

Abstract
Knowledge of all residue-residue contacts within a protein allows determination of the protein fold. Accurate prediction of even a subset of long-range contacts (contacts between amino acids far apart in sequence) can be instrumental for determining tertiary structure. Here we present BCL::Contact, a novel contact prediction method that utilizes artificial neural networks (ANNs) and specializes in the prediction of medium to long-range contacts. BCL::Contact comes in two modes: sequence-based and structure-based. The sequence-based mode uses only sequence information and has individual ANNs specialized for helix-helix, helix-strand, strand-helix, strand-strand, and sheet-sheet contacts. The structure-based mode combines results from 32-fold recognition methods with sequence information to a consensus prediction. The two methods were presented in the 6th and 7th Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiments. The present work focuses on elucidating the impact of fold recognition results onto contact prediction via a direct comparison of both methods on a joined benchmark set of proteins. The sequence-based mode predicted contacts with 42% accuracy (7% false positive rate), while the structure-based mode achieved 45% accuracy (2% false positive rate). Predictions by both modes of BCL::Contact were supplied as input to the protein tertiary structure prediction program Rosetta for a benchmark of 17 proteins with no close sequence homologs in the protein data bank (PDB). Rosetta created higher accuracy models, signified by an improvement of 1.3 Å on average root mean square deviation (RMSD), when driven by the predicted contacts. Further, filtering Rosetta models by agreement with the predicted contacts enriches for native-like fold topologies.
Key words: CASP, computational structural biology, contact prediction, structure prediction
1. Introduction
The contact prediction problem is defined as the identification of all spatially close residue pairs in the tertiary structure of a given protein sequence (conventionally Cβ-Cβ distance ≤8 Å). The motivation to solve this problem is that a complete list of all contacts defines the fold of the protein and allows structure determination using distance geometry methods (Aszodi et al., 1995; Huang et al., 1999). However, even very incomplete lists of long-range contacts can facilitate protein fold prediction by reducing the number of possible topologies sometimes to a unique solution (Li et al., 2004).
It is important to understand that not all contacts within a fold have the same value for protein structure prediction. While local contacts (contacts between amino acids nearby in sequence) are more readily predicted (e.g., within an α-helix or β-hairpin), their ability to constrain the fold space is limited. The challenge is predicting contacts between residues distant in sequence (sequence separations larger than 12 amino acids). Knowing only a few of these contacts frequently allows the fold of a protein to be defined completely (Baker, 2000; Bonneau et al., 2002a; Izarzugaza et al., 2007).
Therefore, contact prediction methods have the potential to improve the speed and the accuracy of de novo protein structure prediction methods in two ways (Izarzugaza et al., 2007): they can be used to enrich for good models in large ensembles of structural models, or they can directly be used to guide de novo folding simulations. Furthermore, contact prediction is useful for fold recognition (Cheng and Baldi, 2006; Olmea et al., 1999) and inferring protein folding rates and pathways (Plaxco et al., 2000; Punta and Rost, 2005b).
Contact prediction methods can be classified into two groups (Cheng and Baldi, 2006): (1) sequence-based and (2) structure-based. Sequence-based methods often use evolutionary correlated mutations (Fariselli et al., 2001a; Gobel et al., 1994; Halperin et al., 2006; Hamilton et al., 2004; Kundrotas and Alexov, 2006; Olmea and Valencia, 1997; Shindyalov et al., 1994; Valencia and Pazos, 2002) and machine learning approaches (Cheng and Baldi, 2007; Fariselli and Casadio, 1999; Fariselli et al., 2001a,b; Lund et al., 1997; Pollastri and Baldi, 2002; Pollastri et al., 2001; Punta and Rost, 2005a) such as artificial neural networks (ANNs), hidden Markov models (HMMs), or support vector machines (SVMs) to predict contacts.
A powerful concept in sequence-based contact prediction is use of evolutionary correlated mutations (Altschuh et al., 1988; Gobel et al., 1994; Pollock et al., 1999). From multiple sequence alignments, residue pairs are identified that are mutated concurrently between sequences in the alignment throughout evolution. Often spatially close residues are mutated to complement the initial mutation and maintain the protein's structure and/or function (Gobel et al., 1994). Therefore, identification of such residue pairs yields potential residue-residue contacts. Halperin et al. (2006) reviews use of correlated mutations for predicting inter-protein and intra-protein contacts, and concludes that correlated mutations by themselves can predict contacts with up to 20% accuracy (Halperin et al., 2006). In comparison, SAM_T06 by Shackelford and Karplus (2007) implements a hybrid approach where information from correlated mutations, along with various additional descriptors, are used to train ANNs for predicting contacts with accuracies ranging up to ∼60% for certain difficult targets while averaging ∼25% for long-distance contacts (Izarzugaza et al., 2007). PROFCON (Punta and Rost, 2005a), which ranked as one of the top groups in CASP6, also uses ANNs with descriptors, including evolutionary profiles and secondary structure prediction. SVMCON uses similar descriptors with SVMs instead of ANNs, and is reported to achieve 27.7% accuracy for ≥12 residue sequence separation contacts (Cheng and Baldi, 2007). A recent report by Wu and Zhang (2008) introduces SVM-SEQ, a sequence-based contact predictor, and SVM-LOMETS, a structure template-based predictor based on previously reported LOMETS (Wu and Zhang, 2007) meta-threading server which uses predictions from nine different threading algorithms. In their analysis of predictions for an independent data set, accuracy of SVM-LOMETS is 39% and accuracy of SVM-SEQ is 23%. However, when only new fold targets in CASP7 are considered, SVM-SEQ outperforms SVM_LOMETS and reaches an accuracy slightly better than of SAM_T06.
On the other hand, structure-based methods generally cluster best energy models generated by structure prediction techniques and pick the contacts that are observed most abundantly across the clusters (Chivian et al., 2005; Lee and Skolnick, 2008; Sali and Blundell, 1993; Shackelford and Karplus, 2007; Shao and Bystroff, 2003; Skolnick et al., 2004; Wu and Zhang, 2007, 2008; Zhang and Skolnick, 2004). PROSPECTOR_3.5 (Lee and Skolnick, 2008) implements a template-based approach, where it collects the contacts found in the tertiary models produced by TASSER_2.0 (Lee and Skolnick, 2008) and picks the ones that are commonly observed across tertiary models. SVM-LOMETS (Wu and Zhang, 2008), as described before, uses a similar approach but instead depends on LOMETS meta-server. As expected and as reported (Wu and Zhang, 2008), structure-based methods outperform sequence-based methods, especially if proteins of similar fold (templates) are available in the PDB and hence the predicted structural models are of high quality (Wu and Zhang, 2008). However, in de novo protein structure prediction, applicability of structure-based methods is limited due to the absence of highly similar and complete structural templates. Further, the computational intensity of protein structure prediction prior to contact prediction requires significant time and resources.
