

Abstract
A highly efficient photocoupling agent, based on perfluorophenylazide (PFPA)-conjugated polyallylamine (PAAm), was developed for the efficient immobilization of polymers, nanoparticles, graphene, and small molecules. The conjugate, PAAm-PFPA, was synthesized, and the percent of the photoactive moiety, PFPA, can be controlled by the ratio of the two components in the synthesis. By treating epoxy-functionalized wafers with PAAm-PFPA, photoactive surfaces were generated. Compared with the PFPA surface, these polymer-based photocoupling matrix resulted in significantly enhanced immobilization efficiencies, especially for nanomaterials and small molecules. Thus polystyrene nanoparticles (PS NPs) and alkyl-functionalized silica nanoparticles (SNPs) were successfully immobilized on the PAAm-PFPA surface, resulting in high material density. Graphene flakes patterned on the PAAm-PFPA surface showed improved feature resolution in addition to a higher material density compared to those immobilized on the PFPA surface. Furthermore, 2-O-α-D-mannopyranosyl-D-mannopyranose (Man2) immobilized on the PAAm-PFPA surface exhibited significantly enhanced signals when treated with the lectin Concanavalin A (Con A).
Introduction
The ability to precisely control surface structure and properties is of paramount importance in applications ranging from sensor technologies1–5 and separations,6–8 to medical implants and electronic devices.9–11 Surface chemistry is often the bottleneck in the advancement of these fields. Requirements for surface modification and the coupling chemistry include the precise control of the structure, functionalities, lateral features, and topography. In applications involving molecular recognition, ligand density, orientation and spatial presentation are also important and the control of which can impact the outcome of the analyses.12
Nanomaterials such as nanoparticles and graphene have demonstrated increasing potentials as the materials of choice in nanoelectronics, nanobiotechnology and nanomedicine.13–18 As such, the ability to manipulate and control the order and packing density of nanomaterials becomes highly relevant. For nanoparticles, various methods have been developed to create closely-packed and ordered particle arrays by modulating the evaporation process.19–24 Covalent attachment of nanomaterials offers high stability, and is necessary in situations where fluidic conditions are involved. In this respect, however, challenges remain, and effective immobilization techniques are still needed.
We developed a general photocoupling chemistry based on functionalized PFPAs.25–27 Upon photochemical or thermal activation, PFPA is converted to a singlet perfluorophenyl nitrene, which subsequently forms a robust covalent linkage with neighboring molecules via a CH insertion or C=C cycloaddition reaction. We have demonstrated that the technique is highly general and versatile, enabling the immobilization of polymers,28–30 biomolecules,31–33 and carbon materials including graphene.34–36 The photocoupling chemistry can thus accommodate diverse molecular structures and offer additional benefit of simplicity as well as adaptivity to various substrate materials. However, the method is not without limitation. We found that it was difficult to immobilize nanoparticles on the PFPA-surface, and on flat surfaces, small molecules were attached with low conjugation efficiency.
In the present work, we aim at developing the second-generation photocoupling surface that is based on a polymer matrix. The polymer-PFPA surface has several advantages over the PFPA surface. Polymers carry a large number of functional groups, which can be used to attach multiple PFPA moieties. For small molecules, high surface concentration of PFPA is necessary since the photocoupling reaction is dependent on the size of the molecule to be immobilized. Large molecules such as polymers can be efficiently immobilized even on low density PFPA surface,26 whereas for small molecules, the coupling yield drastically decreases with the surface PFPA concentration.37 Polymers, being a soft material, allow closer and conformal contact of the materials with the surface. This is essential in the photocoupling reaction involving solid materials that requires the CH bonds to be in close proximity with the surface PFPA groups.38 The soft polymer matrix could also reduce steric hindrance, and when a biological ligand is involved, allow proteins to better retain their native conformation when approaching the ligands on the surface.
In this article, a polymer-based PFPA matrix, PAAm-PFPA, will be synthesized. PAAm-PFPA-functionalized surfaces will be generated and used to immobilize polymers, nanoparticles, graphene, and small molecules.
