

Abstract
ABC transporters are a large and important family of membrane proteins involved in substrate transport across the membrane. The transported substrates are quite diverse, ranging from monatomic ions to large biomolecules. Consequently, some ABC transporters are involved in biomedically relevant situations, from genetic diseases to multidrug resistance. The most conserved domains in ABC transporters are the nucleotide binding domains (NBDs), which form a dimer responsible for the binding and hydrolysis of ATP, concomitantly with substrate translocation. To elucidate how ATP hydrolysis structurally affects the NBD dimer, and consequently the transporter, we performed a molecular dynamics study on the NBD dimer of the HlyB ABC exporter. We have observed a change in the contact surface between the monomers after hydrolysis, even though we have not seen dimer opening in any of the five 100 ns simulations. We have also identified specific regions that respond to ATP hydrolysis, in particular the X-loop motif of ABC exporters, which has been shown to be in contact with the coupling helices of the transmembrane domains (TMDs). We propose that this motif is an important part of the NBD-TMD communication in ABC exporters. Through nonequilibrium analysis, we have also identified gradual conformational changes within a short time scale after ATP hydrolysis.
Keywords: exporters, nucleotide-binding domains, conformational changes, molecular modeling, subtraction technique
Introduction
The transport across biological membranes is an essential function to the cell. ABC transporters are a large and important protein family performing that function (their genes accounting for 4.9% of the Escherichia coli genome1), capable of transporting a myriad of molecules through the lipid membrane against the concentration gradient, while coupled with the binding2 and hydrolysis of ATP.3 Their importance is well evidenced by the plethora of medical issues associated with them, from genetic diseases4 to pathogenesis5 and multidrug resistance in several organisms6 (particularly in cancer cells7). Therefore, a better understanding of how they function would be of undeniable value.
ABC transporters are classified as a superfamily of membrane proteins8 composed by four main domains [Fig. 1(A)]: two transmembrane domains (TMDs), through which the substrate is transported, and two nucleotide binding domains (NBDs), responsible for the ATP binding and hydrolysis; other domains may be present. Although the NBDs have several conserved sequence motifs [Fig. 1(B)], such as the Walker A and LSGGQ motifs that sandwich the nucleotide or the Walker B and H-loop motifs, suggested to participate in ATP hydrolysis,10 the TMDs can vary considerably in sequence, length and number of helices, despite always presenting coupling helices that interact with the NBDs. Furthermore, the NBDs possess a conserved fold, containing one helical and one catalytic sub-domains [Fig. 1(B)], and a conserved dimer topology in full-length transporters structures11 that suggests a common mechanism of transport for all ABC transporters. Nonetheless, the currently known structures of complete transporters allow us to identify three apparently unrelated TMD folds,12 which could hint on different mechanisms for different groups of transporters. Moreover, unlike ABC importers, ABC exporters present contacts between a coupling helix of the TMD and the opposing NBD, as well as a rather conserved region which is in contact with the coupling helices of the TMDs,13 the X-loop [TIVGEQG in HlyB; purple in Fig. 1(B)]. A controversial issue is the NBD dimer arrangement during the ATP hydrolysis catalytic cycle. Structures of full transporters have been determined in states equivalent to some of the possible catalytic steps of the cycle, where the NBD dimer was found in a closed arrangement in a mutant ATP-bound state and in an ATP-bound equivalent state (AMPPNP),14,15 and in an open arrangement in a nucleotide-free state.15–18 The controversy here starts with the physiological relevance of a nucleotide-free state in an ATP-saturated environment,12 keeping also in mind that the NBD dimer was still found in a closed arrangement in the intermediate ADP-bound state of Sav1866 exporter.13 Further studies on this subject using spin-labeled electron paramagnetic resonance on the MsbA exporter have also shown contradictory results. There is a study reporting NBD distances that differ between states, supporting the open dimer nucleotide-free state in crystallographic studies,19 whereas another study suggests that the LSGGQ region remains buried within the NBD structure throughout the ATP hydrolysis catalytic cycle, which is contrary to the existence of an open arrangement.20 Additionally, there are simulation results showing the opening of the NBD dimer in the nucleotide-free state,21 whereas separate simulations of posthydrolysis states with ADP or ADP and inorganic phosphate (IP) have contradictorily shown both a partially opened arrangement22–24 and a closed arrangement.25 Despite this abundance of biochemical, structural, and simulation data already available, the mechanism of transport by these proteins has not yet been clarified, even though some models have been proposed.26–28 More importantly, the structural details of how ATP hydrolysis in the NBDs is coupled to the mechanism of substrate transport by the TMDs are still unknown.
