

Abstract
We have performed a detailed analysis of streptavidin variants with altered specificity towards desthiobiotin. In addition to changes in key residues which widen the ligand binding pocket and accommodate the more structurally flexible desthiobiotin, the data revealed the role of a key, non-active site mutation at the base of the flexible loop (S52G) which slows dissociation of this ligand by approximately sevenfold. Our data suggest that this mutation results in the loss of a stabilizing contact which keeps this loop open and accessible in the absence of ligand. When this mutation was introduced into the wild-type protein, destabilization of the opened loop conferred a ∼10-fold decrease in both the on-rate and off-rate for the ligand biotin-4-fluoroscein. A similar effect was observed when this mutation was added to a monomeric form of this protein. Our results provide key insight into the role of the streptavidin flexible loop in ligand binding and maintaining high affinity interactions.
Keywords: streptavidin, in vitro compartmentalization, loop, dissociation kinetics
Introduction
The protein streptavidin (SA) is one of the most widely used proteins in molecular biology, biotechnology, and more recently, nanotechnology. The interaction between SA and its natural ligand, biotin, is one of the strongest noncovalent interactions in biology1–5 (Kd ∼10−14). As a result, this protein–ligand couple has been the subject of numerous investigations to understand the nature of high affinity protein interactions3,6–9 as well as the target of multiple engineering efforts to alter its specificity and/or binding properties.
In a previous work, we utilized in vitro compartmentalization (IVC) to identify streptavidin mutants with altered specificity for the biotin analog desthiobiotin.10 All selected variants contained three active site mutations (T90S, W108V, and L110T) and at least two additional mutations outside this region introduced by error-prone PCR. One of the selected mutants (R4-6) contained two mutations outside active site (F29L and R53S), while the mutant R7-2 contained one additional mutation when compared with R4-6 (S52G). The best variants displayed improved binding properties toward desthiobiotinylated oligonucleotides (DTB-T10) with dissociation rates ∼50 times slower than the wild-type protein. Interestingly, functional analysis of variants R4-6 and R7-2 indicated that the S52G mutation was responsible for a approximately sevenfold improvement in the off rate. We suggested that this mutation might play a critical role in the function of the “flexible-loop,” which caps the biotin binding pocket when ligand is bound and is essential for maintaining high affinity binding.9,10 More recently, this mutation was exploited in a rational design approach to decrease the off rate for biotin from the wild-type protein by almost 10-fold.11 To obtain insight regarding the structural and kinetic reasons underlying the altered binding specificity of our streptavidin mutants and to better understand the specific contribution of the single mutation, S52G, that caused a marked decrease in a dissociation rates, we have performed crystallographic, thermodynamic, and detailed kinetic analysis of the engineered streptavidin mutants. Our results not only elucidate some of the molecular changes to the binding pocket that lead to the altered specificity of the selected mutant variants but shed light on the role of the S52G mutation, and enhance our understanding of the role of the flexible loop in maintaining high-affinity ligand binding.
Results
Association rate constants
The association rates of streptavidin variants were determined by monitoring protein tryptophan quenching on ligand binding,12 and the data is summarized in Table I. The association rate for desthiobiotin and the mutant R4-6 did not show any improvement when compared with the wild-type protein. Interestingly, a 10-fold increase in kon is observed when this ligand is displayed on a short oligonucleotide (DTB-T10), a result which may reflect the fact that a desthiobiotinylated deoxyribonucleic acid (DNA) was used as the target for the selection instead of free desthiobiotin. When similar experiments were conducted with biotin as the ligand, R4-6 revealed a threefold decrease in the association rate when compared with the wild type, indicating an alteration in the protein's specificity away from this ligand. The association rates for the R7-2 variant decreased for every ligand tested, including biotin, desthiobiotin, a biotinylated oligonucleotide (B-T10), and DTB-T10.
Table I.
Streptavidin Association and Dissociation Rates
| Wild-type SA | kon (M−1 sec−1) | koff (sec−1) | Kd (M) | Ref. |
|---|---|---|---|---|
| Biotin* | 6.7 × 107 ± 7 × 104 | 4.4 × 10−6 ± 5.5 × 10−7 | 6.5 × 10−14 ± 8.2 × 10−15 | 2 |
| Biotin T10* | 8.5 × 107 ± 8 × 106 | 1.1 × 10−6 ± 1.3 × 10−7 | 1.3 × 10−14 ± 1.9 × 10−15 | 10 |
| Desthiobiotin | 2.7 × 107 ± 6.4 × 104 | 1.8 × 10−2 ± 4.9 × 10−3 | 6.6 × 10−10 ± 1.8 × 10−10 | |
| Desthiobiotin T10* | 5.1 × 106 ± 9.3 × 105 | 4.4 × 10−4 ± 3 × 10−5 | 8.2 × 10−11 ± 1.7 × 10−11 | 10 |
| R4–6 SA | ||||
| Biotin | 2.5 × 107 ± 1.2 × 104 | ND | ND | |
| Biotin T10* | ND | 7.7 × 10−5 ± 5.5 × 10−6 | ND | 10 |
| Desthiobiotin | 2.8 × 107 ± 2.5 × 104 | 1.2 × 10−2 ± 4 × 10−3 | 4.3 × 10−10 ± 1 × 10−10 | |
| Desthiobiotin T10* | 4.0 × 107 ± 3 × 104 | 9.4 × 10−5 ± 8.3 × 10−6 | 2.3 × 10−12 ± 2 × 10−13 | 10 |
| R7–2 SA | ||||
| Biotin* | 5.5 × 106 ± 1.2 × 105 | 7.5 × 10−4 ± 3.3 × 10−5 | 1.4 × 10−10 ± 6.7 × 10−12 | 10 |
| Biotin T10* | 1.3 × 107 ± 1.4 × 104 | 1.5 × 10−5 ± 8.3 × 10−7 | 1.2 × 10−12 ± 6.3 × 10−14 | 10 |
| Desthiobiotin | 7.1 × 106 ± 2.4 × 104 | 3.5 × 10−3 ± 6 × 10−4 | 4.9 × 10−10 ± 8.5 × 10−11 | |
| Desthiobiotin T10* | 1.3 × 107 ± 6.1 × 104 | 1.4 × 10−5 ± 1.6 × 10−6 | 1.0 × 10−12 ± 1.2 × 10−13 | 10 |
Dissociation rates obtained from the literature.
