

Abstract
The solution structure of the hypothetical phage-related protein NP_888769.1 from the Gram-negative bacterium Bordetella bronchoseptica contains a well-structured core comprising a five-stranded, antiparallel β-sheet packed on one side against two α-helices and a short β-hairpin with three flexibly disordered loops extending from the central β-sheet. A homology search with the software DALI identified two Protein Data Bank deposits with Z-scores > 8, where both of these proteins have less than 8% sequence identity relative to NP_888769.1, and one has been functionally annotated as a lambda phage tail terminator protein. A sequence-homology analysis then confirmed that NP_888769.1 represents the first three-dimensional structural representative of a new protein family that was previously predicted by the Joint Center for Structural Genomics, which includes so far about 20 prophage proteins encoded in bacterial genomes.
Keywords: protein structure, automated solution NMR, phage tail-terminator protein, new PFAM protein family, automated NOE assignment
Introduction
The NP_888769.1 protein sequence had been tentatively annotated as a phage-related sequence in the genome of the Gram-negative bacterium B. bronchiseptica,1 which causes infectious bronchitis in small mammals and is closely related with other health-threatening human pathogens, such as B. pertussis. NP_888769.1 was initially classified by the JCSG bioinformatics core as a member of a proposed new family BIG_19, based on some low-sequence correspondence with proteins in the PFAM database and was, thus, believed to belong to a large, structurally uncharacterized protein family. As attempts at structure determination by X-ray crystallography were not successful, we followed the JCSG target assignment strategy of trying to fill this gap with nuclear magnetic resonance (NMR) structure determination and NMR-based functional insights that might corroborate the bateriophage origin of NP_888769.1.
The present project draws additional interest from the fact that proteins with more than 30% sequence identity to NP_888769.1 in other bacterial species, such as P. aeruginosa, P. syringae, P. putida, D. acidovorans and Y. pestis, have also been related to phage genomes. Overall, the presence of phage-related sequences is a recurrent feature in bacterial gene annotation, with a significant portion of the published genomes containing extended fragments of chromosome-integrated phage deoxyribonucleic acid (DNA), which can exceed 15% of the bacterial genome and include sequences from several different phages.2 The identification of such phage elements is important for distinguishing bacterial-encoded proteins but often tends to be quite involved. For example, use of gene recognition strategies may be limited due to ambiguity in defining phage-specific integrases, and sequence-based approaches may fail because of diversity of prophage forms, due to high frequency of deletions, insertions and rearrangements. In this context, the present structure-based functional annotation of NP_888769.1 provides an example for the use of tools developed by structural genomics initiatives to expand the pool of well-characterized, phage-related proteins, which then promises to facilitate future bacterial gene annotation.
Results and Discussion
The JCSG target NP_888769.1 was expressed as the uniformly 15N- and 13C,15N-labeled protein in BL21(DE3) cells that had been freshly transformed with the pSpeedET-NP_888769.1 vector. For the NMR structure determination, we used a 1.5 mM protein solution in NMR buffer (20 mM phosphate at pH 6.0, 50 mM sodium chloride, 4.5 mM NaN3, 5% 2H2O). These solution conditions were chosen based on a screen of variable pH-values and ionic strengths, using microcoil NMR equipment that required only microgram amounts of 15N-labeled NP_888769.1 for the acquisition of 1D 1H NMR and 2D [15N,1H]-HSQC spectra.3
Resonance assignment of the polypeptide backbone was based on automated projection spectroscopy (APSY)-NMR experiments,4 which provided the input for the automated assignment software UNIO-MATCH.5 UNIO-MATCH yielded 75% of the assignments, which were then interactively validated and extended with the use of a 15N-resolved [1H,1H]-NOESY spectrum. A chemical shift list for 15N, 1HN, 13Cα, 1Hα, and 13Cβ was, thus, obtained for the complete polypeptide chain, with the following exceptions: all resonances of G1 and M2, W139 13Cα and 1Hα, P119 13Cα and 1Hα, P140 13Cβ. Side chain resonance assignments were obtained with input from the same three 3D heteronuclear-resolved [1H,1H]-NOESY datasets that subsequently provided the NOE distance constraints for the structure calculation. The automated analysis of the NOE data with the software UNIO-ATNOS/ASCAN6,7 yielded 70% of the assignments, which were then interactively checked and extended to achieve assignments for 85% of the side-chain hydrogen atoms (for details see biological magnetic resonance data bank (BMRB) deposit 17124).