Experimental Section
Materials
All solvents, N-hydroxysuccinimide (NHS), polyallylamine hydrochloride (MW 15,000 and 70,000), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) hydrochloride, 3-aminopropyltrimethoxysilane (APTMS), D-(+)-mannose (Man) were purchased from TCI America. Methyl pentafluorobenzoate, sodium azide, styrene, dodecyl trimethoxysilane (DTMS), poly(2-ethyl-2-oxazoline) (PEOX, MW 500,000), poly(vinyl pyrrolidone) (PVP, MW 1,300,000), sodium dodecyl sulfate (SDS), potassium persulfate, FITC (fluorescein isothiocyanate isomer I, 90%), FITC conjugated Con A (lectin from Canavalia ensiformis (Jack bean), Type IV) (FITC-Con A), bovine serum albumin (BSA), tetraethylorthosilane (TEOS), dodecyltrimethoxysilane, and 3-glycidyloxypropyltrimethoxysilane (GOTMS) were purchased from Sigma-Aldrich. Concentrated H2SO4, H2O2 (35%), ammonia (30%), toluene, 1,2-dichlorobenzene (DCB), and N-methylpyrrolidone (NMP) were purchased from Fisher. Polystyrene standard (MW 979,800) was purchased from Scientific Polymer Products Inc. (Ontario, NY). 2-O-α-D-Mannopyranosyl-D-mannopyranose (Man2) was obtained from V-Labs Inc. (Covington, Louisiana). The phosphate buffer solution was prepared by dissolving a phosphate buffered saline tablet (Sigma) in Milli-Q water (200 mL) to yield pH 7.4 phosphate buffer (0.01 M) containing KCl (0.0027 M) and NaCl (0.137 M). Dialysis tubes (G-Biosciences Tube-O-dialyzer, 15K, medium) were purchased from VWR International.
Silicon wafers with a 35 Å native or a 2700 Å thermally-grown oxide layer were purchased from WaferNet, Inc. (San Jose, CA). The later was utilized for the graphene immobilizaiton. The long-pass optical filter (280-nm) was purchased from Schott Glass Technologies, Inc. (Fullerton, CA).
Instrumentation
Surface images were obtained on an atomic force microscope (Nanoscope III, Veeco), optical microscope (Olympus BHM), or fluorescent microscope. TEM images were obtained on a JEOL 100CX transmission electron microscope (TEM) operating at an accelerating bias voltage of 100 kV. The specimens were prepared by dropping nanoparticle suspensions (10 μL) onto a 200-mesh copper grid (coated with carbon supporting film, Electron Microscopy Sciences). Dynamic light scattering (DLS) experiments were carried out on a Horiba LB-550 Dynamic Light Scattering Nano-Analyzer.
Film thickness were measured on a Gaertner Model L116A ellipsometer (Gaertner Scientific Co.) with He/Ne laser (632.8 nm, 2 mW, Melles Griot) at an incident angle of 70° in the manual mode. The real and imaginary parts of the refractive index of the silicon wafer used in the experiments were 3.870 (Ns) and −0.018 (Ks), respectively. The following refractive indices (nf) were used to determine the thickness of various film layers: SiO2 1.465, PFPA-silane 1.503, PS 1.592, PEOX 1.520, and PVP 1.530. Static contact angles were measured on a contact angle goniometer (model 250, Ramé-Hart Instrument Co., Netcong, NJ).
Synthesis of PAAm-PFPA
PFPA-NHS (see Figure 1 for structure) was synthesized following the literature procedure.39 Briefly, methyl pentafluorobenzoate was treated with NaN3 to yield methyl 4-azido-2,3,5,6-tetrafluorobenzoate, which was then hydrolyzed to give 4-azido-2,3,5,6-tetrafluorobenzoic acid. PFPA-NHS was subsequently synthesized by coupling the acid with NHS using EDAC followed by purification by recrystallization. PAAm-PFPA was prepared by mixing an aqueous solution of PAAm hydrochloride (10 mg) in D.I. water (2 mL) with a solution of PFPA-NHS (20 mg) in pyridine (2 mL), and stirring at room temperature for 24 hrs (Figure 1). The resulting product, PAAm-PFPA, was obtained as a clear solution.
Figure 1.

Synthesis of PAAm-PFPA, and immobilization on PAAm-PFPA surface.