BCL::Contact introduces a novel hybrid approach where the sequence-based mode only relies on sequence information and utilizes individual ANNs for each distinct contact type. The structure-based mode combines results from various fold recognition servers using a single ANN. Here, we present evaluations and comparisons of both modes of BCL::Contact on predicting contacts. In particular, the value of fold recognition for contact prediction in the CASP hard fold recognition and new fold categories are evaluated. The objective of this work is to evaluate if consensus fold recognition results improve contact prediction even if no sequence homologs were unambiguously detected by the underlying fold recognition methods. Further, the impact of contact prediction on de novo tertiary structure determination is measured by testing the ability of predicted contacts to (a) enrich for native-like models in a set of decoys or (b) directly guide protein folding simulations using the Rosetta de novo protein folding algorithm (Simons et al., 1997).
2. Methods
2.1. Contact definitions and contact types
We use a Cβ-Cβ distance of ≤8 Å as a threshold for defining two amino acids as being in contact. A minimum sequence separation of 12 residues is required to exclusively focus on non-local contacts. Furthermore, the sequence-based mode uses five distinct contact types between secondary structure elements in the order as they appear in the protein sequence: helix-helix, helix-strand, strand-helix, strand-strand, and sheet-sheet. This distinction was introduced to test the ability of the ANN to specialize for specific types of interactions between secondary structure elements. It is restricted to the sequence-based mode due to the limited amount of training and test data available for the structure-based methods.
2.2. Protein data sets and training procedures
For the sequence-based mode, a non-redundant (<20% sequence similarity) 1834 protein subset of the Protein Data Bank (PDB) was selected using the PISCES server (Wang and Dunbrack, 2003). Ten percent of the structures were selected as an independent dataset and removed prior to training the ANNs. With the remaining 90%, 10 ANNs for each of the five contact types were trained in a cross-validation setup using a different non-overlapping 10% of the data as a monitoring data set.
For the structure-based mode, 545 proteins that served as targets during LIVEBENCH7, LIVEBENCH8, and LIVEBENCH9 experiments (Rychlewski and Fischer, 2005) were used as training dataset. Twelve percent (66) of these proteins were withheld for independent testing. Independent ANNs were trained in a 10-fold cross-validation setup with non-overlapping monitoring data sets.
For both modes, sequence-based and structure-based, the average output from the 10 ANNs is reported as the prediction result. All ANNs were trained in a “balanced” fashion with 50% contacts and 50% non-contacts by under-sampling the non-contacts. In sequence-based mode, the 50% non-contacts were a mixture of “true non-contacts” and “wrong-contacts” (contacts between other types of secondary structure elements). The large ratio of non-contacts to contacts would otherwise bias the ANN towards predicting non-contacts.
2.3. Numerical representation
In the sequence-based mode of BCL::Contact, for every residue pair (i, j), two sequence windows centered around these residues are used to generate input. The length of the window is chosen as five amino acids (two neighbors on each side of the amino acid of interest) for β-strands and nine amino acids (four neighbors on each side of the amino acid of interest) for α-helices. Both windows cover approximately 12 Å or two periods of the secondary structure element type.
Input to the ANNs (Fig. 1) starts with three position descriptors: (1) number of residues N-terminal to I; (2) number of residues between i and j; and (3) number of residues C-terminal to j. These global descriptors are followed by the following descriptors for each amino acid in the two windows: JUFO three-state secondary structure prediction (www.meilerlab.org, three numbers per amino acid) (Meiler and Baker, 2003), amino acid property profiles (seven numbers per amino acid: sterical parameter, polarizability, volume, hydrophobicity, isoelectric point, helix probability, and strand probability) (Meiler et al., 2001), as well as position-specific scoring matrices from PSIBLAST (20 numbers per amino acid) (Altschul et al., 1997). Hence, the five ANNs had a variable number of inputs determined by the associated window lengths: helix-helix 543, helix-strand and strand-helix 423, strand-strand, and sheet-sheet 303. All five ANNs had 16 hidden neurons, and one output neuron with an output range of [0, 1], with 0 being “non-contact” and 1 being “contact.” A consensus output is obtained from these five ANNs by weighing their prediction with the secondary structure predictions of both residues i and j as follows:
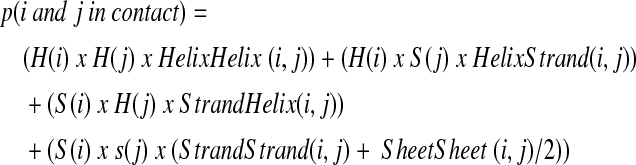 |
(1) |
FIG. 1.

Scheme for sequence-based and structure-based ANN contact prediction. (A) For a given sequence, contact predictions are calculated for every residue pair i and j. Sequence windows around positions i and j are taken into account in the sequence-based mode. (B) The numerical representation for both methods consists of sequence descriptors, single residue, and pair residue descriptors. The sequence descriptors include number of residues N-terminal to i, number of residues between i and j, and number of residues C-terminal to j. The sequence-based mode uses sequence windows centered on residues i and j of length 5 residues (2 neighbors on each side) for β-strands or 9 residues (4 neighbors on each side) for α-helices. (C) The numerical representations are fed to ANNs. The structure-based mode reports the output of the single ANN while for sequence-based mode, the outputs from the five specialized ANNs for individual contact types is obtained using equation (1).
where H(x) is the secondary structure prediction α-helix probability of residue x, and S(x) is the secondary structure prediction β-strand probability of residue x. HelixHelix(x,y), HelixStrand(x,y), StrandHelix(x,y), StrandStrand(x,y), and SheetSheet(x,y) are predicted probabilities of contact for the residue pair (x,y) from each individualized ANN.
In the structure-based mode, the fold recognition results of 32 servers (Bujnicki et al., 2001; Chivian and Baker, 2006; Debe et al., 2006; Fischer, 2003; Fischer et al., 2003; Ginalski et al., 2003a,b; Ginalski and Rychlewski, 2003; Jaroszewski et al., 2005; Jones, 1999; Karplus and Hu, 2001; Karplus et al., 2005; Lundstroem et al., 2001; McGuffin and Jones, 2003; Russell et al., 1998; Shi et al., 2001; Skolnick and Kihara, 2001; Tomii et al., 2005; Torda et al., 2004; Zhang et al., 2008) that participated in the LIVEBENCH7, LIVEBENCH8, and LIVEBENCH9 experiments (Rychlewski and Fischer, 2005) were used as input (Table S1; see Supplementary Material at www.liebertonline.com). The predictions were downloaded for 545 target proteins from the metaserver homepage (www.bioinfo.pl) (Ginalski et al., 2003a). The initial design of this method included only 24 servers, but no significant reduction in accuracy was observed. Nonetheless, reduction of number of servers used below a critical number or selective removal of the best fold-recognition servers is expected to have a negative effect on the accuracy of the method.