For the NMR structure determination, the automated seven-cycle ATNOS/CANDID/CYANA8 combined NOE assignment and structure calculation protocol6,9 was used as implemented in the UNIO software package (see Materials and Methods). This procedure yielded 1972 meaningful upper limit distance constraints to generate a bundle of 20 conformers representing the NMR structure of NP_888769.1. The statistics on the structure calculation, in particular the residual CYANA8 target function value of 1.95 ± 0.37 Å2 and the backbone global root-mean-square deviation (RMSD) value for the core residues of 0.71 ± 0.09 Å (Table I) are representative of a high-quality NMR structure determination, as is also apparent from the structure visualizations in Figure 1(A,C).
Table I.
Input for the Structure Calculation and Characterization of the Ensemble of 20 Energy-Minimized CYANA Conformers used to Represent the NMR structure of NP_888769.1
| Quantities | Valuea |
|---|---|
| NOE upper distance limits | 1972 |
| Intraresidual | 526 |
| Short-range | 605 |
| Medium-range | 275 |
| Long-range | 566 |
| Dihedral angle constraints | 559 |
| Residual target function value (Å2) | 1.95 ± 0.37 |
| Residual NOE violations | |
| Number ≥ 0.1 Å | 24 ± 5 |
| Maximum (Å) | 0.15 ± 0.04 |
| Residual dihedral angle violations | |
| Number ≥ 2.5 (°) | 1 ± 1 |
| Maximum (°) | 3.14 ± 1.35 |
| Amber energies (kcal/mol) | |
| Total | −5064.1 ± 97.2 |
| Van der Waals | −375.7 ± 17.9 |
| Electrostatic | −5836.4 ± 90.2 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.0077 ± 0.0002 |
| Bond angles (°) | 1.96 ± 0.04 |
| RMSD to the mean coordinates (Å)b | |
| bb (3–136) | 1.87 ± 0.36 |
| ha (3–136) | 2.25 ± 0.36 |
| bb (3–50, 66–136) | 0.71 ± 0.09 |
| ha (3–50, 66–136) | 1.18 ± 0.11 |
| Ramachandran plot statistics (%)c | |
| Most favored regions | 77.4 |
| Additional allowed regions | 19.8 |
| Generously allowed regions | 1.5 |
| Disallowed regions | 1.3 |
Except for the top six entries, which describe the input generated in the final cycle of the ATNOS/CANDID/CYANA calculation, the entries refer to the 20 best CYANA conformers after energy minimization with OPALp (see text). Where applicable, the average value for the bundle of 20 conformers and the standard deviation are given.
bb indicates the backbone atoms N, Cα, and C′; ha stands for all heavy atoms. Numbers in parentheses indicate the residues for which the RMSD was calculated.
As determined by PROCHECK.10
Figure 1.