Preparation of PAAm-PFPA surface
The silicon wafers were cleaned in the piranha solution (hydrogen peroxide/sulfuric acid =3/7) at 80–90 °C for 1 hour (Caution: the piranha solution reacts violently with organic solvents.), washed in boiling water three times for 60 min. each, and dried with nitrogen. The wafers were treated with a solution of GOTMS in toluene (12.6 mM) at room temperature for 5 hrs,40 washed with toluene several times and dried with nitrogen. The wafers were then immersed in the solution of PAAm-PFPA, and heated at 50 ºC for 5 hrs. The modified wafers were washed with 0.1 M HCl followed by sonication in pyridine for three times to remove the excess PAAm-PFPA. Finally, the wafers were dried with nitrogen gas.
For the comparison study, PFPA-functionalized silicon wafers were prepared following our previously reported procedure by treating piranha-cleaned wafers with PFPA-silane (see Figure 1 for structure). PFPA-silane was prepared by reacting PFPA-NHS with ATPMS followed by purification by column chromatography.25
Preparation of nanoparticle and graphene solutions
PS NPs were synthesized as follows.41,42 Sodium dodecyl sulfate (800 mg, 2.8 mmol) and potassium persulfate (150 mg, 0.55 mmol) were dissolved in D. I. water (100 mL). After the solution was heated at 80 ºC, a solution of distilled styrene (1 mL) in 1-butanol (100 μL) was dropped slowly into the aqueous solution while stirring using a mechanical stirrer at 200 rpm for 1 h. More styrene (4 mL) was then added and the reaction was continued for 2 hrs. After cooling, the solution was filtered to remove the aggregates and the product was purified with a dialysis membrane.
The FITC-doped silica nanoparticles (FSNPs) were synthesized following a previously described protocol.43,44 Briefly, FITC isothiocyanate was treated with APTMS in absolute ethanol to yield the FITC-silane precursor, which was then co-condensed with TEOS following the classic Stöber protocol to yield FSNPs as a bright yellow colloidal solution. By varying the concentrations of TEOS and NH4OH, different sizes of FSNPs were obtained. The dodecyl-protected FSNPs were prepared by treating FSNPs with a solution of DTMS in ethanol (10 mM) at 78 ºC overnight. After repeated washing and centrifugation in ethanol, dispersed solutions were collected.
Graphene flakes were prepared by sonicating graphite particles (50 mg) in DCB (20 mL) for 1 h using a sonication probe (SONICS, VCX130). The mixture was settled for 1 week, and the supernatant was centrifuged at 4,600 rpm for 30 min. The upper solution was collected and used for immobilization.
Immobilization on PAAm-PFPA and PFPA-silane surfaces
The immobilization of polymers, nanoparticles, graphene, and carbohydrate followed a general procedure shown in Figure 1.
Polymers
A solution of PS, PEOX, or PVP in chloroform (10 mg/mL) was spin-coated at 2000 rpm for 60 s on the PAAm-PFPA surface. The sample was covered with a 280-nm long-path optical filter, and was irradiated for 7 min with a 450-W medium pressure Hg lamp (Hanovia Ltd.). The 280-nm filter was used to remove the deep-UV light that can generate free radicals in the polymers.45 The wafers were then washed 3 times in chloroform by sonication.
Nanoparticles
An aqueous solution of PS NPs or FSNPs (1.0 μL) was dropped with a pipette tip onto the PAAm-PFPA or PFPA-silane surface. After drying the samples in vacuum, the wafers were irradiated for 10 min with the 450-W medium pressure Hg lamp in the presence of a 280-nm optical filter. Finally, the wafers were sonicated in water and ethanol for 3 min each, and dried with nitrogen.
Graphene
The graphene solution in DCB was dropped on the PAAm-PFPA or PFPA-silane surface and dried in air. This drop-and-dry procedure was repeated several times until the surface was completely covered with graphene flakes. The wafers were then irradiated with the 450-W medium pressure Hg lamp in the presence of a photomask, which was tightly held to the sample with a scotch tape. The excess graphene was removed by sonication in NMP.