Isothermal titration calorimetry
The thermodynamic binding parameters are presented in Table II. As expected, the titration for the wild-type protein resulted in a greater enthalpic contribution on biotin binding as compared with desthiobiotin. However, the analysis of enthalpy of binding on R4-6 and R7-2 active sites indicated a considerable change in the proteins' specificities towards desthiobiotin, with greater experimental −ΔH values when compared with biotin. Additionally, titration with R7-2 indicated a higher degree of static interactions to both ligands tested when compared with R4-6. The entropic penalty for ligand binding to the wild-type active site is larger for biotin binding when compared with desthiobiotin. In contrast, the entropic penalties for both mutant proteins are markedly different from that of the wild type, with the penalty for desthiobiotin greater than biotin for R7-2 and the binding enthalpies for R7-2 larger than for R4-6. As the only difference between these two variants lies at the loop residue S52G, we suggest that this mutation has introduced a higher degree of loop freedom in the unbound form of the protein, whose stabilization upon ligand binding produces a higher entropic penalty on the R7-2 mutant.
Table II.
Thermodynamics Parameters Obtained from Calorimetric Titration of Streptavidin (Determined at 300 K)
| Wild-type SA | −ΔH (kcal−1 mol−1) | −ΔG (kcal−1 mol−1) | −TΔS (kcal−1 mol−1) |
|---|---|---|---|
| Biotin | 29.4 ± 0.2 | 18.0 ± 0.1 | 11.4 ± 0.2 |
| Desthiobiotin | 21.7 ± 0.1 | 12.4 ± 0.2 | 9.3 ± 0.2 |
| R4–6 SA | |||
| Biotin | 20.4 ± 0.3 | ND* | ND* |
| Desthiobiotin | 22.8 ± 0.2 | 12 ± 0.1 | 10.8 ± 0.2 |
| R7–2 SA | |||
| Biotin | 21.75 ± 0.3 | 13.40 ± 0.03 | 8.3 ± 0.3 |
| Desthiobiotin | 24.2 ± 0.3 | 12.7 ± 0.1 | 11.6 ± 0.3 |
−ΔG and −TΔS values not calculated, since Kd values for R4–6 biotin were not determined.
Desthiobiotin dissociation kinetics measurements
The measurement of dissociation rates for the mutants R4-6 and R7-2 by the biotin-4-fluorescein method was not feasible, as no fluorescence quenching was observed on ligand binding. Therefore, we have measured [3H]-desthiobiotin dissociation rates allowing us to calculate and compare the dissociation constants of wild type as well as the R4-6 and R7-2 mutants. A representative graph is illustrated in Supporting Information Figure 4, and the data is summarized in Table I. As expected, we observed a fivefold and fourfold decrease in off rates for the R7-2 when compared with wild type and R4-6, respectively.
Figure 4.
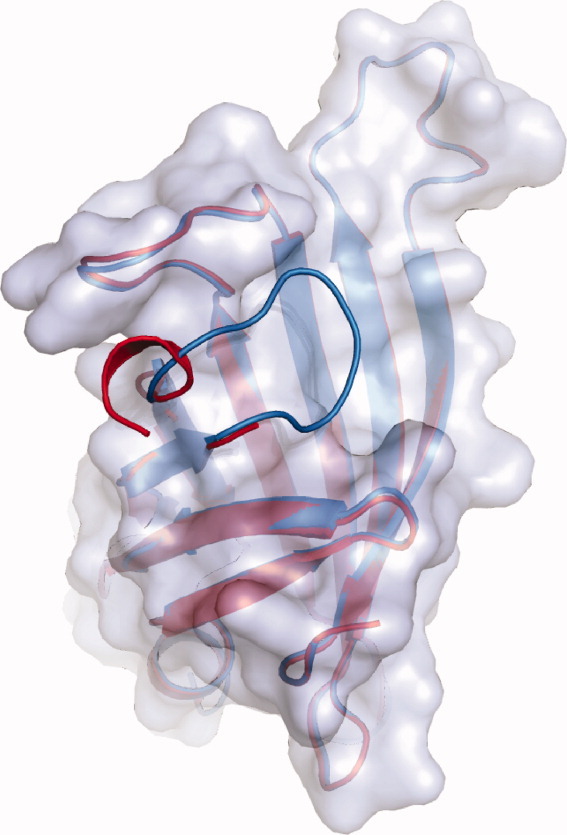
Comparison of the overlaid loop structures of variants R4-6 and R7-2 (both in gray). The structures were derived from protein crystals formed in the absence of any ligand (structures R4-6 [2] and R7-2 [6]). The R4-6 loop (red) is open disordered and only partially visible. The R7-2 loop (blue) is closed and ordered.
Crystallization of R4-6 and R7-2 streptavidin
Crystallization of proteins purified under native conditions yielded structures with endogenous biotin bound (structure R7-2[1], Supporting Information Table I). Therefore, in order to obtain biotin-free structures, all proteins were subsequently purified under denaturing conditions. Interestingly, R4-6 and R7-2 crystals formed from low molecular weight polyethylene glycol (PEG) solutions revealed structures with PEG molecules bound in the biotin-binding site. The crystallographic data demonstrated that the selected variants produced a wider binding pocket when compared with the wild-type protein (Fig. 1). The observed widening of the binding pocket appears to be a direct consequence of the W108V, L110T, and T90S mutations, although the backbone of those residues remained virtually identical to the wild-type protein (Fig. 2). Additionally, we observed that the selected mutations (T90S, W108V, L110T, F29L, S52G, and R53S) did not modify the arrangement of the other important biotin binding residues, such as N23, Y43, S27, S45, and N128, as can be observed by the Global root mean square deviation (RMSD) of 0.533Å when compared to the wild-type protein.