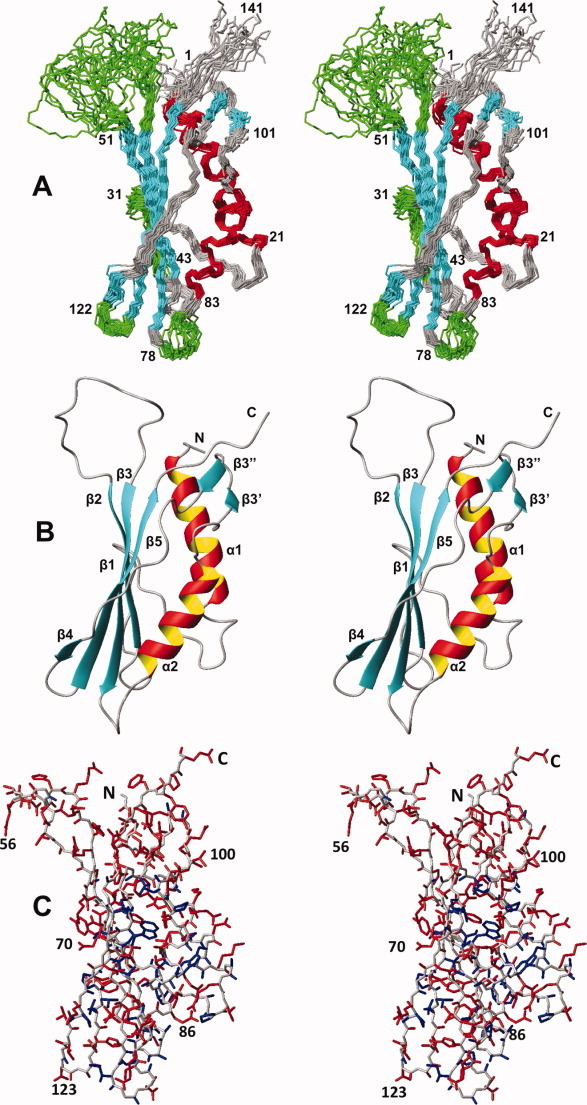
Stereo views of the NMR structure of the protein NP_0888769.1. (A) Polypeptide backbone of residues 1–141 represented as a bundle of 20 energy-minimized CYANA conformers superimposed for best fit of the backbone atoms of residues red, β-strands in cyan, and segments of nonregular structure in grey and green, where green coloring highlights four loops with per-residue global displacements, calculated for the atoms N, Cα, and C′, above the average value of 1.2 Å (see text). Reference sequence positions are indicated. (B) Ribbon representation of the conformer in (A), which is closest to the mean coordinates. The regular secondary structure elements and the N- and C- termini are indicated (the notation β3′ and β3″ for the strands of a short antiparallel β-sheet is used to aid in subsequent comparisons with homologous proteins). (C) All-heavy-atom presentation of the conformer in panel (B). The backbone is represented in grey, and side chains with per-residue global displacements calculated for all backbone and side-chain heavy atoms ≤1.0 Å are colored blue, and those with values >1.0 Å are in red.
NMR structure of NP_888769.1
The NMR structure of NP_888769.1 includes seven β-strands and two α-helices arranged in the sequential order α1–β1–β2–β3–α2–β3′–β3″–β4–β5. The core of the protein consists of the strands β1, β2, β3, β4, and β5, which form an antiparallel β-sheet, and the helices α1 and α2, which are both on the “inner” side of the twisted β-sheet [Fig. 1(A,B)]. β3′ and β3″ form a short hairpin that extends the major β-sheet by associating with the C-terminal tripeptide segment of β5 and adds contacts between the five-stranded β-sheet and helix α1 [Fig. 1(B); see also Fig. 3 below]. The protein has a small core of well-defined side chains, but consistent with the increased structural disorder of polypeptide backbone segments [Fig. 1(A)], the side chains at both ends of the core-β-sheet have high structural plasticity in solution [Fig. 1(C)]. Heteronuclear 15N{1H}-NOE experiments further showed that the less well-defined segments in the structure have increased subnanosecond time scale mobility when compared to the protein core (Fig. 2). This is clearly seen for the surface-exposed loops between β1 and β2, β2 and β3, β3 and α2, and β4 and β5. The rather outstandingly large proportion of structurally disordered polypeptide segments in NP_888769.1 [Fig. 1(A,C)] provides a likely rationale for lack of success with crystal structure determination.
Figure 3.