Carbohydrate
An aqueqous solution of Man2 (150 mM) was printed onto the PAAm-PFPA or PFPA-silane surface using a robotic printer (BioOdyssey Calligrapher miniarrayer; Bio-Rad Laboratories, Inc.). The wafers were then spin-coated with a solution of PEOX in chloroform at 2000 rpm for 1 min. After irradiating for 7 min with the 450-W medium pressure Hg lamp in the presence of the 280-nm optical filter, the wafers were washed with chloroform followed by water. Finally, the wafers were soaked in a solution of FITC-Con A in pH 7.4 PBS buffer (0.275 mg/mL) for 1 hr, rinsed with water and dried.
Results and Discussion
Synthesis of PAAm-PFPA and Preparation of PAAm-PFPA Surfaces
We attempted to synthesize PAAm-PFPA by treating an aqueous solution of PAAm•HCl (MW 15,000) containing potassium carbonate with a solution of PFPA-NHS in ethanol. The mole ratio of PFPA-NHS to allylamine (AAm) was 1:4 in this case. However, the reaction solution turned cloudy under this condition, most likely due to the poor solubility of PFPA-NHS in the solvent used. When the mole percent of PFPA-NHS was increased further or a higher molecular weight of PAAm•HCl (MW 70,000) was used, the product precipitated immediately from the solution. Additionally, when the cloudy solution was subsequently used to treat the epoxy surface (Figure 1), the generated surface was rough (Figure S1a, Supporting Information). In order to improve the solubility of the product and to avoid agglomeration, several solvents and different reaction conditions were tested. A mixed solvent system of water/pyridine gave the best results where pyridine served both as a good solvent for PFPA-NHS and as a base to neutralize HCl (Figure 1). Even at a mole ratio of 1:1 PFPA-NHS/AAm or when a high molecular weight PAAm•HCl 70,000 was used, clear solutions could be obtained. Furthermore, a smoother surface was observed after treating the epoxy surface with the resulting solution (Figure S1b, Supporting Information).
The synthesized PAAm-PFPA was characterized with FTIR. Figure 2 shows the spectrum of PAAm-PFPA synthesized from PAAm•HCl (MW 70,000) at 1:1 mole ratio of PFPA-NHS/AAm. The peak at 2130 cm−1, corresponding to the asymmetric stretching absorption of the azido group, appeared in the product (Figure 2). Also appeared was the amide carbonyl absorption at 1690 cm−1 (Figure 2), which shifted from the ester carbonyl absorption in PFPA-NHS (1730 cm−1, see Figure S2, Supporting Information). These results confirmed the successful synthesis of PAAm-PFPA. The grafting density of the PFPA can be controlled by changing the mole ratio of PFPA-NHS and PAAm•HCl (see Figure S3, Supporting Information). In the subsequent studies, PAAm-PFPA synthesized from 1:1 mole ratio of PFPA-NHS/AAm was used for the preparation of PAAm-PFPA surfaces.
Figure 2.

FTIR spectra of PAAm-PFPA (solid line) and PAAm (dotted line).
To prepare the PAAm-PFPA surface, piranha-cleaned silicon wafers were treated with a solution of GOTMS in toluene followed by curing at room temperature overnight to give an epoxy surface of 14.2±0.2 Å in layer thickness. The resulting epoxy-fuctionalized wafers were incubated in the PAAm-PFPA solution at 50 ºC for 5 h to covalently attach the polymer. The thickness of the PAAm-PFPA layer was measured to be 19.2±3.7 Å by ellipsometry. The static water contact angle of the epoxy surface was 59±1º, which agreed with the previously reported vaule.40 The contact angle decreased to 47±2.5º after the surface was treated with PAAm-PFPA (Figure 3). Compared with the contact angle of the PAAm alone (32±2º, Figure 3), the contact angle of the PAAm-PFPA surface was higher. This is likely due to the presence of PFPA in the PFPA-grafted PAAm polymer, which made the resulting PAAm-PFPA surface more hydrophobic.
Figure 3.

The static water contact angle values of various surfaces.