The input to ANN for the structure-based mode utilizes information from the models provided by these 32 servers in addition to sequence descriptors similar to the ones used by the sequence-based mode. A global agreement (GA) of the server predictions is calculated for each given target sequence as fraction of contacts jointly predicted by all servers over the number of all predicted contacts. For every residue i and j, the input to the ANN consists of six global descriptors: (1) number of residues N-terminal to i, (2) number of residues between i and j, (3) number of residues C-terminal to j, (4) number of valid models from servers where coordinates for i and j were defined (NS), (5) number of such models in which i and j were found to be in contact (NC), and (6) the global agreement value GA for this given sequence. These global descriptors are followed by JUFO three-state secondary structure prediction (three values per amino acid) and amino acid property profile (seven values per amino acid) for i and j. For each of the 32 servers, two values are input: (1) the inverse of the minimum distance observed between i and j in the 10 models available for each server (MD), (2) the agreement of this server's predictions for i and j with all other servers (AG - if i and j predicted to be in contact by this server S1, iterate over every other server S2 that also predicts i and j to be in contact and sum over the ratio of contacts S1 and S2 share). This process is illustrated in Figure 1. The ANN had 90 inputs, 32 hidden neurons, and one output neuron. The output range is [0, 1], with 0 being “non-contact” and 1 being “contact.”
2.4. ANN training and ROC curve analysis
The training algorithm was back-propagation of errors. The ANNs were trained until the root mean square deviation (RMSD) of the monitoring dataset was minimized (approximately 10,000 training periods). Training takes about 24 h on a single typical PC processor.
The predictions from both methods were analyzed using receiver operating characteristics (ROC) curves. For all ROC curves, area under curve (AUC) values are reported to quantify the improvement over random predictor.
All methods for training, analysis, and contact prediction are implemented in the BioChemical Library (BCL), an in-house developed C++ programming library.
2.5. Rosetta model building guided by BCL::Contact
Improving accuracy of protein structure prediction is one the most important aims behind development of contact prediction methods. Thus, in order to further analyze performance of BCL::Contact, contact predictions from BCL::Contact have been used as additional input to the protein structure prediction program Rosetta (Simons et al., 1997).
Rosetta was modified to include an additional contact prediction score. Disregarding predictions below a certain threshold, Rosetta assigns bonuses in the energy function during folding process for structures in which residue pairs predicted to be in contact are found within 8 Å (Cβ-Cβ distance). Variations on the threshold were systematically tested on the benchmark set of proteins, and 0.2 was found to give optimum performance.
A subset of 17 structures was selected from all targets released in LIVEBENCH7, LIVEBENCH8, CASP5, and CASP6. The selection was based on having a size of less than ∼150 residues (limitations of Rosetta for de novo folding) (Bonneau et al., 2002b) and being a hard fold recognition or de novo target without a known template (3D Jury J score lower than 50; http://bionfo.pl [Ginalski et al., 2003a]). The rationale for choosing hard fold recognition targets was to realistically test the impact of low confidence fold recognition results on de novo protein structure determination. The resultant subset was formed of the following structures: 1hjz, 1j1t, 1j26, 1l3p, 1lxj, 1mzb, 1nek, 1oh1, 1ojg, 1owx, 1oz9, 1p0z, 1p57, 1roc, 1sou, 1uan, and 1v32. None of these structures was used in training any of the ANNs used by BCL::Contact.
For all 17 proteins, 10,000 structural models were generated using Rosetta's unaltered de novo folding protocol. The runs were then repeated for each protein with contact predictions from the sequence-based mode and with contact predictions from the structure-based mode as additional inputs.
2.6. Enrichment of native-like de novo models
To test the ability of predicted contacts to select for native-like models and discriminate incorrect fold topologies, enrichment values were computed among the 10,000 models generated with Rosetta's unmodified de novo folding protocol. The enrichment values of low RMSD de novo models are calculated as follows:
 |
(2) |
where n is the total number of models (∼10,000), and m is the number of models in the top 10% by RMSD that can also be found in the top 10% by the newly implemented Rosetta sequence-based and structure-based contact scores, respectively.
2.7. RMSD and MAXN% distributions of de novo models
The Rosetta models generated with and without the use of contact prediction as input were compared by their distributions of RMSD and MAXN% (percentage of residues that can be superimposed to the native within 4 Å) (Ortiz et al., 2002) for all models generated for 17 benchmark proteins. Both of these values are computed within Rosetta.
The top, 10th percentile, and average values for RMSD and MAXN% are reported in Tables 1 and 2 for all 17 proteins. For cases where improvements are observed, p-values are calculated from one-tailed t-tests to assess the statistical significance of improvements. In addition, the distributions are presented in histogram plots in Figure 4 below.
Table 1.
Root Mean Square Deviation (RMSD) (Å) Distributions for Rosetta Folding Runs for All 17 Benchmark Targets
| |
RMSD (Å) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
No-contact |
Sequence-based |
Structure-based |
||||||||
| pdb id | Top | 10% | Avg | Top | 10% | Avg | p-value | Top | 10% | Avg | p-value |
| 1hjz | 10.0 | 15.6 | 18.0 | 9.9 | 15.4 | 18.0 | 5.24E-02 | 9.5 | 13.9 | 16.7 | 1.13E-139 |
| 1j1t | 14.9 | 19.4 | 21.4 | 14.0 | 19.3 | 21.3 | 1.18E-06 | 15.3 | 19.2 | 21.0 | 1.22E-32 |
| 1j26 | 9.6 | 15.7 | 18.1 | 9.1 | 15.3 | 17.9 | 2.74E-19 | 8.4 | 13.2 | 16.7 | 3.10E-121 |
| 1l3p | 4.3 | 9.8 | 12.7 | 3.8 | 9.9 | 12.9 | Not improved | 3.6 | 5.1 | 7.6 | 0.00 |
| 1lxj | 7.5 | 11.7 | 14.3 | 8.1 | 11.6 | 14.2 | 1.70E-02 | 8.2 | 12.8 | 15.2 | Not improved |
| 1mzb | 6.5 | 11.8 | 14.3 | 5.8 | 11.4 | 14.0 | 5.54E-29 | 5.4 | 9.0 | 12.5 | 1.39E-182 |
| 1nek | 6.9 | 10.2 | 13.4 | 6.6 | 10.5 | 13.6 | Not improved | 6.5 | 9.0 | 11.3 | 0.00 |
| 1oh1 | 7.9 | 12.3 | 14.4 | 7.9 | 12.2 | 14.2 | 2.19E-10 | 5.6 | 10.1 | 13.2 | 4.06E-117 |
| 1ojg | 6.2 | 11.6 | 14.5 | 6.5 | 11.7 | 14.4 | 1.02E-07 | 5.7 | 10.8 | 13.8 | 2.17E-35 |
| 1owx | 12.7 | 16.5 | 18.2 | 12.0 | 16.3 | 18.0 | 1.01E-17 | 9.2 | 14.6 | 16.9 | 2.74E-190 |
| 1oz9 | 7.8 | 13.8 | 16.5 | 7.5 | 13.8 | 16.4 | 4.76E-01 | 6.3 | 11.8 | 15.0 | 1.11E-136 |
| 1p0z | 4.5 | 10.7 | 13.9 | 5.3 | 10.2 | 13.7 | 7.66E-15 | 4.3 | 7.0 | 12.1 | 4.13E-104 |
| 1p57 | 10.8 | 14.2 | 15.7 | 11.0 | 14.0 | 15.7 | 9.69E-05 | 10.5 | 13.3 | 15.1 | 1.72E-70 |
| 1roc | 13.3 | 16.6 | 19.1 | 10.0 | 16.5 | 19.0 | 5.34E-05 | 12.0 | 16.0 | 18.6 | 1.02E-30 |
| 1sou | 11.2 | 16.6 | 19.1 | 10.4 | 16.7 | 19.0 | 1.30E-02 | 10.5 | 15.6 | 18.4 | 7.62E-45 |
| 1uan | 15.3 | 19.3 | 21.4 | 15.6 | 19.1 | 21.3 | 1.45E-06 | 14.6 | 18.2 | 20.5 | 4.80E-93 |
| 1v32 | 7.5 | 11.5 | 13.5 | 7.6 | 11.3 | 13.4 | 2.81E-06 | 6.5 | 9.2 | 11.6 | 5.34E-284 |
| Avg | 9.2 | 14.0 | 16.4 | 8.9 | 13.8 | 16.3 | – | 8.4 | 12.3 | 15.1 | – |
RMSD (Å) distributions for Rosetta folding runs for all 17 benchmark targets with no additional input and with input from sequence-based and structure-based modes of BCL::Contact. The top model, 10th percentile, and average (Avg) RMSD values are reported. For improved cases, p-values from a one-tailed t-test are also reported.