Figure 1.
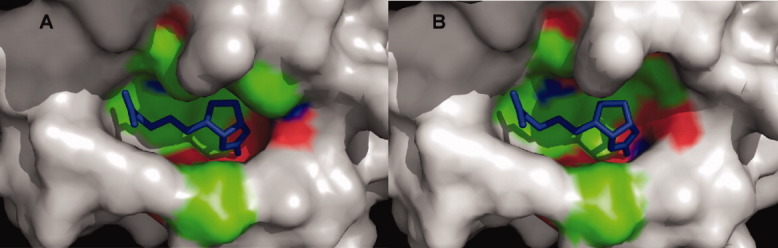
Active sites of wild-type streptavidin (panel A) and mutant R7-2 (panel B) bound to biotin. The R7-2 active site residues appear to shape a wider active site as compared to the wild-type protein.
Figure 2.
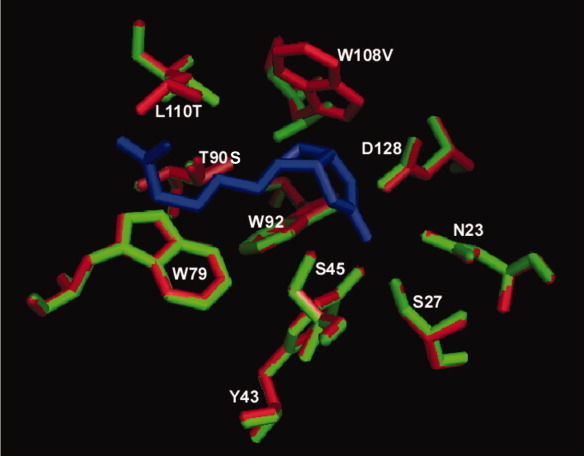
Comparison of binding pocket residues between wild-type (red) streptavidin and R7-2 (green). Biotin is represented as dark blue sticks. The position of most residues remained virtually identical to the wild-type protein, including the backbone residues of the active site mutations W108V and L110T that produced a broadening effect on the binding pocket of the selected variants.
To investigate the dynamic properties of the flexible loop, we compared structures of unbound, partially bound and fully occupied molecules. For R4-6, we examined structures in which the binding pocket was either completely empty (structure R4-6[2]) or occupied by a disordered PEG molecule (structure R4-6[1]). The flexible loop appeared open and disordered in both structures. In the case of R7-2, we performed crystallizations in both the presence and absence of ligands. As expected, in the presence of either biotin or desthiobiotin, the flexible loop was closed and well ordered (structures R7-2[1], R7-2[2], and R7-2[3]). The presence of PEG in the binding pocket also yielded structures with an ordered and closed loop (R7-2[4] and R7-2[5]). Interestingly, when crystals were generated in the absence of PEG, we observed crystals of the unliganded form of R7-2, where the flexible loop also appeared closed and well ordered even in the absence of any ligand (R7-2[6]), although the observed average temperature factors are higher than in other ligand-bound structures. Unlike a previously reported structure in which crystal packing contacts resulted in the closure of this loop in the absence of ligand,9,13 in the unliganded form of R7-2, the binding loop does not make any contacts (within a 3.5 Å cutoff) with symmetry related molecules. For a comparison of the loop structures of R7-2 in the presence of biotin, desthiobiotin and in the absence of any ligand (see Supporting Information Fig. 5).
S52G Association Rates
The association rate data is summarized in Table III. These measurements were performed at 37°C for comparison with dissociation rates measured by the biotin-4-fluorescein method that required higher temperatures to reach at least three half-lives of dissociation. Accordingly, the S52G mutation introduced in wild-type streptavidin also showed a fivefold and 10-fold decrease in association rates of biotin and biotin-4-fluorescein, respectively.
Table III.
Kinetic Parameters of Streptavidin S52G mutant
| kon (M−1 sec−1) | koff (sec−1) | koff *(sec−1) | |
|---|---|---|---|
| wt streptavidin | |||
| Biotin | 6.8 × 107 ± 5.0 × 106 | ND | – |
| Biotin-4-fluorescein | 5.5 × 107 ± 1.0 × 106 | 3.3 × 10−5 ± 3 × 10−7 | – |
| S52G streptavidin | |||
| Biotin | 1.4 × 107 ± 1.0 × 106 | ND | ND |
| Biotin-4-fluorescein | 5.9 × 106 ± 1.0 × 105 | 2 × 10−4 ± 2 × 10−6 (20%) | 3 × 10−6 ± 5 × 10−8 (80%) |
koff corresponds to the values for the single exponential fit (wild type) or the first exponential of a double exponential fit (S52G mutant).
Koff* is the value for the second exponential of double exponential fit for S52G mutant. The amplitude in percentage of the first or second phase for the second exponential fit of S52G mutant is reported in parentheses.
Measurement of S52G dissociation rates
To gather more information about the contributions of S52G mutation to the streptavidin dissociation kinetics, biotin dissociation rate constants of the wild type, and the mutant were measured using a biotin-4-fluoroscein fluorescence assay.14 Differently from the wild type, the mutant shows a biphasic dissociation curve (Supporting Information Fig. 6), with an initial fast rate followed by a second steady slow rate of dissociation. The data is summarized in Table III. As the second steady slow rate of dissociation for S52G mutant corresponded to about 80% of biotin dissociation, we have interpreted this as the major dissociation event. In this case, we can observe an 11-fold decrease in koff of biotin-4-fluorescein for the S52G mutant when compared with the wild type.