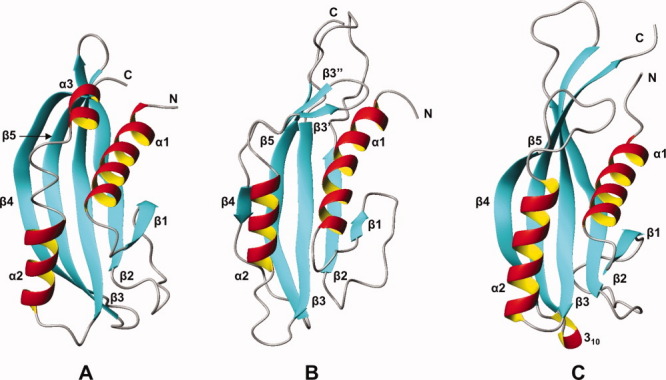
Comparison of the NMR structure of NP_888769.1 with two proteins with similar structures, as identified by DALI.10 Ribbon representations are shown with the same color code as in Figure 1(B). (A) Tail terminator protein gpU from bacteriophage lambda, Z = 9.2 (PDB id 3FZB, conformer A). (B) NP_888769.1 conformer of Figure 1(B) after counter-clockwise rotation by 90° about a vertical axis. (C) Protein of unknown function from S. typhimurium, Z = 9.6 (PDB id 2GJV, conformer A). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
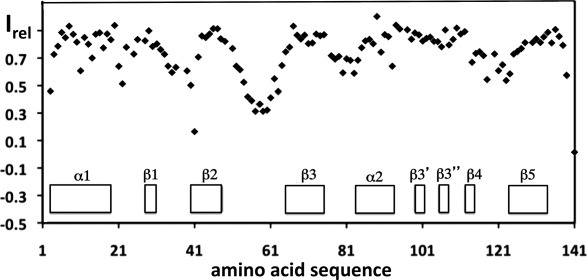
High-frequency mobility in the NMR structure of NP_0888769.1. Relative 15N{1H}-NOE intensities, Irel, are plotted versus the amino-acid sequence. Diamonds represent experimental measurements at a 1H frequency of 600 MHz and T = 298 K. The locations of the regular secondary structures are indicated at the bottom.
Three-dimensional homology search provides link to phage-related protein
A three-dimensional structure similarity search with the algorithm DALI11 yielded two similar structures with a Z score > 8.0 (Fig. 3), both of which share less than 8% sequence identity with NP_888769.1. The top hit, with a Z-score of 9.1 and a backbone RMSD of 3.7 Å, is encoded in the Salmonella typhimurium genome, and its hexameric crystal structure has been deposited as a protein of unknown function (PDB id 2GJV). Subsequently, two different putative functional annotations have been proposed for this sequence, that is, a cytoplasmic protein12 and a phage-related protein.13 The second hit, with a Z-score of 8.3 and a backbone RMSD of 3.2 Å, corresponds to the crystal structure of the phage lambda tail terminator protein (gpU, PDB id 3FZ2), for which a NMR structure is also available (PDB id 1Z1Z). gpU is essential for phage assembly, by capping the fast polymerizing phage tail once it reaches the appropriate length.14
Similar to NP_888769.1, gpU exhibits a high proportion of structurally poorly defined polypeptide segments, which is apparent from the superposition of a bundle of 20 NMR conformers in the presentation of the gpU solution structure,15 and can also be inferred from comparison of the 12 molecules in the crystal asymmetric unit.16,17 The disordered polypeptide segments in the NMR structure of gpU15 are involved in the oligomerization interfaces seen in crystal structure,16 where they form a series of hydrogen bonds with other subunits. In solution, formation of the hexamers is promoted by increasing concentrations of Mg2+, and was abolished by designed replacement of some negatively charged residues.15 Although none of the identified key acidic residues seem to be conserved in NP_888769.1, the overall structure similarity with gpU and the presence of several solvent-accessible glutamates and aspartates in the helices α1 and α2 of NP_888769.1 prompted us to study the effect of addition of variable concentrations of Mg2+ on the [15N,1H]-HSQC spectra of NP_888769.1 (Fig. 4). In apparent contrast to the observations with gpU,16 our results do not show either decay of [15N,1H]-HSQC peak intensities or line broadening at high Mg2+ concentrations. Although there is, thus, no indication for oligomerization of NP_888769.1 at high concentrations of Mg2+, significant Mg2+-induced shifts are seen for a discrete set of residues [Fig. 4(A)]. The largest shifts are observed for residues in helix α2 and strands β4 and β5, which are in close spatial proximity to each other [Figs. 1(B) and 4(B)], and there are indications that Mg2+ induces structural rearrangements in the two structurally disordered surface-exposed loops linking β3 with α2 and β4 with β5 [Figs. 1(A) and 4(B)].