Immobilization of polymers
To test the effectiveness of PAAm-PFPA in immobilizing polymers, a solution of PS in toluene was spin-coated on the PAAm-PFPA surface and the sample was irradiated with UV. After removing the excess PS by sonicating the sample in toluene, a thin film of 131±5 Å in thickness remained on the surface. After immobilization of PS, the contact angle of the surface increased considerably to ~87º (Figure 3). This value is similar to the contact angle of PS films immobilized on wafers functionalized with PFPA-silane (Figure 3).25 To further confirm that PAAm-PFPA was responsible for the covalent immobilization of PS, control samples were prepared where PS was spin-coated on GOTMS- or PAAm-treated surfaces. After UV irradiation and solvent extraction, no PS films were obtained, and the contact angles of the resulting surfaces were the same as those of the original surfaces. These results suggested that PAAm-PFPA should indeed be responsible for the covalent attachment of PS. In addition to PS, other polymers were also efficiently immobilized on the PAAm-PFPA surface. There include PVP and PEOX, which yielded films with the same contact angles as those prepared on wafers treated with PFPA-silane (Figure 3).
Immobilization of nanoparticles
We attempted to immobilize alkyl-protected silica NPs (~120 nm in diameter) by depositing the particles on wafers functionalized with PFPA-silane followed by irradiating with UV light. Very few particles remained on the surface when examined by AFM (Figure 4a). In order for the CH insertion reaction to occur, the material needs to be within the bond formation distance to the surface azido group.38 The “hard” silica NPs may not form conformal contact with the equally “hard” PFPA surface. The spherical geometry of particles also reduces their contact area with the PFPA surface. The PAAm-PFPA matrix, however, is softer and could greatly enhance the contact between the PAAm-PFPA surface and the nanoparticles. Indeed, when the PAAm-PFPA surface was used, the same silica NPs were successfully immobilized with high particle density (Figure 4b). In addition to the 120-nm particles, silica NPs of 470 nm and 35 nm in diameter (Figures 4c and 4d) as well as PS NPs (Figure 4e) were also successfully immobilized on the PAAm-PFPA surface. In all cases, the sizes of the immobilized particles observed by AFM (Figure 4) were consistent with the diameters of the corresponding NPs measured by DSL (see Figure S4, S5, Supporting Information). It appears that the immobilized particle densities are higher for smaller particles than the larger ones (Figure 4). This can be attributed to an increase in the contact area with the surface as the particles become smaller and less spherical. It can be seen from the TEM images that the smaller silica NPs were irregular in shape whereas the larger ones were more spherical (Figure 5Sa, Supporting Information). While a perfectly spherical particle would have only one contact point with a flat surface, more contact points are possible as the particles become irregular. This would increase the probability of the particles to be attached and thus higher immobilization yield and particle density. In principle, only a monolayer of particles should result, however, aggregates were visible in all cases. This is likely due to particle agglomeration which often occurs in nanoparticle samples.
Figure 4.
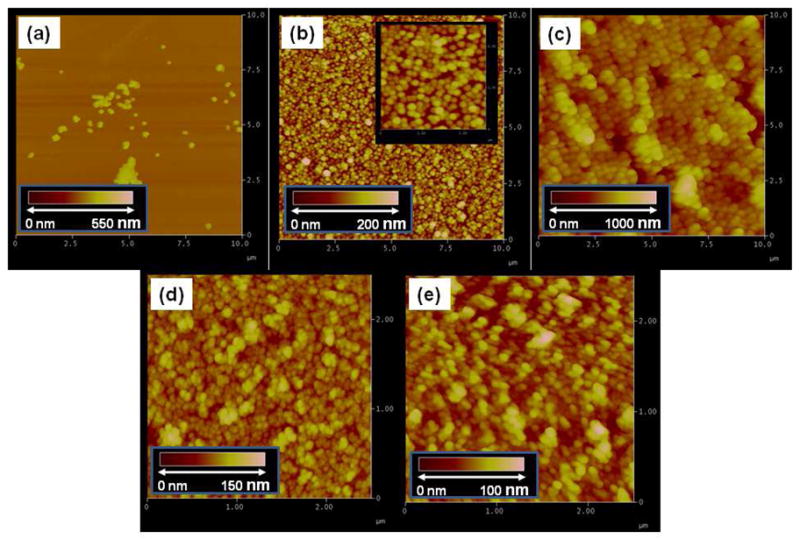
AFM images of immobilized particles: (a) silica NPs (120 nm), (b) silica NPs (120 nm), (c) silica NPs (470 nm), (d) silica NPs (35 nm), (e) PS NPs (28 nm). Samples were prepared on the PAAm-PFPA surface except for (a), which was prepared on PFPA-silane surface. The inserts in all graphs are topographic scale bars.