SPR analysis of the monomeric streptavidins
Based on the experimental data that supports improvement in dissociation kinetics by the S52G mutation, we have introduced these mutations into a monomeric streptavidin construct.15 The kinetic properties of these mutants were analyzed by surface plasmon resonance (SPR) experiments and the data is summarized in Table IV. The introduction of the S52G mutation into monomeric streptavidin conferred a 10-fold decrease in the respective association and dissociation rates, with no additional effect on Kd. The introduction of an additional mutation, R53S or R53D, to the S52G mutant, did not produce any further significant change on the dissociation constant. Additionally, the simple comparison of the respective sensograms, shows a clear contribution of the S52G mutation on the individual patterns of association and dissociation (Fig. 3).
Table IV.
Kinetic Parameters of Monomeric Streptavidins Determined by SPR
| kon (M−1 sec−1) | koff (sec−1) | Kd (M) | |
|---|---|---|---|
| mSA | 1.2 × 105 | 1.3 × 10−2 | 1.0 × 10−7 |
| mSA S52G | 1.6 × 104 | 2.0 × 10−3 | 1.3 × 10−7 |
| mSA S52G, R53S | 1.9 × 104 | 2.0 × 10−3 | 1.0 × 10−7 |
| mSA S52G, R53D | 1.5 × 104 | 2.5 × 10−3 | 1.7 × 10−7 |
Figure 3.
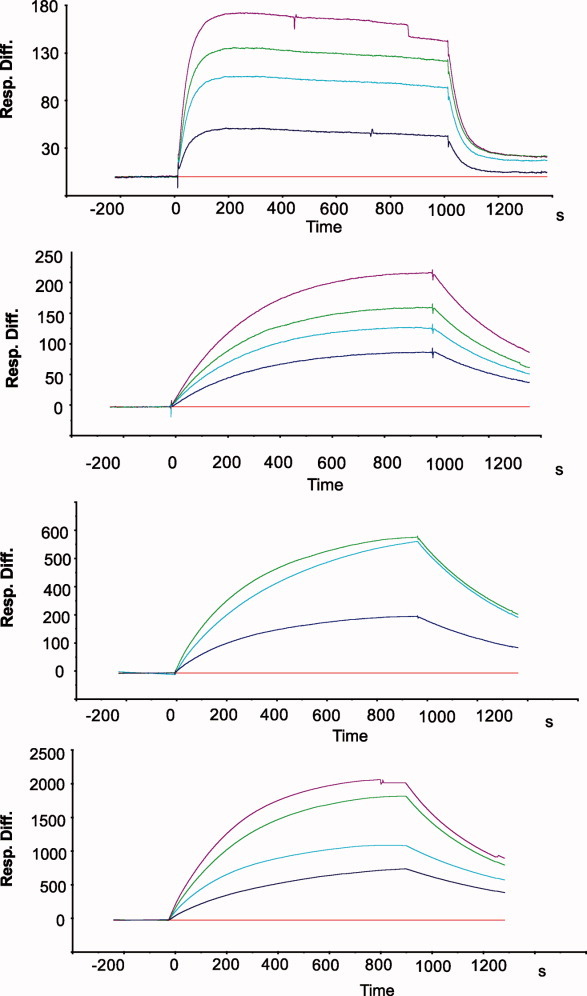
SPR sensograms showing the interaction of immobilized biotin and monomeric streptavidin variants. The traces represent experimental data in response units at 0 nM (red), 25 nM (blue), 50 nM (cyan), 100 nM (green), and 125 nM (magenta) of streptavidin variants.
Discussion
The selected streptavidin mutants evolved improved binding properties toward desthiobiotin
The selected mutants that presented improved binding properties toward desthiobiotin contained three mutations present in the biotin binding pocket (T90S, W108V, and L110T).10 The analysis of the crystal structures revealed that those substitutions supported a wider active site (Fig. 1), apparently tailored to accommodate desthiobiotin, which requires an extra degree of rotational freedom due to the lack of the thiophene ring. The analysis of the thermodynamic parameters for wild-type and mutant proteins reiterated the strong preference of biotin binding over desthiobiotin in the wild-type binding pocket but indicated a significant change in both the enthalpic and entropic contributions for binding in R4-6. Perhaps most marked is the 8.7 kcal mol−1 decrease in the enthalpy for biotin binding, likely a consequence of the strong selective pressure during the selection to maintain desthiobiotin binding even in the presence of excess biotin.10 Analysis of the equilibrium enthalpy for binding desthiobiotin reveals that the evolved mutants favor this ligand by 2.4kcal mol−1. When compared with the wild-type protein, the R4-6 enthalpic contribution for desthiobiotin binding is 1.1 kcal mol−1 higher, further suggesting that after four rounds of selection, the binding pocket had at least in part reoptimized to better accommodate desthiobiotin binding. These observations are further supported by the off-rate analyses of the selected mutants, which demonstrate slower off rates than the wild type for desthiobiotin and a DTB-T10 than for biotin or B-T10.
Also interesting is the clear preference of the selected mutants for ligands (biotin and desthiobiotin) displayed on short oligonucleotides, as exemplified by a >200-fold decrease in DTB-T10 Kd when compared with free desthiobiotin for R4-6 and R7-2. It is interesting to compare this result with that observed in another directed evolution experiment in which single chain dimeric streptavidin variants expressed in E. coli for binding to biotin-4-fluorescein evolved a marked preference (>104) for biotin-4-fluorescein over free biotin.16 Taken together, these results not only support the adage, “you get what you select for” but, perhaps more importantly, demonstrate the potential to “delocalize” the specificity of ligand binding by this protein to a region outside of the core biotin binding pocket. However, in the case of our streptavidin variants, in the absence of crystallographic data for these ligands, we have not yet identified the molecular factors involved in this binding improvement.