Figure 4.
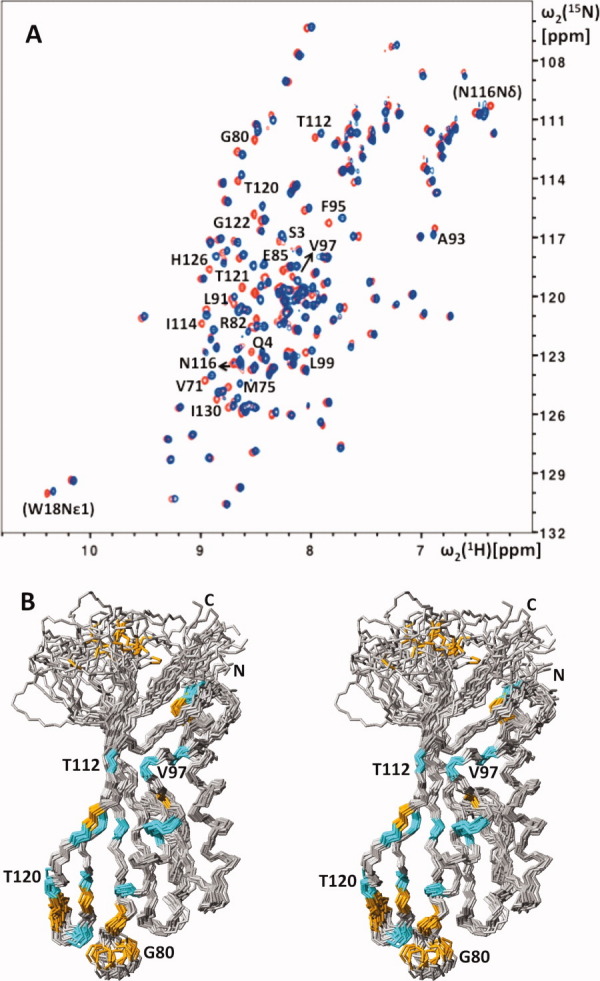
Effect of Mg2+ on the solution structure of NP_888769.1 and aggregation state of the protein. (A) 2D [15N,1H]-HSQC spectra of 1 mM NP_888769.1 in the absence (red) and presence (blue) of 200 mM MgCl2. The cross peaks of residues with Δδav = {0.5[Δδ(1HN)2 + (0.2Δδ(15N))2]}1/2 > 0.06 ppm for the backbone amide groups are identified with the one-letter amino acid symbol and the sequence number, and the same information is given in parentheses for residues with Δδav > 0.06 ppm for side-chain resonances. (B) Same stereo view of NP_888769.1 as in Figure 1(A), but now color-coded to indicate residues with large Mg2+-induced chemical shift changes of the backbone amide groups: orange, Δδav > 0.1 ppm; cyan, 0.1 ppm ≥ Δδav > 0.06 ppm.
Sequence homology search with NP_888769.1 leads to creation of a new PFAM family of phage-related proteins
To explore the use of the aforementioned putative three-dimensional structure-based functional annotation of NP_888769.1 for classifying other polypeptide sequences into protein families, we performed a homology search against the PFAM-24.0 database,18 which revealed that NP_888769.1 was not yet a member of any PFAM family. A second search for remote sequence homologues with the HHpred software19 suggested that NP_888769.1 is related to the phage minor tail protein U family (PF06141), which includes gpU. However, the sequence similarity with other PF06141 members is below the threshold of 10% that would enable it to include NP_888769.1 in this family. Therefore, we evaluated NP_888769.1 as a potential founder of a new PFAM family, which might include sequences that were previously classified in the hypothetical family BIG-19. Closely related proteins were then identified using BLAST, which yielded 22 hypothetical proteins with sequence identity above 30% (representative sequences are shown in Fig. 5). None of these hits have so far been classified as members of a PFAM family, and they are bacterial proteins of unknown function, phage-related proteins, or proteins encoded in bacteriophage genomes. Overall, a high degree of conservation is observed for polypeptide segments corresponding to regular secondary structures in the protein core of NP_888769.1 and, in particular, strands β2, β3, and β5, and some positions in helices α1 and α2, while structurally less well-defined regions show increased sequence diversity (Fig. 5). The overall high-sequence identity among the 20 bacterial proteins, and specifically the extensive sequence conservation in polypeptide segments forming regular secondary elements in the core of NP_888769.1, suggest that all these proteins exhibit a similar fold and that they all might share a bacteriophage origin.