Control experiments were carried out where silica NPs (120 nm) were deposited on surfaces that were functionalized with GOTMS and PAAm, and the samples were treated under the same conditions as the PAAm-PFPA surface. Almost no particles remained on the surfaces after sonication in water and ethanol. Furthermore, the immobilized particles were highly stable, and could withstand extensive sonication in water, acids (HCl), and organic solvents. These results are strong evidences that the particles were covalently attached to the PAAm-PFPA surface.
While the PS NPs can be readily immobilized on the PAAm-PFPA surface, the alkyl group appears necessary for the successful immobilization of silica NPs. We found that when the same procedure was used to immobilize un-functionalized silica NPs or silica NPs functionalized with APTMS, few particles remained on the surface. These results were consistent with the photocoupling chemistry of PFPA which requires CH groups for the covalent bond formation. Therefore, organic particles or NPs rich in surface hydrocarbons are ideal for this immobilization method.
Immobilization of graphene
In a previous study, we were able to covalently immobilize graphene flakes on wafers functionalized with PFPA-silane.36 In this case, however, gaps and defects were observed between the graphene flakes. Similar to nanoparticles, graphene is a solid material that is difficult to form conformal contact with the hard wafer surface. In the case of HOPG (highly oriented pyrolytic graphite), pressure was applied to firmly press the HOPG disc to the PFPA surface, and fragmented graphene films were obtained under these conditions. To test whether the PAAm-PFPA surface might be more efficient than the PFPA surface in immobilizing graphene, solution-exfoliated graphene flakes were spin-coated on the two surfaces followed by UV irradiation in the presence of a photomask. The optical images of the resulting samples show the successful immobilization of graphene on both surfaces (Figure 5). Close examination of the images at a higher magnification revealed a higher density of graphene flakes on the PAAm-PFPA surface than that of the PFPA-silane surface on the entire sample surfaces. In addition, the graphene patterns on the PAAm-PFPA surface were well-resolved whereas on the PFPA-silane surface, the patterns were rough and the edges were ill-defined. This is consistent with the hypothesis that PAAm-PFPA improved the conformal contact with solid materials such as the graphene flakes.
Figure 5.

Optical microscope images of graphene flakes immobilized on (a) PAAm-PFPA, (b) PFPA-silane surfaces. The images on the right were taken at a higher magnification.
Immobilization of small molecules
A disaccharide, Man2, was used to test the effectiveness of the PAAm-PFPA surface for the immobilization of small molecules. In the experiment, an aqueous solution of Man2 was spotted on the PAAm-PFPA surface using a robotic printer followed by spin coating a solution of PEOX in chloroform. PEOX was introduced to reduce the non-specific adsorption of proteins in the subsequent binding assays.46 Because Man2 is insoluble in organic solvents, PEOX can be spin-coated from the chloroform solution without disturbing the spotted Man2 structures. The sample was then irradiated with UV light to covalently attach both Man2 and PEOX on the PAAm-PFPA surface. After sonicating in chloroform and water to remove unattached PEOX and Man2, the resulting sample was treated with FITC-Con A. Con A is a plant lectin that exhibits specific affinity to α-D-mannose structures; the solution dissociation constant (Kd) of Con A with Man2 was measured as 24 μM.47,48 After incubating with FITC-Con A, the Man2 spots on the surface were brightly fluorescent (Figure 6a). The same procedure was then repeated on surface that was treated with PFPA-silane (Figure 1). After binding with FITC-Con A, very weak signals were observed (Figure 6b). The markedly increased signals from the PAAm-PFPA surface were likely due to a combination of higher Man2 immobilization density and enhanced interactions with proteins when a polymer matrix is used.
Figure 6.

Fluorescence images of spotted Man2 after treating with FITC-Con A. Man2 was immobilized on (a) PAAm-PFPA, and (b) PFPA-silane surface, respectively.