The S52G mutation causes opened binding loop destabilization
Our previous studies reported a approximately fivefold decrease in the off rate (koff) of DTB-T10 for R4-6 when compared with the wild-type protein, and another approximately sevenfold decrease in koff for the dominant mutant R7-2 variant.10 Interestingly, variant R7-2 only acquired one additional mutation (S52G) when compared with R4-6. This mutation lies at the base of the flexible binding loop (residues 45 to 52), which is essential for high-affinity biotin binding.9,13 In the ligand-free, apo-form of the protein, the flexible binding loop is thought to adopt an open conformation.1,13 This hypothesis is supported by numerous crystallographic studies in which the loop of the apo-protein is open and often disordered.1,13,17 Detailed structural studies of the loop have suggested that the open conformation is stabilized by the formation of a hydrogen bond at the base of this loop between residues S45 and S52.1,13 In most reported structures, when biotin is present, the streptavidin binding loop is found to be ordered and closed like a lid over the binding pocket.1,5,13 The stably opened loop, followed by a structured and closed loop over biotin on binding appears to be an important structural feature of this high-affinity interaction system, and it has been proposed that the breakage of S45–S52 hydrogen bond takes place as biotin binds within the binding pocket forming a hydrogen bond between S45 and its ureido nitrogen.9 Only two indications of a closed loop in the ligand-free form of the protein have been reported: one due to crystal packing interactions13 and the other due to the presence of sulfate ions localized in the binding pocket.18 As the only difference between R4-6 and R7-2 lies in residue S52, we reasoned that the approximately sevenfold decrease in koff between these proteins could be caused by destabilization of the opened-loop due to the lack of the hydrogen bond between the S52G mutation and S45. We hypothesized that in the absence of this hydrogen bond stabilizing the open-loop, the loop might be freer to adopt a closed conformation, even in the absence of the ligand. Such a mutation would have two potential effects. First, it would slow the koff, consistent with our previously observed results, and second it would slow kon. Accordingly, we can verify a fourfold decrease in kon for all ligands tested on R7-2 binding when compared with R4-6. Indeed, this observation is in agreement with another recently published streptavidin variant, “traptavidin,” which harbors the S52G as well as an additional mutation at R53D and is reported to show a ∼10-fold decrease in koff when compared with wild type.11 Further agreement with our hypothesis of opened-loop destabilization caused by the S52G substitution is observed in the loop behavior for the R4-6 and R7-2 mutants crystallized in the presence or absence of different ligands (Supporting Information Table I). In accordance with previous data for the wild-type streptavidin, the presence of ligand renders both mutant proteins with ordered, closed loop residues. Interestingly, in the case of R7-2, even the presence of PEG in the active site resulted in a closed and ordered loop. Moreover, in crystals generated in the absence of ligand and PEG, the ligand-free form of R7-2 mutant is also observed to have a closed and ordered loop (Fig. 4). However, in the high-salt, ligand-free R7-2 structure, the average temperature factor is higher than in the ligand bound other structures. In contrast, like most reported structures of the wild-type protein, the R4-6 loop appears opened and not visible in the ligand-free structures (Fig. 4). Our open-loop destabilization hypothesis is further supported by our calorimetric data, where we observed a larger entropic penalty on ligand binding in the R7-2 pocket when compared with R4-6 for both biotin and desthiobiotin, suggesting that the unliganded form of R7-2 is less ordered than R4-6. This increase in conformational entropy is likely a direct result of the absence of a hydrogen bond between S52 and S45, which in the wild-type protein extends the β-strand-like structure of the open loop and likely stabilizes the 310 helical character observed for residues 49 through 52.13 Interestingly, engineered versions of streptavidin with improved affinity for the Strep-tag II peptide also contained loop mutations (S45, A46, and V47). However, these mutations were shown to induce a fixed opened conformation of the loop at the protein's binding site.19
The S52G mutation caused decreased koff and kon rates in wild-type streptavidin
To further investigate the individual role of S52 in streptavidin dissociation and association kinetics, we introduced S52G mutation into the wild-type protein (wtS52G). Differently from the wild type, a kinetic analysis of biotin-4-fluorescein dissociation by the wtS52G protein shows a biphasic dissociation curve (Supporting Information Fig. 6), with a fast initial dissociation rate of 2 ×10−4 sec−1 corresponding to 20% of total ligand dissociation and a second steady slow rate of 3 × 10−6 sec−1 corresponding to 80% total ligand dissociation (Table III). The streptavidin variant, traptavidin, also showed biphasic dissociation kinetics with a 2% fast initial biotin dissociation followed by a steady slow dissociation of 4.2 × 10−5 sec−1.11 Such observations imply that at least two distinguishable steps are occurring during dissociation. Although we cannot precisely pinpoint reasons underlying the biphasic behavior, this appears to have been introduced by just the S52G mutation suggesting that the flexibility of the loop is involved in this effect. Analysis of the biotin and biotin-4-fluoroscein association rates for the wtS52G mutant revealed a decrease in the on rate by fourfold and 10-fold when compared with the wild type (Table III), in agreement with the penalty on binding observed for R4-6 and R7-2.
The S52G mutations introduced into monomeric streptavidin produced variants with improved dissociation kinetics
Streptavidin is a tetramer that binds one molecule of biotin per subunit, which is advantageous in some applications, such as multimerization and improvement of binding by avidity effects. However, in some cases, tetrameric valency may affect the desired function and limit applications. Therefore, intensive efforts have also been made to extend the use of streptavidin technology by engineering monomeric variants with high affinity for biotin binding.11,15,20,21 However, one of the important contributions for the high-affinity interaction of biotin binding is provided by interactions with tryptophan 120 from the neighboring subunit,8,22 and attempts to produce a monomeric streptavidin have generated variants with greatly reduced binding affinities ranging from 10−7 to 10 −9 M−1.15,20 Given the improvement in dissociation kinetics provided by the S52G mutation, we decided to investigate the impact of this mutation into monomeric streptavidin constructs.