Figure 5.

Sequence alignment of NP_888769.1 (B. bronchiseptica, accession number Q7TTM0_BORBR) with sequence homologues identified by BLAST. Protein multiple-sequence alignment was performed using ClustalW2 and covered sequences from B. pertussis (Q7VTX6_BORPE), P. fluorescens (Q4KA76_PSEF5), P. putida (B0kJS3_PSEPG), P. mirabilis (B4EV39_PROMH), Y. pestis (Q7CIE9_YERPE), D. acidovorans (A9C0F3_DELAS), Methylotenera sp. (accession number B8IIV8_METNO), R. loti (Q984G5_RHILO), and Bacteriophage T1 (Q6XQC8_BPT1). At the top, the sequence numbering for NP_888769.1 and the locations of the regular secondary structures in the NMR structure are indicated. At the bottom is a histogram-type presentation of the extent of conservation along the sequence, with the highest bar at position 70 representing 100% conservation among the 11 species. Amino acids are colored according to conservation and biochemical properties, following ClustalW conventions.
Based on the results presented in this article, the JCSG Bioinformatics Core in collaboration with the PFAM database has created a new PFAM family, PF13554. It should be emphasized that without the availability of the presently determined three-dimensional structure of NP_888769.1, its bacteriophage origin could only have been suggested as a rather far-fetched hypothesis from the remote sequence similarity with other phage-related proteins. In contrast, the structure-based functional annotation of NP_888769.1 enabled identification and characterization of an entire group of prophage sequences encoded in different bacterial genomes that now represent the new protein family PF13554, with NP_888769.1 as its first three-dimensional structure representative.
Materials and Methods
Protein purification and NMR sample preparation
Using the pSpeedET-NP_888769.1 vector obtained from the JCSG Crystallomics Core, expression of the uniformly 13C,15N-labeled protein was carried out by growing freshly transformed BL21(DE3) cells in M9 minimal medium containing 1g/L 15NH4Cl and 4 g/L [13C6]-D-glucose as the sole nitrogen and carbon sources, respectively. Cell cultures were grown at 37°C with vigorous shaking to 0.6 optical density at 600 nm. The temperature was then lowered to 18°C and, after induction with 1 mM isopropyl-β-D-1- thiogalactopyranoside (IPTG), the cells were grown for 20 h, harvested by centrifugation, resuspended in extraction buffer [pH 7.5, 20 mM sodium phosphate, 200 mM sodium chloride, 10 mM imidazole, complete ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail tablets (Roche)] and lysed by sonication. The cell debris was removed by centrifugation (20,000g for 30 min) and the supernatant loaded onto a Ni+2-affinity column (HisTrap HP column; Amersham) equilibrated with buffer A (20 mM phosphate at pH 7.5, 200 mM sodium chloride, 10 mM imidazole). The imidazole concentration was stepwise increased, first to 30 mM to remove nonspecifically bound proteins, and then to 500 mM to elute NP_888769.1. After digestion with 0.6 mg/mL tobacco etch virus (TEV) protease overnight at 4°C, the proteins were loaded onto a desalting column (HiPrep™ 26/10; Amersham) and eluted with buffer A. The protein fractions were again loaded onto a Ni+2 affinity column (HisTrap HP column; Amersham) equilibrated with buffer A to remove the TEV protease and the cleaved Histag from NP_888769.1. Using a flow rate of 2 mL/min, fractions containing NP_888769.1, as determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), were pooled and loaded onto a size exclusion column (Superdex™ 75 Hiload™ 26/60; Amersham) equilibrated with buffer B (20 mM phosphate at pH 6.0, 50 mM sodium chloride) and eluted with the same buffer. The fractions containing NP_888769.1 were concentrated to a final volume of 550 μL for a final protein concentration of 1.5 mM, using 3 kDa-cutoff centrifugal filter devices (Millipore). The NMR sample was supplemented with 5% 2H2O (v/v) and 4.5 mM NaN3.