Figure 1.
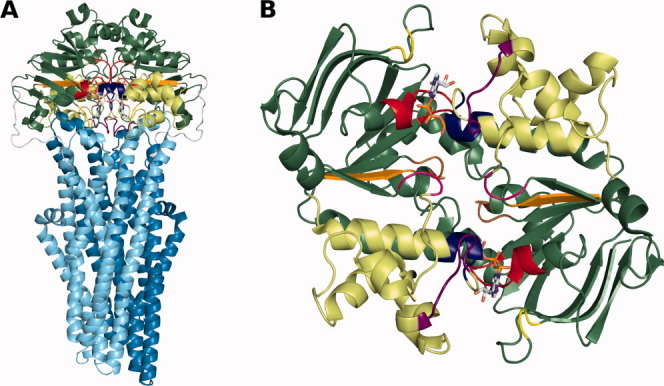
General representation of Sav1866 full transporter and a bottom view of the HlyB NBD dimer with the NBD conserved motifs highlighted. A: The Sav1866 full transporter (PDB code: 2HYD) is shown in cartoon representation. The ATP is in stick representation. Each TMD monomer is in cyan or marine blue and the NBDs are in green, with the helical sub-domains in a lighter green than the catalytic sub-domains. The NBD conserved motifs are shown in the following color scheme: Walker A, red; Walker B, orange; LSGGQ motif, blue; Q-loop, magenta; H-loop, brown; A-loop, yellow; X-loop, purple. B: The HlyB NBD dimer (PDB code: 2FGK) is represented in the same color scheme as in (A). This figure was prepared with PyMOL.9 [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The determination of structures of full transporters in other stages of the transport cycle has been suggested as a possible solution for unveiling this mystery,11,12 but as these states have been difficult to obtain, molecular modeling and simulation techniques29 emerge as an alternative to study and characterize them, as has been shown in previous works on posthydrolysis states.22–25,30 In two of them,24,25 we have studied several possible steps of the catalytic cycle of ATP and identified how each of them affected the structure of MJ0796 NBDs, and consequently the TMDs, or the structure of Sav1866 full transporter. Because this kind of studies have been focused on ABC importers mainly, in this work, we used molecular dynamics simulations to study the NBDs of HlyB, a bacterial ABC exporter responsible for transporting haemolysin A,31 both in prehydrolysis and posthydrolysis states. From these studies, we were able to identify structural differences in the helical sub-domains of the NBDs, particularly in the X-loop, as a result of ATP hydrolysis.
Results and Discussion
The wild-type system is structurally similar to the mutant and shows stability throughout time
Because the HlyB NBD dimer starting structure is from a mutant and we intended to study the wild-type system, a validation of the mutation reversal was necessary. We performed five 10 ns simulations for each system, mutant and wild type, and analyzed the root mean square deviation (RMSD) from the X-ray structure, the root mean square fluctuations (RMSF), and the average structure. This analysis revealed a similar behavior for the two systems (results not shown), as we observed previously for the MJ0796 NBD dimer.25 From this step onward, we could focus only on the wild-type system.