Table 2.
MAXN% Distributions for Rosetta Folding Runs for All 17 Benchmark Targets
| |
MAXN% |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
No-contact |
Sequence-based |
Structure-based |
||||||||
| pdb id | Top | 10% | Avg | Top | 10% | Avg | p-value | Top | 10% | Avg | p-value |
| 1hjz | 58.3 | 30.7 | 24.5 | 57.8 | 30.7 | 24.5 | Not improved | 59.4 | 39.1 | 28.7 | 1.22E-133 |
| 1j1t | 27.5 | 16.7 | 13.9 | 28.8 | 17.2 | 14.0 | 6.37E-03 | 24.9 | 18.0 | 14.6 | 1.03E-33 |
| 1j26 | 74.1 | 38.4 | 30.8 | 69.6 | 38.4 | 31.5 | 2.39E-15 | 73.2 | 45.5 | 35.7 | 1.04E-160 |
| 1l3p | 92.2 | 50.0 | 38.5 | 92.2 | 49.0 | 37.4 | Not improved | 100.0 | 85.3 | 64.9 | 0.00 |
| 1lxj | 80.8 | 50.0 | 41.7 | 80.8 | 48.1 | 40.4 | Not improved | 66.4 | 46.2 | 41.8 | 2.07E-01 |
| 1mzb | 73.5 | 45.6 | 34.3 | 80.9 | 47.1 | 35.0 | 4.42E-10 | 82.4 | 64.0 | 49.5 | 0.00 |
| 1nek | 64.6 | 43.4 | 33.0 | 69.9 | 42.5 | 32.3 | Not improved | 69.0 | 48.7 | 36.9 | 2.04E-86 |
| 1oh1 | 59.6 | 43.1 | 33.5 | 64.2 | 41.3 | 32.0 | Not improved | 83.5 | 50.5 | 40.5 | 1.21E-264 |
| 1ojg | 71.3 | 47.8 | 38.5 | 73.5 | 47.8 | 38.2 | 1.30E-03 | 83.1 | 50.7 | 41.4 | 1.43E-54 |
| 1owx | 64.5 | 37.2 | 30.2 | 57.9 | 37.2 | 30.2 | Not improved | 77.7 | 46.3 | 36.2 | 1.74E-209 |
| 1oz9 | 68.0 | 42.0 | 32.8 | 72.0 | 41.3 | 32.4 | Not improved | 76.7 | 48.0 | 38.1 | 1.12E-171 |
| 1p0z | 93.1 | 53.4 | 45.4 | 86.3 | 54.2 | 44.9 | Not improved | 94.7 | 65.7 | 52.3 | 1.62E-167 |
| 1p57 | 48.3 | 29.8 | 24.9 | 56.1 | 30.7 | 25.2 | 1.70E-05 | 44.7 | 32.5 | 26.5 | 1.52E-54 |
| 1roc | 36.1 | 22.6 | 18.9 | 38.1 | 23.2 | 19.2 | 6.67E-11 | 38.7 | 24.5 | 19.9 | 9.78E-34 |
| 1sou | 44.3 | 29.4 | 23.2 | 48.5 | 28.4 | 22.6 | Not improved | 43.8 | 31.4 | 24.3 | 2.24E-21 |
| 1uan | 34.4 | 21.6 | 17.8 | 41.0 | 22.0 | 18.0 | 1.33E-10 | 40.5 | 25.6 | 20.3 | 4.35E-148 |
| 1v32 | 72.3 | 45.5 | 35.9 | 69.3 | 45.5 | 36.1 | 1.58E-02 | 84.2 | 67.3 | 50.1 | 0.00 |
| Avg | 62.5 | 38.1 | 30.5 | 63.9 | 37.9 | 30.2 | – | 67.2 | 46.4 | 36.6 | – |
MAXN% (the percentage of residues that can be superimposed to the native within 4 Å) distributions for Rosetta folding runs for all 17 benchmark targets with no additional input and with inputs from sequence-based and structure-based modes of BCL::Contact. The top model, 10th percentile, and average (Avg) MAXN% values are reported. For improved cases, p-values from a one-tailed t-test are also reported.
FIG. 4.

RMSD and MAXN% histograms for Rosetta folding runs with and without BCL::Contact prediction as input. For proteins 1v32, 1uan, 1l3p, and 1j26 the (A) RMSD distributions with a bin size of 1 Å and (B) MAXN% percentage distributions with a bin size of 4% are provided as histograms. For each protein, distributions are reported for folding with no contact prediction input, with input from sequence-based contact prediction mode, and with input from structure-based contact prediction mode.