NMR spectroscopy
NMR experiments were recorded at 298 K on Bruker AVANCE 600 MHz and AVANCE 800 MHz spectrometers equipped with 5-mm bruker 5 mm triple-resonance 3-axis gradient probe (TXI-HCN) z- or xyz- gradient probes. The backbone assignments were based on the three experiments 4D APSY-HACANH, 5D APSY-HACACONH, and 5D CBCACONH,4 and the side-chain resonance assignments were based on 3D 15N-resolved, 3D 13C(aliphatic)-resolved, and 3D 13C(aromatic)-resolved [1H,1H]-NOESY experiments recorded with τm = 60 ms. In addition, a 2D [15N,1H]-HSQC spectrum20 and a heteronuclear 2D 15N{1H}-NOE experiment21 were recorded at 600 MHz.
Proton chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonic acid sodium salt (DSS). The 13C and 15N chemical shifts were referenced indirectly to DSS using the absolute frequency ratios.22
Chemical shift assignments
The APSY-NMR experiments were analyzed with the software GAPRO.23 The resulting peak list was used as input for the software UNIO-MATCH5 for automated polypeptide backbone assignment. The backbone assignments were then completed interactively, using a 3D 15N-resolved [1H,1H]-NOESY spectrum. The completed backbone assignments and aforementioned three NOESY experiments were used as input for automated side-chain assignments with the software UNIO-ATNOS/ASCAN,6,7 which were then further extended and validated interactively.
Structure calculation and analysis
The input for the structure calculation was obtained from the aforementioned 3D heteronuclear-resolved [1H,1H]-NOESY experiments. The software UNIO-ATNOS/CANDID6,9 was used in combination with the torsion angle dynamics algorithm CYANA-3.08 for automated NOESY peak picking, NOE cross-peak identification, NOE assignment and structure calculation. In the second and subsequent cycles of the standard seven-cycle protocol,6,9 the intermediate protein structures were used as an additional guide for the interpretation of the NOESY data. Backbone ϕ and ψ dihedral angle constraints derived from the 13Cα chemicals shifts were used to supplement the NOE upper distance constraints in the final input for the structure calculation. The 40 conformers with the lowest residual CYANA target function values obtained from the seventh ATNOS/CANDID/CYANA cycle were subjected to energy minimization in a water shell with the program OPALp,24,25 using the AMBER force field.26 After structure validation (see below), 20 conformers were selected to represent the NMR structure, and the program MOLMOL27 was used to analyze this ensemble.
Structure validation
The stereochemical quality of the molecular structure was analyzed using the Joint Center for Structural Genomics protein structure validation site (http://smb.slac.stanford.edu/jcsg/QC/), the PSVS server (http://psus-1_4-dev.nesg.org) and the PDB validation server (http://deposit. pdb.org/validate).
Accession numbers
The chemical shifts and the atomic coordinates of the bundle of 20 conformers used to represent the solution structure of NP_888769.1 have been deposited in the Biological Magnetic Resonance Bank (http://www.bmrb.wisc.edu/) and in the Protein Data Bank (PDB; http://www.rcsb.org/pdb/), with the accession codes 17124 and 2L25, respectively.