We started by analyzing the conformational drift and stability of the NBD dimer during the simulations. By calculating the Cα RMSD to the X-ray structure throughout time for both the ATP and ADP-IP states (Supporting Information Fig. S1), it was observed that there was little conformational drift after 5 ns of simulation, varying on average no more than 1 Å after that time. Additionally, to assure that the NBD dimer did not lose its native structure, we followed the secondary structure content throughout all simulation time using the DSSP program,32 and we did not observe major loss of structure in any simulation (results not shown).
NBD dimer remains closed in the posthydrolysis state within the simulated time scale
The NBD dimer full opening was first suggested based on the available crystal structures of NBDs, which show dimerization in the presence of ATP, but remain monomeric in its absence.33 In fact, dimer full opening is taken as an important step in the transport mechanism in some proposed models,27,34 with its open–closed alternation directly coupled with a change in the inward–outward orientation of the TMDs. As mentioned before, there is controversy around NBD dimer opening during the ATP hydrolysis catalytic cycle. Regardless of that controversy, there must be some sort of conformational change in the NBD dimer that allows the ADP substitution for ATP to restart the catalytic cycle, possibly arising right after the ATP hydrolysis. To elucidate this question, we calculated the distances between the monomers, and between opposite helical and catalytic sub-domains. We observed that distances between NBD monomers and its sub-domains are maintained in both the ATP and ADP-IP states (Supporting Information Fig. S2), showing no evidence of either dimer full or partial opening. To further investigate the effect of hydrolysis in possible opening of the dimer through an effect on the interaction of the monomers, we calculated the contact surface between monomers for each sampled conformation, in both ATP and ADP-IP states. This contact surface is the difference between the sum of the solvent accessible surfaces of the two isolated NBD monomers and the solvent accessible surface of the NBD dimer. In this approach, instead of looking at the property in a time dependent manner, we took all the conformations from the last 90 ns of each simulation and looked at the property in the ensemble of conformations by building a histogram, in this case, of the contact surface for each state (Fig. 2). It is clear that the pattern of the ATP state [Fig. 2(A)] is different from the ADP-IP state [Fig. 2(B)]. In fact, the average total contact surface increases from 33.78 ± 0.63 nm2 in the ATP state to 35.44 ± 2.48 nm2 in the ADP-IP state. This is consistent with the calculation of distances mentioned above, and contrary to dimer opening. Interestingly, the pattern observed in the histogram and the difference in the standard deviations show that, even though the contact surface increases on average, there is broader contact flexibility in the ADP-IP state than in the ATP state, suggesting that ATP “glues” the monomers together. Both distances and contact surface calculations are in agreement with our previous results for MJ079625 and disagree with other studies with MJ079623 and with results for MalK.22 The situation may be different in the simulation of full length transporters; in our recent study on Sav186624 inserted into a simulated bilayer, despite that most replicates did not show any dimer separation, this event was seen in one of the simulations. Note that the simulation time in this study is longer than in all the mentioned previous studies (100 ns vs. ∼20 to 70 ns22,23,25,30), with an equal or larger number of simulations, which should also provide better sampling. We should also keep in mind that some conditions in these previous works could promote or accelerate dimer opening, like the high temperature and pressure23 or the presence of regulatory domains.22 The presence of the complete protein can also have an effect on this process, and the use of simpler models, such as the NBD dimers may not capture all the features of this phenomenon. Additionally, we cannot exclude at this stage of force field development, that some of these results may be force-field dependant.35,36 Finally, we cannot rule out that this difference in behavior could be due to differences in mechanism for these transporters (MJ0796, MalK, HlyB, and Sav1866), particularly between exporters and importers, even though we cannot see a pattern of behavior in either of the groups. The hydrolysis step simulated in this work could be just a precursor for dimer opening in longer time-scales or in the following steps of the mechanism.
Figure 2.