A series of four mutants was constructed based on the monomeric SA variant: M4 initially reported by Wu and Wong,15 but with specific substitutions at positions 52 and 53, which included S52G, S52G and R53S, or S52G R53D. The mutation at R53S had been identified as part of the six mutations in R7-2 mutant that accounted for improved dissociation kinetics, whereas the R53D mutation has been utilized by Chivers et al.11 in the development of a streptavidin variant with a 10-fold improvement of biotin dissociation, increased mechanical strength and improved thermostability.
The proteins monomeric nature was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Supporting Information Fig. 7) and binding parameters were determined by SPR (Table IV). As we had observed previously with the tetrameric proteins, the SPR binding results using the monomeric variants containing that S52G mutation introduced a 10-fold decrease in both kon and koff (Table IV), with no net effect on Kd.
Material and Methods
Protein expression, purification, and refolding
The wtSA and mutants R4-6 and R7-2 were constructed and cloned as previously described.10 Recombinant plasmids were transformed into Escherichia coli strain (KRX) cells (Promega, Madison, WI). Cells were inoculated into Terrific Broth (TB) media and grew until reach optical density (OD) of 1.5 followed by induction with 0.1% of rhamnose at 37°C for 20 h. Cell pellets were suspended in 50mM Tris-HCl, pH 8.0, 0.1% (v/v) Triton X-100, 100 mM NaCl, and cells were disrupted by sonication. The insoluble cell fraction was suspended in 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, and centrifuged at 10,000g for 45 min. Cell pellets were solubilized with 6M guanidine hydrochloride, 20 mM phosphate, pH 1.5, for 30 min at room temperature. The insoluble debris was removed by centrifugation, and the pH of the solution was adjusted to 8.0. Ni-NTA Resin (Genescript, Piscataway, NJ) was mixed with the crude protein extract, and resin was washed with 10-column volumes of 6M guanidine hydrochloride, 10 mM Tris-HCl, pH 8.0, followed by 25 column volumes of 8M urea, 10 mM tris-HCl, 10 mM immidazole, pH 8.0. The proteins were eluted in 8M urea, 10 mM Tris-HCl, 250 mM immidazole, pH 8.0, and refolded by dialysis against tris-buffered saline tween-20 (TBST). Any insoluble debris was removed by centrifugation.
Site directed mutagenesis of the wild-type streptavidin
The single point mutation, S52G, was introduced into plasmid pCR2.1-TOPO::SA10 using Megawhop PCR as previously described23 using a mutagenic primer (5′-GGAATGCGGAGGGCCGT TATGTGC-3′) and the M13 reverse primer (5′-CAGGAAACAGCT ATGAC-3′) that is complementary to the upstream region of the vector. The resultant mixture was DpnI treated and transformed into E. coli Top10 cells. Single colonies were screened for the presence of the desired mutation by DNA sequencing.
Site directed mutagenesis of the monomeric streptavidin
The gene for the monomeric streptavidin was based on the sequence reported by Wu and Wong,15 except that our construct contained an additional mutation (W120G) that was designed to further prevent tetramerization of the protein, as it has been shown that W120 is part of the neighboring biotin-binding pocket.8 The gene was assembled as previously described10 and cloned into pCR2.1-TOPO. Using a sequence verified clone, three additional monomeric variants were made using the Megawhop PCR protocol.23
Association rate measurement
Stopped-flow fluorescence measurements were measured at 25 or 37°C (as indicated) using a rapid mixing system (Kintek, Austin, TX) coupled to a Fluoromax 3 (Horiba, Ann Arbor, MI) spectrophotometer. The reaction was followed by monitoring the intrinsic tryptophan fluorescence of streptavidin, which decreases by about 40% on biotin binding.12 Because of the rapid association rate, the reactions were followed under second-order conditions using equimolar reactants at 1 μM as previously described.6 In short, 200 μL of protein and ligand solutions at 2 μM were injected for each run, giving a final concentration of 1 μM. Fluorescence was excited at 280 nm and emission was monitored at 340 nM, and each curve was the average of at least five shots. The dead time of the instrument was ∼3 msec. Data were analyzed using the Hi-Tech software using a second-order model.
Biotin-4-fluoroscein dissociation rate measurement
The kinetics of biotin 4-fluoroscein dissociation for the wild-type and S52G mutant was performed using a biotin-4-fluoroscein fluorescence assay.14 Measurements were performed at 37°C over three half-lives of dissociation and monitored using an ABI 7300 Real Time PCR System. Solutions of streptavidin (1μM) and biotin-4-fluoroscein (1 μM) were assembled in phosphate buffered saline (PBS) and incubated at room temperature for 1 h. After binding, a 500-fold excess of free biotin was added to the mixture, and the increase in fluorescence due to dissociation was monitored over time. The concentration of competing biotin was saturating, as reducing the biotin concentration 10-fold produced indistinguishable dissociation rates. The fraction of bound ligand was calculated using either a single or double exponential decay curve, as illustrated by Eqs. (1) and (2), respectively.
| (1) |
| (2) |
Desthiobiotin dissociation rate measurement
All desthiobiotin dissociation rates were measured at 25°C using a radiometric competition assay as described previously.7 Briefly, 1-mL reaction volumes were assembled in TBST containing 100 nM of protein and 10 nM of [3H] desthiobiotin (ViTrax, Placentia, CA). The dissociation experiment was initiated by the addition of 5 μL of 10 mM nonradioactive desthiobiotin and 40 μL aliquots were removed at different time points and precipitated by ZnSO4/NaOH precipitation.10 Samples were centrifuged at 14,000g for 15 min, and 10 μL aliquots of the supernatant were combined with 3 mL of Ultima Gold scintillation fluid (Perkin Elmer, Waltham, MA). The amount of free ligand was determined by scintillation counting using a Tri Carb Liquid Scintilation Counter (Perkin-Elmer). Rates represent the average of two independent experiments consisting of a minimum of 15 time points. The fraction of free [3H] desthiobiotin was plotted against time and fit to a single exponential rise according to the Eq. (1).
