceptor SNAREs and a cis-SNARE complex is formed (Fig. 7, top). This results in the replacement of the fluorescently labeled syb-2 (49–96) fragment and is directly followed by membrane fusion. Thus a signal is generated upon binding of the two receptors. This approach has been used instead of using a labeled liposome to separate the receptor interaction from the following process of liposome fusion. The result of a thermophoresis experiment as a function of the concentration of (unlabeled) syb-2 liposomes is shown in Figure 7 (bottom). The concentration of labeled acceptor SNARE liposomes has been kept constant. The 50% point of the binding curve is found at about 450 nM. The change in thermophoretic amplitude shows the dissociation of the syb-2 (49–96) fragment. Since this dissociation is irreversible, the result reflects point at which 50% of active acceptor SNAREs have bound. The binding curve that is obtained (in equilibrium) shows a relatively strong change from the region of very high concentrations of (unlabeled) syb-2 liposomes toward low concentrations where the MST signal change is only small because little of the syb-2 (49–96) is dissociated. As a control, plain liposomes containing no synabtobrevin have been titrated to the labeled liposomes. As expected no change of the thermophoretic signal is observed. This experiment demonstrates that even complexes with a size of several 100 nm can be analyzed with MST. The use of liposomes allows the measurement of membrane-associated proteins and trans-membrane proteins at conditions that are, in comparison to other approaches, close to native conditions.
Fig. 7.

Membrane–receptor interactions. The interaction of two receptors incorporated into liposomes (Synaptobrevin and Syb49-96 Complex) is measured with MST. The synaptobrevin containing liposomes are titrated to a solution containing liposomes with the labeled Syb49-96-Complex. Upon receptor–receptor interaction, a fluorescently labeled peptide is released (black diamonds). The process of liposome docking is followed by liposome fusion. In this experiment, a competitive approach (i.e., labeled peptide replacement) has been used instead of incorporating a label in a liposome to focus on the interaction between the membrane receptors and not the following process of liposome fusion. As a control plain liposomes, containing no receptor have been used (black triangles). Error bars of synaptobrevin liposomes represent standard error of n=3 measurements.
Conclusion and Outlook
MST is an equilibrium method in bulk solution for the analysis of a broad range of molecular interactions. It measures the mobility of a fluorescently labeled molecule in a temperature field (Thermophoresis) and the sensitivity of fluorescence yield on temperature (MST T-Jump). The approach is sufficiently sensitive to measure the interactions of low-molecular-weight compounds to proteins as well as the interactions of proteins and liposomes. The measurement in free solution grants high flexibility in assay design so that even interaction in the presence of multiple cofactors can be measured.
Beside interaction studies, it is also possible to analyze the activity of enzymes by measuring product formation in enzymatic assays. In the future this will allow functional studies in addition to direct binding studies, using the very same platform.
Since the only prerequisite for the analysis of molecule thermophoresis with an MST instrument is a method to monitor concentration changes, the method can be extended, at least for some interaction studies, to the label free realm by using tryptophane fluorescence. This approach can be used for a wide range of interaction studies as well as thermal stability studies.
Future work will reveal more insight in the processes of MST T-Jump, thermophoresis, and the relevance of structural and conformational changes. Also, the effect of conformational changes induced by IR-Laser heating will be further elucidated by future studies and might provide further insight in protein function.
Abbreviations
- BSA
bovine serum albumin
- DSB
double-strand breaks
- EMSA
electrophoretic mobility shift assay
- FA
fluorescence anisotropy
- FCS
fluorescence correlation spectroscopy
- HTS
high-throughput screening
- IR
infrared
- ITC
isothermal titration calorimetry
- MST
microscale thermophoresis
- PKA
protein kinase A
- PKI
stable protein kinase inhibitor
- SPR
surface plasmon resonance
- T-Jump
temperature jump
Acknowledgments
The authors want to express their gratitude to the following individuals and institutions:
Prof. Iliakis (Universitätsklinikum Essen, Essen, Germany) for providing the data for the DNA–protein interaction experiment (Fig. 6); Karsten Meyenberg and Prof. Ulf Diederichsen (Institut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Göttingen, Germany), Geert van den Bogaart, and Reinhard Jahn (Max-Planck-Institut für biophysikalische Chemie, Göttingen, Germany) for the liposome experiments (Fig. 7); and Krishna Saxena (Johann Wolfgang Goethe-Universität, Frankfurt, Germany) for the p38 inhibitor and tracer binding studies (Figs. 3 and 4). The catalytic subunit of PKA and the heat stable inhibitor PKI were kind gifts from F.W. Herberg (University of Kassel, Kassel, Germany) and B. Zimmermann (Biaffin GmbH & Co KG, Kassel, Germany) (Fig. 5) supported by the EU FP7 collaborative project Affinity Proteome (contract 222635).