3. Results and Discussion
3.1. Sequence-based mode correctly predicts 42% of native contacts with a 7% false positive rate, while structure-based mode correctly predicts 45% of native contacts with a 2% false positive rate
The sequence-based mode was tested with 183 proteins excluded from the training sets (10%). ROC curves for the average outputs for each contact type–specific ANN and merged values (Fig. 1) are shown in Figure 2A along with the AUC values. The helix-helix ANN achieves an AUC of 0.796. Helix-strand and strand-helix ANNs have AUC values of 0.834 and 0.831, respectively. Sheet-sheet contacts (0.784) and strand-strand contacts (0.789) are hardest for our method to predict correctly, because, in contrast to all other classes of contacts, distinguishing these contact types is not possible by predicted secondary structure. The consensus prediction method has an AUC value of 0.835. The significant deviations from the random predictor (the diagonal) for all ANNs indicate that the sequence-based mode is able to identify a substantial fraction of the non-local contacts correctly. With merged predictions and a threshold of 0.4, the sequence-based mode was able to correctly predict 42% of native contacts while identifying falsely 7% of non-contacts as contacts (Table S2).
FIG. 2.

Receiver Operator Characteristics (ROC) curves for sequence-based and structure-based modes (A) The ROC curves for sequence-based mode using the independent data set of 184 proteins are plotted. Individual curves are presented for all 5 ANNs specialized for individual contact types, the merged predictions, and the random predictor (diagonal). The helix-strand and strand-helix ANNs are represented with only one curve since they are virtually identical. The inset provides a magnification for the high confidence prediction region. AUC (Area under curve) values for these curves are 0.796 (helix-helix), 0.834 (helix-strand), 0.831 (strand-helix), 0.789 (strand-strand), 0.784 (sheet-sheet), and 0.835 (merged). (B) Plot shows ROC curve (same as (A)) for the structure-based mode benchmark on 66 LIVEBENCH targets excluded from the training and monitoring data sets. In addition, curves for the sequence-based mode for the same 66 targets and the random predictor are provided. The insert provides a magnification for the high confidence prediction region. The AUC values for these curves are 0.860 (structure-based) and 0.795 (sequence-based).
The structure-based mode has been benchmarked with 66 LIVEBENCH (Rychlewski and Fischer, 2005) targets excluded from training. Figure 2B shows ROC curves displaying the average predictions for the independent dataset along with predictions from the sequence-based mode for the same dataset and corresponding AUC values. The structure-based mode (0.860) outperforms sequence-based mode (0.795) for these targets. The inset shows a clear differentiation between sequence and structure-based modes in the region corresponding to higher predictions.
When predictions above a threshold of 0.7 are identified as contacts, 45% of native contacts and 2% of non-contacts in the independent data set are predicted to be contacts (Table S2). Since only 5–8% of residue pairs in proteins are found to be in contact, absolute numbers for false and true positives are roughly equal at this cutoff.
In order to facilitate comparison of BCL::Contact with other methods, accuracy of highest L, L/2, and L/5 predictions were calculated for each protein in the independent data set where L is the length of the protein of interest (Table S3). The sequence-based mode achieved accuracies of 12.2%, 15.4%, and 20.9%, while the structure-based mode achieved accuracies of 67.4%, 72.7%, and 77.0% when highest L, L/2, and L/5 predictions are considered.
3.2. Structure-based mode has been ranked as one of the three best methods in CASP6
The structure-based mode has participated in CASP6. The analysis done by Graña et al. (2005) placed the method as one the top three groups (out of 16 groups) in terms of accuracy and coverage. In analysis of 11 new fold targets (sequences with no structural homologues), the method achieved a mean accuracy of 16% and a mean coverage of 8%. BCL::Contact was also one of the few methods that predicted the non-local sheet topology for target 273 (PDB code 1WDJ) correctly (Fig. 1f in Graña et al., 2005). Figure 3B shows the tertiary structure and the contact map with the predictions from the structure-based mode for protein 1V5P. The contact map indicates a significant overlay of native and predicted contacts in particular for within β-sheet topology, while the non-local contacts within the β-sheet are correctly identified.
FIG. 3.

BCL::Contact predictions mapped on tertiary structures and shown as contact maps. The contact maps are colored according to the scale shown from blue (contact probability of 0.0) to red (contact probability of 1.0) (A) Tertiary structure for CASP7 target T0345_1 (pdb code 2HE3) with residues corresponding to L/5 highest confidence BCL::Contact predictions in sequence-based mode highlighted in dark blue and the contact map corresponding to the predictions submitted in CASP7 for the same protein. The highlighted residues in the structure correspond to strand pairings between LEU61-PRO67 to ALA30-GLU34 and ALA30-GLUE34 to LYS156-ILE159. The magnified insets on the contact map correspond to these strand pairings. (B) The tertiary structure and the contact map with the predictions from the structure-based mode for target with LIVEBENCH id of 25864 (PDB CODE 1V5P). The high confidence predictions (red color) overlay with most of the native contacts (black boxes). The predictions lead to a true positive rate of 87% and false positive rate of 6%. The highlighted residues in the structure correspond to the strand pairing between PHE20-GLU24 and ARG97-ALA102. The magnified inset on the contact map corresponds to this strand pairing and indicates a nearly perfect identification of these crucial non-local contacts.
3.3. Sequence-based mode predicted long-distance contacts in CASP7 with up to 40% accuracy
The sequence-based mode of BCL::Contact has participated in CASP7 in the contact prediction category along with 16 other methods. The results were analyzed in detail by Izarzaguza et al. (2007) based on predictions for 19 selected targets composed of 15 free modeling targets and four template-based modeling targets. Predictions were submitted for BCL::Contact for 18 targets out of these 19. In 14 of them, our predictions met the criteria of having at least L/5 (length of given sequence divided by 5) number of predictions for long-distance contacts (>24 residue sequence separation). Due to the lack of a large subset of common targets for which most groups have submitted predictions, no clear ranking of all groups was obtained (Izarzugaza et al., 2007).
The sequence-based method achieved an average accuracy of 4.6% and an average coverage of 2.4% for long-distance contacts over 14 targets included in this analysis. However, in 50% of these targets none of the L/5 long-range contact predictions were correct. Our method achieved its best ranking (4th out of 10) for target T0356_3 (PDB code 2IDB chain C) out of this set of targets, with an accuracy of 20.8% and coverage of 14.8%. When L/10 instead of L/5 highest confidence predictions are considered for this target, the accuracy reaches 34%. When all targets (including the template-based modeling targets) are considered, our method predicted most accurately for target T0345_1 (PDB code 2HE3), which is not a free modeling target. For this target, our method achieved 40.5% accuracy and 5.5% coverage for L/5 highest predictions, while these values rise up to 61.1% and 26.7%, respectively, when L/10 highest predictions are considered. Figure 3A illustrates structure of 2HE3 with residues corresponding to L/5 highest predictions highlighted in purple and the contact map that shows predictions submitted for this target. The accurate prediction of non-local contacts within the β-sheet is remarkable.