Isothermal titration calorimetry
ITC experiments were performed using a specific Isothermal Titration Calorimeter from Microcal (VP-ITC) microcalorimeter (Microcal, Inc., Northampton, MA). All measurements were carried out at 25°C in PBS Buffer. Enzyme preparations were dialyzed extensively against the above buffer, and all ligand solutions were prepared in the final dialysate. In individual titrations, injections of 4 μL of either biotin (0.5 mM) or desthiobiotin (0.5 mM) were made into the enzyme solution (cell volume = 1.43 mL) via a computer-controlled microsyringe at an interval of 4 min while being stirred at 310 rpm. The instrument was calibrated using the calibration kit supplied by the manufacturer. The experimental data were fit with a theoretical titration curve using software supplied by Microcal (GE Healthcare, Piscataway, NJ), with ΔH (the binding enthalpy change in kcal mol−1), Ka (the binding constant in M−1), and n (the number of binding sites per monomer) as adjustable parameters. We have used the ITC titration curve to determine the binding enthalpy, ΔH. The thermodynamic parameters ΔG and ΔS were calculated from Eq. (3), with the given that Ka has been determined by other methods,
| (3) |
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding. T is the absolute temperature, and R = 1.98 cal mol−1 K−1.
Crystallization of the mutant and wild-type streptavidin
The concentrations of R4-6 and R7-2 streptavidin for crystallization were 15 and 18 mg mL−1 in phosphate buffered saline containing 0.1% Tween-20, respectively. Diffraction quality crystals were grown using the sitting drop vapor diffusion method by mixing 1 μL of protein and 1 μL of reservoir solution and equilibrating the samples against the corresponding reservoir solution. The compositions of the reservoir solutions are listed in the Supporting Information Table I.
X-ray diffraction data collection and crystallographic refinement
Crystals of R4-6 and R7-2 streptavidin with overall dimensions 0.3 × 0.3 × 0.3 mm3 were mounted in cryo-loops directly from the crystallization droplet and flash-cooled in liquid nitrogen. Before freezing, 20% glycerol was added (as a cryo-protectant) to the droplets. Diffraction data were collected on a Quantum 315 CCD detector (Area Detector Systems Corporation, Poway, CA) with 1.08 or 0.979 Å wavelength radiation on the X29A beamline (National Synchrotron Light Source, Brookhaven National Laboratory, NY). Intensities were integrated using the HKL2000 program and reduced to amplitudes using the TRUNCATE program (see Supporting Information Table II).24,25 Structures were determined using the molecular replacement method with PHASER.26 Model building and refinement were performed with the programs REFMAC.24,27 The quality of the final structures were verified with composite omit maps, and stereochemistry was checked with the programs WHATCHECK28 and PROCHEK.29 LSQKAB, and SSM algorithms were used for structural superpositions.25,30
SPR analysis of the monomeric streptavidins
All the measurements were performed at 25°C. Evaluation of kinetic parameters was determined by SPR using a BIAcore 3000 biosensor system (GE Healthcare, Piscataway, NJ). Biotin pentylamine (Pierce, Rockford, IL) was immobilized on a research grade CM5 sensor chip using EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide) according to standard protocols. Solutions of streptavidin at different concentrations (25–125 nM) in 20 mM KHPO4, pH 7.4, 130 mM KCl, 3.4 mM EDTA were injected at a flow rate of 15μL min−1. The kinetic rate constants (kon and koff) as well as dissociation constant (Kd) were determined by global fit using the Biaevaluation software (Biacore System).
Conclusions
Flexible loops functioning as opened gates to optimize ligand association followed by gate closure to prevent dissociation (as in the case of the streptavidin-biotin) are important structural elements of high-affinity interactions, and a deep understanding of these features is of major importance for protein engineering purposes. Evolution ultimately selects the preferred balance between entropic penalties and the benefits upon loop closure to maximize function.
Previous studies of the streptavidin-biotin interaction by phage-display using a shot gun scanning mutagenic analysis have revealed three long-range nonactive site residues providing important contributions for the binding interaction of this high- affinity ligand pair.31 Here, we have uncovered the molecular basis for the contribution of an additional specific nonactive site residue, S52, previously identified by directed evolution. The S52G mutation results in ∼5–10-fold decrease in kon and koff of every ligand tested with only a small overall change in the respective Kd. From a practical perspective, the reduction in association is of less importance (considering kon is still extremely fast and close to diffusion limit) when compared with the advantages of a decrease in dissociation, which may have a greater impact for biotechnological purposes. Our studies not only clarified the molecular reasons for the ∼10-fold decrease in koff as previously reported,11 but also shed light on an important molecular feature of this high-affinity system: stabilization of an opened gate before ligand binding.
Acknowledgments
The authors thank Huiyong Cheng for assistance with SPR experiments, Rafael Toro for assistance with protein crystallization and data collection, Amy C. Yan for her assistance editing this manuscript and the Albert Einstein Cancer Center (3P30CA013330) for support.