Sergey Tarasov (NCI Frederick) is thanked for fruitful discussions of biomolecular interaction studies.
The experiments detailed in Fig. 6 were carried out in the Institute of Medical Radiation Biology, University of Duisberg-Essen, Essen, Germany.
The work was supported by the Federal Ministry of Education and Research, Germany, Hightech Strategie “KMU-Innovativ: Biotechnologie-Biochance” (Grant No: FKZ 0315376).
Center for Nanoscience (CeNS) and the Nanosystems Initiative Munich (NIM) supported this work.
Disclosure Statement
S.D. and P.B. are founders of the LMU spin out company NanoTemper Technologies GmbH, which provides services and devices based on thermophoresis.
References
- 1.Duhr S. Braun D. Why molecules move along a temperature gradient. PNAS. 2006;103:19678–19682. doi: 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baaske P. Wienken CJ. Reineck P. Duhr S. Braun D. Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew Chem Int Ed. 2010;49:2238–2241. doi: 10.1002/anie.200903998. [DOI] [PubMed] [Google Scholar]
- 3.Wienken CJ. Baaske P. Rothbauer U. Braun D. Duhr S. Protein binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:10. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- 4.Wienken CJ. Baaske P. Duhr S. Braun D. Thermophoretic melting curves quantify the conformation and stability of RNA and DNA. Nucleic Acids Res. 2011;10:1093. doi: 10.1093/nar/gkr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raether H. Advances in research, development. In: Hass G, editor; Francombe MH, editor; Hoffman RW, editor. Physics of Thin Films. Academic Press; New York: 1977. pp. 145–261. [Google Scholar]
- 6.Frey BL. Jordan CE. Kornguth S. Corn RM. Control of the specific adsorption of proteins onto gold surfaces with Poly(L-lysine) Monolayers Anal Chem. 1995;67:4452–4457. [Google Scholar]
- 7.Faegerstam LG. A non label technology for real time biospecific interactions analysis. In: Villafranca JJ, editor. Techniques in Protein Chemistry II. Academic Press, Inc.; San Diego, CA: 1991. pp. 65–71. [Google Scholar]
- 8.Myszka DG. Jonsen MD. Graves BJ. Equilibrium analysis of high affinity interactions using BIACORE. Anal Biochem. 1998;265:326–330. doi: 10.1006/abio.1998.2937. [DOI] [PubMed] [Google Scholar]
- 9.Folker ES. Baker BM. Goodson HV. Interactions between CLIP-170, tubulin, and microtubules: implications for the mechanism of Clip-170 plus-end tracking behavior. Mol Biol Cell. 2005;11:5373–5384. doi: 10.1091/mbc.E04-12-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiseman T. Williston S. Brandts JF. Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 11.Freire E. Mayorga OL. Straume M. Isothermal titration calorimetry. Anal Chem. 1990;62:950–959. [Google Scholar]
- 12.Ladbury JE. Counting the calories to stay in the groove. Structure. 1995;3:635–639. doi: 10.1016/s0969-2126(01)00197-6. [DOI] [PubMed] [Google Scholar]
- 13.Schwille P. Haustein E. Fluorescence Correlation Spectroscopy. A tutorial for the Biophysics Textbook Online (BTOL) Biophysical Society; Rockville, MD: 2002. [Google Scholar]
- 14.Ludwig C. Diffusion zwischen ungleich erwärmten Orten gleich zusammengesetzter Lösungen. Sitzungsber Akad Wiss Wien Math-Naturwiss. 1856;20:539. [Google Scholar]
- 15.Emery AH., Jr. Drickamer HG. Thermal diffusion in polymer solutions. J Chem Phys. 1955;23:2252–2257. [Google Scholar]
- 16.Ham J. Kinetic theory of thermal diffusion in dilute polymer solutions. J Appl Phys. 1960;31:1853–1858. [Google Scholar]
- 17.Morozov KI. Thermal diffusion in disperse systems. J Exp Theor Phys. 1999;88:944–946. [Google Scholar]
- 18.Schimpf ME. Semenov SN. Mechanism of polymer thermophoresis in nonaqueous solvents. J Phys Chem B. 2000;104:9935–9942. [Google Scholar]