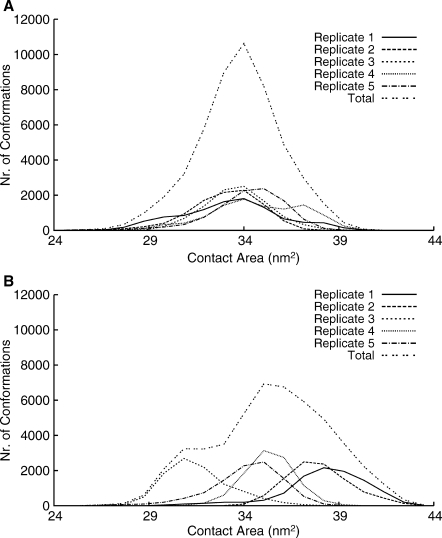
Histograms of the contact surface between the NBD monomers. The contact surface was calculated as the difference between the sum of the solvent accessible surfaces of the two isolated NBD monomers and the solvent accessible surface of the NBD dimer. A: ATP state. B: ADP-IP state.
One final remark is that this result shows the importance of replicate simulations in better exploring different regions of the conformational space. For example, if we had just the first replicate, we would conclude that contact surface increased from the ATP to the ADP-IP state, whereas if we had just the third replicate, we would conclude the opposite.
Outward and inward movements in response to ATP hydrolysis
To identify major differences between the ATP and ADP-IP states in specific zones of the dimer, for each of these states, an average structure was obtained for each individual simulation from the conformations of the last 90 ns, as well as the global average structure [Fig. 3(B) and Supporting Information Movie S3]. Additionally, the RMSD per residue of each ADP-IP state average structure relative to the corresponding replicate ATP state average structure was also calculated [Fig. 3(A)]. When comparing the average structures, it can be seen that the X-loop motif and the α3, α4, and α5 helices, all in the helical sub-domain, move outwards in both NBD monomers in the posthydrolysis state. These changes in the helical sub-domain are also reflected in the RMSD plot, where it is clearly the region most affected by ATP hydrolysis, as is the A-loop. Although using an ADP-only state (no IP), Jones and George observed a rotation of the entire helical sub-domain.30 We have also observed in two previous works24,25 a rotation and higher RMSD of the helical sub-domain. The conformational change here observed is consistent with our previous results and only consistent to a lesser magnitude (and limited to a smaller region in the helical sub-domain) with the results from Jones and George. We must here restate that the higher pressure and temperature used in these simulations might explain the difference in the results. Regarding the comparison with crystallographic studies, our results are also consistent with HlyB crystallographic studies,37 but are distinct from the crystallographic studies of the MJ0796 and MJ1267 NBDs,33,38,39 where, accompanying the rotation of the helical sub-domain, there is also a outward movement of the LSGGQ motif, which we do not observe. Perhaps the simulation of other systems representing states further into the catalytic cycle (such as states without IP) could allow us to see more extensive modifications. Furthermore, there is an inward movement of the A-loop in both monomers, even though the changes in the catalytic sub-domain are not as symmetrical as in the helical one. The effect of these conformational changes may be extrapolated to the TMDs through a structural alignment with the X-ray structure of the Sav1866 full transporter,13 which is an obvious choice due to fact that it is an exporter like HlyB. We can see from the alignment [Fig. 3(C) and Supporting Information Movie S4] that the above mentioned zones are facing the TMDs and move away from it upon ATP hydrolysis. The X-loop, and in particular the exporter conserved glutamate (Glu473 in Sav1866, Glu601 in HlyB), was shown to be in contact with the coupling helices of the TMDs,13 so the fact that it moves away from the TMD may implicate a conformational rearrangement of the coupling helices and consequently of the TMDs.
Figure 3.