Conclusion
We developed a polymer-based photocoupling surface, based on PAAm-PFPA, for the efficient immobilization of a variety of materials including polymers, nanoparticles, graphene, and small molecules. Higher immobilization efficiencies were observed on the PAAm-PFPA surface than on the surface functionalized with PFPA-silane. The polymer matrix offers multiple functional groups and thus a higher density of PFPA leading to enhanced immobilization efficiencies. Also, polymers are soft materials that allow a conformal contact with the materials to be immobilized. This facilitates the photocoupling reaction, especially the solid materials that are difficult to form conformal contact with the hard surface. As a result, nanoparticles of different sizes, which were difficult to immobilize on the PFPA-silane surface, can be covalently attached to the PAAm-PFPA surface in high density. Furthermore, higher immobilization density and improved feature resolution were achieved when graphene was photopatterned on the PAAm-PFPA surface. Finally, in situations where subsequent interactions with proteins are involved, the soft polymer matrix may additionally reduce steric hindrance allowing proteins to better retain their native conformation when approaching the ligands on the surface. This, combined with the increased ligand density, resulted in significantly enhanced interaction signals when an array of Man2 was treated with Con A. Taken together, the second-generation PAAm-PFPA photocoupling matrix and the method developed should greatly facilitate applications where robust attachment and fluidic analysis are required.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of General Medical Science (NIGMS) under NIH Award Numbers R01GM080295 and 2R15GM066279.
Footnotes
Supporting Information Available: AFM images of PAAm-PFPA surfaces, FTIR spectra of PAAm-PFPA, DLS histograms of PS NPs and silica NPs, and TEM images of silica NPs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ulman A. Chem Rev. 1996;96:1533–1554. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 2.Stewart MP, Buriak JM. Adv Mater. 2000;12:859–869. [Google Scholar]
- 3.Veiseh M, Zareie MH, Zhang M. Langmuir. 2002;18:6671–6678. [Google Scholar]
- 4.Balamurugan S, Obubuafo A, Soper S, Spivak D. Anal Bioanal Chem. 2008;390:1009–1021. doi: 10.1007/s00216-007-1587-2. [DOI] [PubMed] [Google Scholar]
- 5.Massad-Ivanir N, Shtenberg G, Zeidman T, Segal E. Adv Funct Mater. 2010;20:2269–2277. [Google Scholar]
- 6.Karim A, Douglas JF, Lee BP, Glotzer SC, Rogers JA, Jackman RJ, Amis EJ, Whitesides GM. Phys Rev E. 1998;57:R6273. [Google Scholar]
- 7.Kalkan AK, Henry MR, Li HD, Cuiffi JD, Hayes DJ, Palmer C, Fonash SJ. Nanotechnology. 2005;16:1383–1391. [Google Scholar]
- 8.Striemer CC, Gaborski TR, McGrath JL, Fauchet PM. Nature. 2007;445:749–753. doi: 10.1038/nature05532. [DOI] [PubMed] [Google Scholar]
- 9.Craighead HG. Science. 2000;290:1532–1535. doi: 10.1126/science.290.5496.1532. [DOI] [PubMed] [Google Scholar]
- 10.Eda G, Fanchini G, Chhowalla M. Nat Nanotechnol. 2008;3:270–274. doi: 10.1038/nnano.2008.83. [DOI] [PubMed] [Google Scholar]
- 11.Park S, Ruoff RS. Nat Nanotechnol. 2009;4:217–224. doi: 10.1038/nnano.2009.58. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Xu B, Tong H, Wang L. Macromolecules. 2010;43:8917–8923. [Google Scholar]
- 13.Roduner E. Chem Soc Rev. 2006;35:583–592. doi: 10.1039/b502142c. [DOI] [PubMed] [Google Scholar]