3.4. BCL::Contact induces up to 5 Å shift in average RMSD distributions and up to 26% shift in average MAXN% distributions when guiding de novo folding
For both modes, sequence-based and structure-based, shifts in RMSD and MAXN% are reported in Tables 1 and 2, and Figure 4. In RMSD plots (Fig. 4A), any improvement on the accuracy of models generated would be signified as a decrease in the RMSD values of models. These shifts are observed clearly for all four targets when using the structure-based contact predictions. The sequence-based mode also leads to a decrease in the RMSD values for 1v32, 1uan, and 1j26, although not as pronounced as in the structure-based mode.
In MAXN% plots (Fig. 4B), in contrast to RMSD plots, improvement would be represented by a shift to the right when inputs from BCL::Contact are supplied to Rosetta. Similar to RMSD plots, usage of the structure-based contact predictions results in distinct shifts, whereas the sequence-based mode improves Rosetta only slightly for targets 1uan and 1j26.
The sequence-based mode slightly improves the RMSD for the best model for 10 proteins, 10th percentile for 13 proteins and average for 15 proteins, while structure-based mode improves the RMSD for the best model for 15 proteins, 10th percentile and average for 16 proteins. A similar improvement of MAXN% values is observed for a similar number of proteins.
The structure-based mode provides an improvement of 1.3 Å in average RMSD values of all models produced, while also providing a 5.8% increase in the MAXN% distributions of the models generated. The sequence-based mode does not lead to any significant shift in the averages of both distributions. The structure-based mode performs exceptionally well for target 1l3p, where it improves the RMSD of models on average by 5.1 Å (from 12.7 to 7.6) while improving the MAXN% of models by 26.4% (from 38.5% to 64.9%). With predictions from the structure-based mode, Rosetta is able to produce the best model with RMSD of 3.6 Å to the native structure and MAXN% value of 100%.
In order to visualize the improvements provided by contact predictions in tertiary structure prediction, the best models by RMSD for 1l3p and 1oh1 are presented in Figure 5. For 1l3p, contacts from both sequence-based mode and structure-based mode result in a more compact packing for the helices, indicated also by the improvements in RMSD from 4.3 Å to 3.8 Å and 3.6 Å, respectively. In particular contacts predicted between amino acids ALA168-PHE184, ALA168-PHE188, and ALA171-PHE184, as well as ILE158-SER244 and ILE161-ILE240 help bring helices closer. In the case of protein 1oh1, sequence-based contact prediction does not result in an improvement of model accuracy. However, structure-based contact prediction results in an RMSD improvement of 2.3 Å. The resultant model is the only model that has a well-defined sheet formation triggered by predicted contacts. The three highest predictions for the whole sheet region (residues 61–92) correspond to native contacts between amino acid pairs LEU67-ILE77, GLU78-LEU89, and ILE78-LEU89.
FIG. 5.

Comparison of best Rosetta models by RMSD in folding runs. (A) The lowest RMSD Rosetta model for protein 1l3p (rainbow coloring scheme) is shown superimposed with the native structure (gray). Panels (B) and (C) display the best models by RMSD when contacts predict by sequence-based and structure-based modes are used as score. The RMSDs of the models are 4.3Å, 3.8Å, and 3.6Å, respectively. The black arrows in panels (B) and (C) indicate strongly predicted contacts between amino acids ALA168-PHE184, ALA168-PHE188, ALA171-PHE184 as well as ILE158-SER244, ILE161-ILE240 facilitate improved helix packing. Panels (D–F) show lowest RMSD models for folding protein 1oh1. The RMSDs for the models are 7.9 Å, 7.9 Å and 5.6 Å, respectively. The black arrows in panel (F) indicate strongly predicted contacts between amino acids LEU67-ILE77, GLU78-LEU89 and ILE78-LEU89 that are responsible for improved in sheet formation. The highest 50 predictions for the same region also correspond to native contacts.
In terms of improving de novo protein structure prediction, structure-based contact predictions are highly valuable as seen by the higher accuracy of models produced when provided as an additional input to folding algorithm. In contrast, the sequence-based mode was able to improve only some of the test cases slightly and was clearly outperformed by the structure-based mode. This result is consistent with the fact that fold recognition results improve tertiary structure prediction, even if no single topology can be unambiguously identified by these methods.
3.5. BCL::Contact enriches for native-like models by factors of up to five
Another possible use of contact prediction is the discrimination of native-like models from the pool of thousands of models produced in de novo protein structure prediction runs. The discriminative power of contact predictions can be measured by enrichment values (Table S4). The sequence-based mode performs poorly for targets 1l3p and 1nek, while providing slight enrichments for the rest of the cases with an average enrichment of 1.3. The structure-based mode achieves an average enrichment of 2.5, performing well for all targets except 1lxj. For example, the enrichment of 5.5 for target 1l3p maintains 548 of the best 1,000 models by RMSD when selecting the top 10% of 10,000 models by contact score, where a random scoring scheming would yield only 100 of the best 1,000 models by RMSD.
3.6. Structure-based contact prediction outperforms sequence-based contact prediction even for hard fold-recognition targets
In all comparisons, the structure-based mode outperforms the sequence-based mode, which is expected since it utilizes tertiary structure prediction results. This holds even for hard fold recognition targets and new folds, demonstrating that even though no template can be confidently identified, some structures found by fold recognition servers have at least partial similarity with the target structure. However, usage of the structure-based mode requires the submission of the sequence to tertiary structure prediction servers. This leads to a long processing time, whereas the sequence-based mode provides contact predictions instantly. The accuracy of the sequence-based mode is currently limited by the lack of descriptors for evolutionary correlated mutations, which has been demonstrated to be one of the most successful approaches in contact prediction methods (Cheng and Baldi, 2007; Fariselli et al., 2001a; Gobel et al., 1994; Halperin et al., 2006; Hamilton et al., 2004; Kundrotas and Alexov, 2006; Olmea and Valencia, 1997; Shindyalov et al., 1994; Valencia and Pazos, 2002). Further, it generates many long-range predictions for residue pairs that reside in different registers of interfaces in a pair of secondary structure elements. A fraction of these false positives could be eliminated by a subsequent filter that limits the number of high probability predictions for each pair of secondary structure elements.
4. Conclusion
In this article, we have presented BCL::Contact, a novel contact prediction method based on ANNs. BCL::Contact competed in both CASP6 and CASP7 experiments. The structure-based mode was ranked as one of the top three groups in CASP6. The sequence-based mode was able to identify crucial long-range contacts in CASP7 for some of the new fold targets. While achieving up to ∼40% accuracy for such contacts, performance was not evaluated for several other targets due to the selection criteria applied prior to evaluation.
In addition to CASP experiments, both modes have been benchmarked for independent data sets. The sequence-based mode, when used with a threshold value of 0.4, was able to predict 42% of contacts correctly while identifying 7% of non-contacts falsely as contacts. The structure-based mode, when used with a threshold value of 0.7, achieved 45% accuracy in predicting contacts while falsely predicting 2% of non-contacts as contacts.