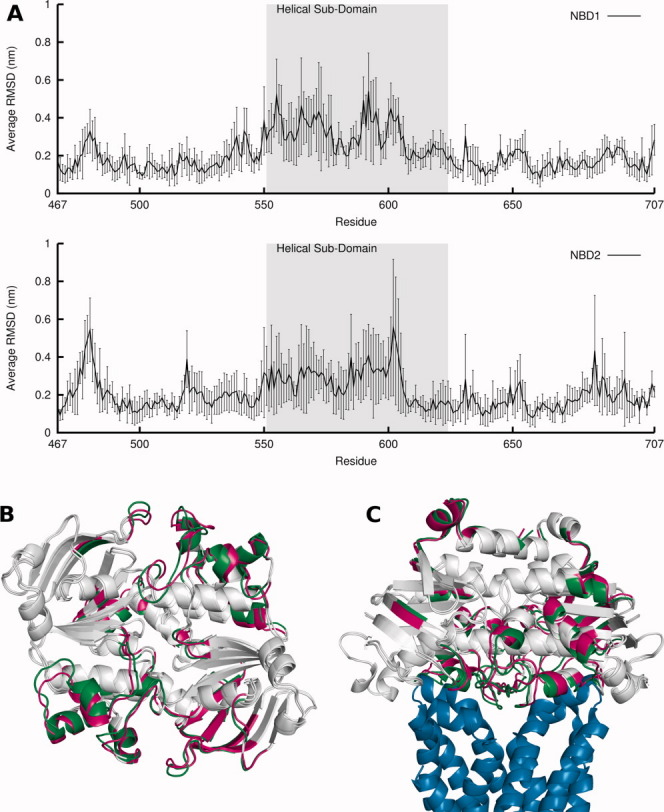
Comparison between ATP and ADP-IP states: RMSD, average structures and relation to the TMDs. A: Average RMSD per residue over all replicates of ADP-IP state in relation to ATP state. The RMSD calculation was done for each ADP-IP state replicate average structure in relation to the average structure of the ATP state replicate from which the ADP-IP state simulation started from, and was averaged over all five simulations, with the standard deviations of the means represented in vertical lines. The helical sub-domain is highlighted in gray. B: Aligned global average structure over all simulations for ATP and ADP-IP states. The average structures are in cartoon representation and in a bottom view. The ATP and ADP-IP state average structure is colored in green and magenta, respectively, with zones that have less than 0.1 nm difference in RMSD between states colored in gray. C: Same as (B), but now aligned with Sav1866 TMDs (in blue) and in a side view. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Glu601 of X-loop fluctuates more in the posthydrolysis state
The previous analysis gave us the average conformational displacement of the ADP-IP state relative to the ATP state. However, this disregards the dynamic information that the used methodology gives us and that would allow us to know if the studied states present a similar flexibility or not. This flexibility is here measured through the RMSF of each residue and was calculated once again for the last 90 ns of each simulation.
Each replicate simulation of each state can be considered a different set of data from which we can calculate the RMSF—the individual RMSF—which will measure the flexibility within that particular replicate of that state (see Supporting Information Fig. S5), and from these individual RMSFs, we can calculate the average RMSF [Fig. 4(A)]. When comparing the average RMSF of each state, no major difference in flexibility between states is observed. Still, if we look at individual RMSFs, there are some differences between states, even though no clear pattern emerges. In fact, the flexibility in the helical sub-domain is very variable between replicates of the same state. To further investigate flexibility, we can consider all replicate simulations of each state as a global set of data from which we can calculate the RMSF—the global RMSF—which will measure the global flexibility in that state [Fig. 4(B)]. From the comparison between the global RMSF of each state, a clear difference stands out: in the region of the X-loop, a motif present in ABC exporters, there is a higher global RMSF in the ADP-IP state than in the ATP state, especially Glu601 (equivalent of Glu473 of Sav1866), which shows consistent global RMSF differences between states in both NBD monomers. That glutamate was shown to interact with both coupling helices of the TMDs in Sav1866,13 so a higher flexibility in the posthydrolysis state may mean a higher destabilization of that interaction and a possible pathway of ATP-hydrolysis/substrate-transport coupling, at least in ABC exporters. In a previous simulation work with Sav1866,24 we have observed the formation of a cytoplasmic gate entrance near the X-loop region in response to hydrolysis, hypothetically meaning that X-loop may be important for substrate accommodation inside the TMD cavity. The global RMSF results from two contributions: the average RMSF [Fig. 4(A)] and the dispersion of the average structures over replicates. Because the average RMSF does not show major differences (as mentioned above), the differences of global RMSF in the X-loop region must arise from the dispersion term. In fact, if one compares the average structures of each replicate simulation, there are differences in the X-loop region (results not shown). We must notice once again the importance of replicate simulations in the observation of these results.