- 14.Gao J, Gu H, Xu B. Acc Chem Res. 2009;42:1097–1107. doi: 10.1021/ar9000026. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Piao Y, Hyeon T. Chem Soc Rev. 2009;38:372–390. doi: 10.1039/b709883a. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Liu LH, Ramström O, Yan MD. Exp Biol Med. 2009;234:1128–1139. doi: 10.3181/0904-MR-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White RJ, Luque R, Budarin VL, Clark JH, Macquarrie DJ. Chem Soc Rev. 2009;38:481–494. doi: 10.1039/b802654h. [DOI] [PubMed] [Google Scholar]
- 18.Pumera M. Chem Soc Rev. 2010;39:4146–4157. doi: 10.1039/c002690p. [DOI] [PubMed] [Google Scholar]
- 19.Xia YN, Gates B, Yin YD, Lu Y. Adv Mater. 2000;12:693–713. [Google Scholar]
- 20.Li W, Yang B, Wang D. Langmuir. 2008;24:13772–13775. doi: 10.1021/la802062c. [DOI] [PubMed] [Google Scholar]
- 21.Govor LV. ACS Appl Mater Interfaces. 2009;1:488–493. doi: 10.1021/am800151e. [DOI] [PubMed] [Google Scholar]
- 22.Teh LK, Yan Q, Wong CC. ACS Appl Mater Interfaces. 2009;1:775–779. doi: 10.1021/am800188z. [DOI] [PubMed] [Google Scholar]
- 23.Tourillon G, Dreesen L, Volcke C, Sartenaer Y, Thiry P, Peremans A. J Mater Sci. 2009;44:6805–6810. [Google Scholar]
- 24.Jeong S, Hu L, Lee HR, Garnett E, Choi JW, Cui Y. Nano Lett. 2010;10:2989–2994. doi: 10.1021/nl101432r. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett MA, Yan M. Adv Mater. 2001;13:1449–1451. [Google Scholar]
- 26.Liu L, Engelhard MH, Yan M. J Am Chem Soc. 2006;128:14067–14072. doi: 10.1021/ja062802l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L-H, Yan M. Acc Chem Res. 2010;43:1434–1443. doi: 10.1021/ar100066t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan M, Bartlett MA. Nano Lett. 2002;2:275–278. [Google Scholar]
- 29.Liu L, Yan M. Angew Chem Int Ed. 2006;45:6207–6210. doi: 10.1002/anie.200602097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L-H, Dietsch H, Schurtenberger P, Yan M. Bioconjugate Chem. 2009;20:1349–1355. doi: 10.1021/bc900110x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al-Bataineh SA, Luginbuehl R, Textor M, Yan M. Langmuir. 2009;25:7432–7437. doi: 10.1021/la900334w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norberg O, Deng L, Yan M, Ramström O. Bioconjugate Chem. 2009;20:2364–2370. doi: 10.1021/bc9003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Ramström O, Yan M. J Mater Chem. 2009;19:8944–8949. doi: 10.1039/B917900C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L-H, Yan M. Nano Lett. 2009;9:3375–3378. doi: 10.1021/nl901669h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L-H, Lerner MM, Yan M. Nano Lett. 2010;10:3754–3756. doi: 10.1021/nl1024744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L-H, Zorn G, Castner DG, Solanki R, Lerner MM, Yan M. J Mater Chem. 2010;20:5041–5046. doi: 10.1039/C0JM00509F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Ramström O, Yan M. Anal Chem. 2010;82:9082–9089. doi: 10.1021/ac102114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graupner RK, Yan M. Langmuir. 2004;20:8675–8680. doi: 10.1021/la0494728. [DOI] [PubMed] [Google Scholar]
- 39.Keana JFW, Cai SX. J Org Chem. 1990;55:3640–3647. [Google Scholar]
- 40.Tsukruk VV, Luzinov I, Julthongpiput D. Langmuir. 1999;15:3029–3032. [Google Scholar]
- 41.Ming W, Zhao J, Lu X, Wang C, Fu S. Macromolecules. 1996;29:7678–7682. [Google Scholar]
- 42.Xu XJ, Chew CH, Siow KS, Wong MK, Gan LM. Langmuir. 1999;15:8067–8071. [Google Scholar]
- 43.Stöber W, Fink A. J Colloid Interface Sci. 1968;26:62–69. [Google Scholar]
- 44.Nyffenegger R, Quellet C, Ricka J. J Colloid Interface Sci. 1993;159:150–157. [Google Scholar]
- 45.Yan M, Harnish B. Adv Mater. 2003;15:244–248. [Google Scholar]
- 46.Wang H, Li L, Tong Q, Yan M. submitted. [Google Scholar]
- 47.Brewer CF, Bhattacharyya L. Glycoconjugate J. 1988;5:159–173. [Google Scholar]
- 48.Mandal DK, Kishore N, Brewer CF. Biochemistry. 1994;33:1149–1156. doi: 10.1021/bi00171a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.