When used in protein folding simulations, the sequence-based mode provided only slight improvements in RMSD distributions of models, while the structure-based mode resulted in a significant reduction of RMSD values observed. It is expected that, with the inclusion of additional descriptors, such as correlated mutations, the sequence-based mode will also be able to provide clear improvements for tertiary structure prediction. Both methods are capable of enriching for native-like folds in a set of protein models created with the Rosetta de novo folding protocol, although the structure-based achieves approximately twice as high enrichment factors.
Despite the improvements in experimental protein structure elucidation field, many proteins of interest with little or no structural information still exist. Contact prediction methods that rely only on sequence information can be beneficial for structure prediction in such cases. Alternatively, with the emergence of new and better de novo tertiary structure predictions, contact prediction methods can increase their accuracy significantly by integration of models produced by such methods. BCL::Contact with both sequence-based and structure-base modes can be utilized in both of these situations. BCL::Contact is available to the scientific community at www.meilerlab.org.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (grant R01-GM080403) and by the National Science Foundation (grant 0742762).
Disclosure Statement
No competing financial interest exists.
References
- Altschuh D. Vernet T. Berti P., et al. Coordinated amino acid changes in homologous protein families. Protein Eng. 1988;2:193–199. doi: 10.1093/protein/2.3.193. [DOI] [PubMed] [Google Scholar]
- Altschul S.F. Madden T.L. Schäffer A.A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszodi A. Gradwell M.J. Taylor W.R. Global fold determination from a small number of distance restraints. J. Mol. Biol. 1995;251:308–326. doi: 10.1006/jmbi.1995.0436. [DOI] [PubMed] [Google Scholar]
- Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- Bonneau R. Ruczinski I. Tsai J., et al. Contact order and ab initio protein structure prediction. Protein Sci. 2002a;11:1937–1944. doi: 10.1110/ps.3790102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R. Strauss C.E.M. Rohl C., et al. De novo prediction of three-dimensional structures for major protein families. J. Mol. Biol. 2002b;322:65–78. doi: 10.1016/s0022-2836(02)00698-8. [DOI] [PubMed] [Google Scholar]
- Bujnicki J.M. Elofsson A. Fischer D., et al. LiveBench-1: continuous benchmarking of protein structure prediction servers. Protein Sci. 2001;10:352–361. doi: 10.1110/ps.40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. Baldi P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics. 2006;22:1456–1463. doi: 10.1093/bioinformatics/btl102. [DOI] [PubMed] [Google Scholar]
- Cheng J. Baldi P. Improved residue contact prediction using support vector machines and a large feature set. BMC Bioinform. 2007;8:113. doi: 10.1186/1471-2105-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivian D. Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivian D. Kim D.E. Malmstrom L., et al. Prediction of CASP-6 structures using automated Robetta protocols. Proteins. 2005;61:157–166. doi: 10.1002/prot.20733. [DOI] [PubMed] [Google Scholar]
- Debe D.A. Danzer J.F. Goddard W.A., et al. STRUCTFAST: protein sequence remote homology detection and alignment using novel dynamic programming and profile-profile scoring. Proteins. 2006;64:960–967. doi: 10.1002/prot.21049. [DOI] [PubMed] [Google Scholar]
- Fariselli P. Casadio R. A neural network based predictor of residue contacts in proteins. Protein Eng. 1999;12:15–21. doi: 10.1093/protein/12.1.15. [DOI] [PubMed] [Google Scholar]
- Fariselli P. Olmea O. Valencia A., et al. Prediction of contact maps with neural networks and correlated mutations. Protein Eng. 2001a;14:835–843. doi: 10.1093/protein/14.11.835. [DOI] [PubMed] [Google Scholar]
- Fariselli P. Olmea O. Valencia A., et al. Progress in predicting inter-residue contacts of proteins with neural networks and correlated mutations. Proteins. 2001b;(Suppl 5):157–162. doi: 10.1002/prot.1173. [DOI] [PubMed] [Google Scholar]
- Fischer D. 3DS3 and 3DS5 3D-SHOTGUN meta-predictors in CAFASP3. Proteins. 2003;53:517–523. doi: 10.1002/prot.10537. [DOI] [PubMed] [Google Scholar]
- Fischer D. Rychlewski L. Dunbrack R.L., Jr., et al. CAFASP3: the third critical assessment of fully automated structure prediction methods. Proteins. 2003;53:503–516. doi: 10.1002/prot.10538. [DOI] [PubMed] [Google Scholar]
- Ginalski K. Elofsson A. Fischer D., et al. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003a;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- Ginalski K. Pas J. Wyrwicz L.S., et al. ORFeus: Detection of distant homology using sequence profiles and predicted secondary structure. Nucleic Acids Res. 2003b;31:3804–3807. doi: 10.1093/nar/gkg504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginalski K. Rychlewski L. Detection of reliable and unexpected protein fold predictions using 3D-Jury. Nucleic Acids Res. 2003;31:3291–3292. doi: 10.1093/nar/gkg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel U. Sander C. Schneider R., et al. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- Grana O. Baker D. MacCallum R.M., et al. CASP6 assessment of contact prediction. Proteins. 2005;61:214–224. doi: 10.1002/prot.20739. [DOI] [PubMed] [Google Scholar]
- Halperin I. Wolfson H. Nussinov R. Correlated mutations: advances and limitations. A study on fusion proteins and on the Cohesin-Dockerin families. Proteins. 2006;63:832–845. doi: 10.1002/prot.20933. [DOI] [PubMed] [Google Scholar]
- Hamilton N. Burrage K. Ragan M.A., et al. Protein contact prediction using patterns of correlation. Proteins. 2004;56:679–684. doi: 10.1002/prot.20160. [DOI] [PubMed] [Google Scholar]
- Huang E.S. Samudrala R. Ponder J.W. Ab initio fold prediction of small helical proteins using distance geometry and knowledge-based scoring functions. J. Mol. Biol. 1999;290:267–281. doi: 10.1006/jmbi.1999.2861. [DOI] [PubMed] [Google Scholar]
- Izarzugaza J.M. Grana O. Tress M.L., et al. Assessment of intramolecular contact predictions for CASP7. Proteins. 2007;69:152–158. doi: 10.1002/prot.21637. [DOI] [PubMed] [Google Scholar]
- Jaroszewski L. Rychlewski L. Li Z., et al. FFAS03: a server for profile–profile sequence alignments. Nucleic Acids Res. 2005;33:W284–W288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Karplus K. Hu B. Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics. 2001;17:713–720. doi: 10.1093/bioinformatics/17.8.713. [DOI] [PubMed] [Google Scholar]