- 19.Voit A. Krekhov A. Enge W. Kramer L. Köhler W. Thermal patterning of a critical polymer blend. Phys Rev Lett. 2005;92:214501. doi: 10.1103/PhysRevLett.94.214501. [DOI] [PubMed] [Google Scholar]
- 20.Duhr S. Braun D. Thermophoretic depletion follows Boltzmann distribution. Phys Rev Lett. 2006;96:168301. doi: 10.1103/PhysRevLett.96.168301. [DOI] [PubMed] [Google Scholar]
- 21.Dhont JK. Wiegand S. Duhr S. Braun D. Thermodiffusion of charged colloids: single-particle diffusion. Langmuir. 2007;23:1674–1683. doi: 10.1021/la062184m. [DOI] [PubMed] [Google Scholar]
- 22.de Groot SR. Mazur P. Nonequilibrium Thermodynamics. North–Holland: Amsterdam; 1969. [Google Scholar]
- 23.Zelent B. Troxler T. Vanderkooi JM. Temperature dependence for fluorescence of β-NADH in Glycerol/Water solution and in Trehalose/Sucrose glass. J Fluoresc. 2007;17:37–42. doi: 10.1007/s10895-006-0146-0. [DOI] [PubMed] [Google Scholar]
- 24.Buschmann V. Weston KD. Sauer M. Spectroscopic study and evaluation of red-absorbing fluorescent dyes. Bioconjug Chem. 2003;14:195–204. doi: 10.1021/bc025600x. [DOI] [PubMed] [Google Scholar]
- 25.Lakowicz JR. Principles of Fluorescence Spectroscopy. second. Kluwer Academic/Plenum Publishers; New York, Boston, Dordrecht, London, Moscow: 1999. [Google Scholar]
- 26.Vaiana A, et al. Fluorescence quenching of dyes by tryptophan: interactions at atomic detail from combination of experiment and computer simulation. J Am Chem Soc. 2003;125:14564–14572. doi: 10.1021/ja036082j. [DOI] [PubMed] [Google Scholar]
- 27.Axelrod D. Koppel DE. Schlessinger J. Elson E. Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sperling R, et al. Size determination of (Bio)conjugated water-soluble colloidal nanoparticles: a comparison of different techniques. J Phys Chem C. 2007;111:11552–11559. [Google Scholar]
- 29.Duhr S, et al. Thermophoresis of DNA determined by microfluidic fluorescence. Eur Phys J E Soft Matter. 2004;15:277–286. doi: 10.1140/epje/i2004-10073-5. [DOI] [PubMed] [Google Scholar]
- 30.Dominguez C. Powers DA. Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr Opin Drug Discov Devel. 2005;8:421–430. [PubMed] [Google Scholar]
- 31.Davies SP. Reddy H. Caivano M. Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MR. Dominguez C. MAP kinase p38 inhibitors: clinical results and an intimate look at their interactions with p38α protein. Curr Med Chem. 2005;12:2979–2994. doi: 10.2174/092986705774462914. [DOI] [PubMed] [Google Scholar]
- 33.Pargellis C, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 34.Zimmermann B. Schweinsberg S, et al. Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem J. 2008;413:93–101. doi: 10.1042/BJ20071665. [DOI] [PubMed] [Google Scholar]
- 35.Ma Y. Lieber MR. DNA length-dependent cooperative interactions in the binding of Ku to DNA. Biochemistry. 2001;40:9638–9646. doi: 10.1021/bi010932v. [DOI] [PubMed] [Google Scholar]
- 36.Blier PR. Griffith AJ, et al. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J Biol Chem. 1993;268:7594–7601. [PubMed] [Google Scholar]
- 37.Rathmell WK. Chu GA. A DNA end-binding factor involved in double strand break repair and V(D)J recombination. Mol Cell Biol. 1994;14:4741–4748. doi: 10.1128/mcb.14.7.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taghva A. Ma Y. Lieber MR. Analysis of the kinetic and equilibrium binding of Ku protein to DNA. J Theor Biol. 2002;214:85–97. doi: 10.1006/jtbi.2001.2446. [DOI] [PubMed] [Google Scholar]
- 39.Schuette CG, et al. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc Natl Acad Sci USA. 2004;101:2858–2863. doi: 10.1073/pnas.0400044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pobbati AV. Stein A. Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]