Acknowledgments
Kurt Wüthrich is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute. We thank the Crystallomics Core of the JCSG for providing the plasmid for NP_888769.1, and Marc-André Elsliger and Ian Wilson for helpful comments on the paper. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- 1.Parkhill JSM, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeño-Tárraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O'Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 2.Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peti W, Page R, Moy K, O'Neil-Johnson M, Wilson IA, Stevens RC, Wüthrich K. Towards miniaturization of a structural genomics pipeline using micro-expression and microcoil NMR. J Struct Funct Genomics. 2005;6:259–267. doi: 10.1007/s10969-005-9000-x. [DOI] [PubMed] [Google Scholar]
- 4.Hiller S, Fiorito F, Wüthrich K, Wider G. Automated projection spectroscopy (APSY) Proc Natl Acad Sci USA. 2005;102:10876–10881. doi: 10.1073/pnas.0504818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volk J, Herrmann T, Wüthrich K. Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR. 2008;41:127–138. doi: 10.1007/s10858-008-9243-5. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 7.Fiorito F, Herrmann T, Damberger FF, Wüthrich K. Automated amino acid side-chain NMR assignment of proteins using 13C- and 15N-resolved 3D [1H,1H]-NOESY. J Biomol NMR. 2008;42:23–33. doi: 10.1007/s10858-008-9259-x. [DOI] [PubMed] [Google Scholar]
- 8.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 9.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 10.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK—a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 11.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 12.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 13.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, Harris D, Clarke L, Whitehead S, Sangal V, Marsh K, Achtman M, Molyneux ME, Cormican M, Parkhill J, Maclennan CA, Heyderman RS, Dougan G. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009;19:2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsura I, Tsugita A. Purification and characterization of the major protein and the terminator protein of the bacteriophage λ tail. Virology. 1977;76:129–145. doi: 10.1016/0042-6822(77)90290-2. [DOI] [PubMed] [Google Scholar]
- 15.Edmonds L, Liu A, Kwan JJ, Avanessy A, Caracoglia M, Yang I, Maxwell KL, Rubenstein J, Davidson AR, Donaldson LW. The NMR structure of the gpU tail-terminator protein from bacteriophage lambda: identification of sites contributing to Mg(II)-mediated oligomerization and biological function. J Mol Biol. 2007;365:175–186. doi: 10.1016/j.jmb.2006.09.068. [DOI] [PubMed] [Google Scholar]
- 16.Pell LG, Liu A, Edmonds L, Donaldson LW, Howell PL, Davidson AR. The X-ray crystal structure of the phage lambda tail terminator protein reveals the biologically relevant hexameric ring structure and demonstrates a conserved mechanism of tail termination among diverse long-tailed phages. J Mol Biol. 2009;389:938–951. doi: 10.1016/j.jmb.2009.04.072. [DOI] [PubMed] [Google Scholar]
- 17.Mohanty B, Serrano P, Jaudzems K, Geralt M, Horst R, Pedrini B, Elsliger M-A, Wilson IA, Wüthrich K. Comparison of NMR and crystal structures for the proteins TM1112 and TM1367. Acta Cryst F. 2010;66:1381–1392. doi: 10.1107/S1744309110020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. The Pfam protein families database. Nucl Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucl Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori S, Abeygunawardana C, Johnson MO, Berg J, van Zijl PCM. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–96. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 21.Zhu G, Youlin X, Nicholson LK, Sze KH. Protein dynamics measurements by TROSY-based NMR experiments. J Magn Reson. 2000;143:423–426. doi: 10.1006/jmre.2000.2022. [DOI] [PubMed] [Google Scholar]
- 22.Wishart D, Bigam C, Yao J, Abildgaard F, Dyson H, Oldfield E, Markley J, Sykes B. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 23.Hiller S, Wider G, Wüthrich K. APSY-NMR with proteins: practical aspects and backbone assignment. J Biomol NMR. 2008;42:179–195. doi: 10.1007/s10858-008-9266-y. [DOI] [PubMed] [Google Scholar]
- 24.Luginbühl P, Güntert P, Billeter M, Wüthrich K. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J Biomol NMR. 1996;8:136–146. doi: 10.1007/BF00211160. [DOI] [PubMed] [Google Scholar]
- 25.Koradi R, Billeter M, Güntert P. Point-centered domain decomposition for parallel molecular dynamics simulation. Comp Physics Commun. 2000;124:139–147. [Google Scholar]
- 26.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 27.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]