- Karplus K. Katzman S. Shackleford G., et al. SAM-T04: what is new in protein-structure prediction for CASP6. Proteins. 2005;61:135–142. doi: 10.1002/prot.20730. [DOI] [PubMed] [Google Scholar]
- Kundrotas P.J. Alexov E.G. Predicting residue contacts using pragmatic correlated mutations method: reducing the false positives. BMC Bioinform. 2006;7:503. doi: 10.1186/1471-2105-7-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.Y. Skolnick J. Benchmarking of TASSER_2.0: an improved protein structure prediction algorithm with more accurate predicted contact restraints. Biophys. J. 2008;95:1956–1964. doi: 10.1529/biophysj.108.129759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Zhang Y. Skolnick J. Application of sparse NMR restraints to large-scale protein structure prediction. Biophys. J. 2004;87:1241–1248. doi: 10.1529/biophysj.104.044750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund O. Frimand K. Gorodkin J., et al. Protein distance constraints predicted by neural networks and probability density functions. Protein Eng. 1997;10:1241–1248. doi: 10.1093/protein/10.11.1241. [DOI] [PubMed] [Google Scholar]
- Lundstroem J. Rychlewski L. Bujnicki J., et al. Pcons: a neural-network -based consensus predictor that improves fold recognition. Protein Sci. 2001;10:2354–2362. doi: 10.1110/ps.08501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J. Jones D.T. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- Meiler J. Baker D. Coupled prediction of protein secondary and tertiary structure. Proc. Natl. Acad. Sci. USA. 2003;100:12105–12110. doi: 10.1073/pnas.1831973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J. Müller M. Zeidler A., et al. Generation and evaluation of dimension reduced amino acid parameter representations by artificial neural networks. J. Mol. Model. 2001;7:360–369. [Google Scholar]
- Olmea O. Rost B. Valencia A. Effective use of sequence correlation and conservation in fold recognition. J. Mol. Biol. 1999;293:1221–1239. doi: 10.1006/jmbi.1999.3208. [DOI] [PubMed] [Google Scholar]
- Olmea O. Valencia A. Improving contact predictions by the combination of correlated mutations and other sources of sequence information. Fold Des. 1997;2:S25–S32. doi: 10.1016/s1359-0278(97)00060-6. [DOI] [PubMed] [Google Scholar]
- Ortiz A.R. Strauss C.E.M. Olmea O. MAMMOTH (Matching molecular models obtained from theory): an automated method for model comparison. Protein Sci. 2002;11:2606–2611. doi: 10.1110/ps.0215902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxco K.W. Larson S. Ruczinski I., et al. Evolutionary conservation in protein folding kinetics. J. Mol. Biol. 2000;298:303–312. doi: 10.1006/jmbi.1999.3663. [DOI] [PubMed] [Google Scholar]
- Pollastri G. Baldi P. Prediction of contact maps by GIOHMMs and recurrent neural networks using lateral propagation from all four cardinal corners. Bioinformatics. 2002;18:S62–S70. doi: 10.1093/bioinformatics/18.suppl_1.s62. [DOI] [PubMed] [Google Scholar]
- Pollastri G. Baldi P. Fariselli P., et al. Improved prediction of the number of residue contacts in proteins by recurrent neural networks. Bioinformatics. 2001;17:S234–S242. doi: 10.1093/bioinformatics/17.suppl_1.s234. [DOI] [PubMed] [Google Scholar]
- Pollock D.D. Taylor W.R. Goldman N. Coevolving protein residues: maximum likelihood identification and relationship to structure. J. Mol. Biol. 1999;287:187–198. doi: 10.1006/jmbi.1998.2601. [DOI] [PubMed] [Google Scholar]
- Punta M. Rost B. PROFcon: novel prediction of long-range contacts. Bioinformatics. 2005a;21:2960–2968. doi: 10.1093/bioinformatics/bti454. [DOI] [PubMed] [Google Scholar]
- Punta M. Rost B. Protein folding rates estimated from contact predictions. J. Mol. Biol. 2005b;348:507–512. doi: 10.1016/j.jmb.2005.02.068. [DOI] [PubMed] [Google Scholar]
- Russell R.B. Saqi M.A. Bates P.A., et al. Recognition of analogous and homologous protein folds—assessment of prediction success and associated alignment accuracy using empirical substitution matrices. Protein Eng. 1998;11:1–9. doi: 10.1093/protein/11.1.1. [DOI] [PubMed] [Google Scholar]
- Rychlewski L. Fischer D. LiveBench-8: the large-scale, continuous assessment of automated protein structure prediction. Protein Sci. 2005;14:240–245. doi: 10.1110/ps.04888805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A. Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Shackelford G. Karplus K. Contact prediction using mutual information and neural nets. Proteins. 2007;69:159–164. doi: 10.1002/prot.21791. [DOI] [PubMed] [Google Scholar]
- Shao Y. Bystroff C. Predicting interresidue contacts using templates and pathways. Proteins. 2003;53:497–502. doi: 10.1002/prot.10539. [DOI] [PubMed] [Google Scholar]
- Shi J. Blundell T.L. Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Shindyalov I.N. Kolchanov N.A. Sander C. Can three-dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Eng. 1994;7:349–358. doi: 10.1093/protein/7.3.349. [DOI] [PubMed] [Google Scholar]
- Simons K.T. Kooperberg C. Huang E., et al. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- Skolnick J. Kihara D. Defrosting the frozen approximation: PROSPECTOR—a new approach to threading. Proteins. 2001;42:319–331. [PubMed] [Google Scholar]
- Skolnick J. Kihara D. Zhang Y. Development and large scale benchmark testing of the PROSPECTOR_3 threading algorithm. Proteins. 2004;56:502–518. doi: 10.1002/prot.20106. [DOI] [PubMed] [Google Scholar]
- Tomii K. Hirokawa T. Motono C. Protein structure prediction using a variety of profile libraries and 3D verification. Proteins. 2005;61:114–121. doi: 10.1002/prot.20727. [DOI] [PubMed] [Google Scholar]
- Torda A.E. Procter J.B. Huber T. Wurst: a protein threading server with a structural scoring function, sequence profiles and optimized substitution matrices. Nucleic Acids Res. 2004;32:W532–535. doi: 10.1093/nar/gkh357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A. Pazos F. Computational methods for the prediction of protein interactions. Curr. Opin. Struct. Biol. 2002;12:368–373. doi: 10.1016/s0959-440x(02)00333-0. [DOI] [PubMed] [Google Scholar]
- Wang G. Dunbrack R.L., Jr PISCES: a protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Wu S. Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 2008;24:924–931. doi: 10.1093/bioinformatics/btn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. Liu S. Zhou Y. SP5: improving protein fold recognition by using torsion angle profiles and profile-based gap penalty model. PLoS ONE. 2008;3:e2325. doi: 10.1371/journal.pone.0002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl. Acad. Sci. USA. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.