Supplementary material
References
- 1.Weber PC, Ohlendorf DH, Wendoloski JJ, Salemme FR. Structural origins of high-affinity biotin binding to streptavidin. Science. 1989;243:85–88. doi: 10.1126/science.2911722. [DOI] [PubMed] [Google Scholar]
- 2.Piran U, Riordan WJ. Dissociation rate constant of the biotin-streptavidin complex. J Immunol Methods. 1990;133:141–143. doi: 10.1016/0022-1759(90)90328-s. [DOI] [PubMed] [Google Scholar]
- 3.DeChancie J, Houk KN. The origins of femtomolar protein-ligand binding: hydrogen-bond cooperativity and desolvation energetics in the biotin-(strept)avidin binding site. J Am Chem Soc. 2007;129:5419–5429. doi: 10.1021/ja066950n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green NM. Avidin. Adv Protein Chem. 1975;29:85–133. doi: 10.1016/s0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- 5.Hendrickson WA, Pahler A, Smith JL, Satow Y, Merritt EA, Phizackerley RP. Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc Natl Acad Sci USA. 1989;86:2190–2194. doi: 10.1073/pnas.86.7.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyre DE, Le Trong I, Merritt EA, Eccleston JF, Green NM, Stenkamp RE, Stayton PS. Cooperative hydrogen bond interactions in the streptavidin-biotin system. Protein Sci. 2006;15:459–467. doi: 10.1110/ps.051970306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klumb LA, Chu V, Stayton PS. Energetic roles of hydrogen bonds at the ureido oxygen binding pocket in the streptavidin-biotin complex. Biochemistry. 1998;37:7657–7663. doi: 10.1021/bi9803123. [DOI] [PubMed] [Google Scholar]
- 8.Chilkoti A, Tan PH, Stayton PS. Site-directed mutagenesis studies of the high-affinity streptavidin-biotin complex: contributions of tryptophan residues 79, 108, and 120. Proc Natl Acad Sci USA. 1995;92:1754–1758. doi: 10.1073/pnas.92.5.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu V, Freitag S, Le Trong I, Stenkamp RE, Stayton PS. Thermodynamic and structural consequences of flexible loop deletion by circular permutation in the streptavidin-biotin system. Protein Sci. 1998;7:848–859. doi: 10.1002/pro.5560070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy M, Ellington AD. Directed evolution of streptavidin variants using in vitro compartmentalization. Chem Biol. 2008;15:979–989. doi: 10.1016/j.chembiol.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chivers CE, Crozat E, Chu C, Moy VT, Sherratt DJ, Howarth M. A streptavidin variant with slower biotin dissociation and increased mechanostability. Nat Methods. 7:391–393. doi: 10.1038/nmeth.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurzban GP, Gitlin G, Bayer EA, Wilchek M, Horowitz PM. Biotin binding changes the conformation and decreases tryptophan accessibility of streptavidin. J Protein Chem. 1990;9:673–682. doi: 10.1007/BF01024762. [DOI] [PubMed] [Google Scholar]
- 13.Freitag S, Le Trong I, Klumb L, Stayton PS, Stenkamp RE. Structural studies of the streptavidin binding loop. Protein Sci. 1997;6:1157–1166. doi: 10.1002/pro.5560060604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebner A, Marek M, Kaiser K, Kada G, Hahn CD, Lackner B, Gruber HJ. Application of biotin-4-fluorescein in homogeneous fluorescence assays for avidin, streptavidin, and biotin or biotin derivatives. Methods Mol Biol. 2008;418:73–88. doi: 10.1007/978-1-59745-579-4_7. [DOI] [PubMed] [Google Scholar]
- 15.Wu SC, Wong SL. Engineering soluble monomeric streptavidin with reversible biotin binding capability. J Biol Chem. 2005;280:23225–23231. doi: 10.1074/jbc.M501733200. [DOI] [PubMed] [Google Scholar]
- 16.Aslan FM, Yu Y, Mohr SC, Cantor CR. Engineered single-chain dimeric streptavidins with an unexpected strong preference for biotin-4-fluorescein. Proc Natl Acad Sci USA. 2005;102:8507–8512. doi: 10.1073/pnas.0503112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freitag S, Le Trong I, Klumb LA, Chu V, Chilkoti A, Stayton PS, Stenkamp RE. X-ray crystallographic studies of streptavidin mutants binding to biotin. Biomol Eng. 1999;16:13–19. doi: 10.1016/s1050-3862(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 18.Katz BA. Binding to protein targets of peptidic leads discovered by phage display: crystal structures of streptavidin-bound linear and cyclic peptide ligands containing the HPQ sequence. Biochemistry. 1995;34:15421–15429. doi: 10.1021/bi00047a005. [DOI] [PubMed] [Google Scholar]
- 19.Korndorfer IP, Skerra A. Improved affinity of engineered streptavidin for the Strep-tag II peptide is due to a fixed open conformation of the lid-like loop at the binding site. Protein Sci. 2002;11:883–893. doi: 10.1110/ps.4150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qureshi MH, Yeung JC, Wu SC, Wong SL. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities. J Biol Chem. 2001;276:46422–46428. doi: 10.1074/jbc.M107398200. [DOI] [PubMed] [Google Scholar]
- 21.Laitinen OH, Hytonen VP, Nordlund HR, Kulomaa MS. Genetically engineered avidins and streptavidins. Cell Mol Life Sci. 2006;63:2992–3017. doi: 10.1007/s00018-006-6288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber PC, Cox MJ, Salemme FR, Ohlendorf DH. Crystallographic data for Streptomyces avidinii streptavidin. J Biol Chem. 1987;262:12728–12729. [PubMed] [Google Scholar]
- 23.Miyazaki K. Creating random mutagenesis libraries by megaprimer PCR of whole plasmid (MEGAWHOP) Methods Mol Biol. 2003;231:23–28. doi: 10.1385/1-59259-395-X:23. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski ZM, W Processing of x-ray Diffraction data collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 26.Breaker RR, Joyce GF. A DNA enzyme that cleaves RNA. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 29.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 31.Avrantinis SK, Stafford RL, Tian X, Weiss GA. Dissecting the streptavidin-biotin interaction by phage-displayed shotgun scanning. Chembiochem. 2002;3:1229–1234. doi: 10.1002/1439-7633(20021202)3:12<1229::AID-CBIC1229>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.