Figure 4.
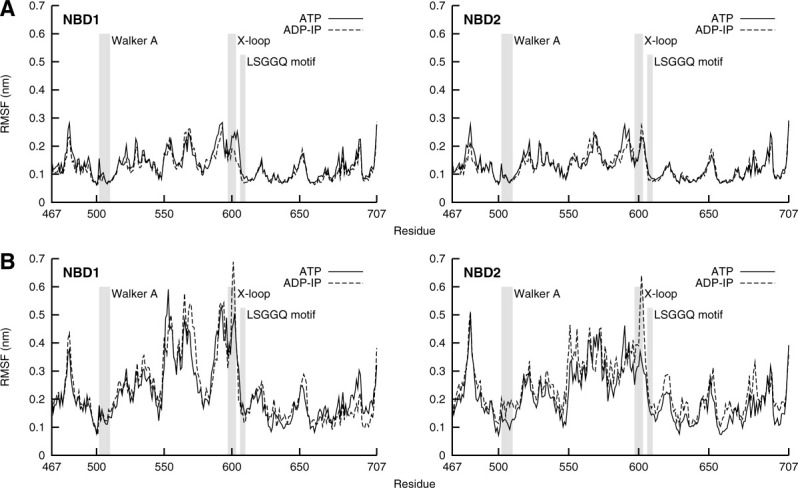
Conformational flexibility of ATP and ADP-IP states assessed with average and global RMSF. Walker A, X-loop and LSGGQ motif are highlighted with gray shadows in the plots. A: Average RMSF, averaged over replicates individual RMSFs of each state (Supporting Information Fig. S5). B: Global RMSF, calculated over conformations from all replicate simulations of each state.
Nonequilibrium analysis shows gradual and slow conformational changes
Besides the equilibrium analysis, we also performed a nonequilibrium analysis to learn more about the fast conformational events that occur right after ATP hydrolysis takes place. This particular analysis will allow us to have a better understanding of the time dependence of these conformational events by canceling the noise coming from the intrinsic fluctuations of the system, allowing us to observe the direct response of the dimer to the perturbation event and possibly understand the underlying mechanism in a reliable time-dependent manner. In practice, we used the subtraction technique to perform RMSD calculations between the ATP and ADP-IP states in the 100 ps after the hydrolysis. In the first picosecond (Fig. 5, Supporting Information Fig. S8 and Movie S9), the Walker A and LSGGQ motif have a higher RMSD than any other part of the NBDs, showing an immediate conformational rearrangement in response to a change in the molecule they bind. These regions change very little in the following picoseconds, especially Walker A, whose RMSD in the first picosecond is comparable to the results found in the long simulations. We assessed the integrity of the binding pockets during the hydrolysis, as well as the interactions between the nucleotides and the residues of the binding pockets, through the calculation of the average structure for each picosecond (Supporting Information Fig. S6 and Movie S7). The hydrogen bonds between the Ser504, Ser506, Thr510, Ser607, and His662 residues, and the phosphate moiety of the nucleotides are lost during the first 5 ps, only to be regained after the ADP and IP are fully grown. In contrast, the interaction between Ser509 and the magnesium ion is lost after hydrolysis, not to be formed again. In the picoseconds that follow the growth of ADP and IP, we observe a gradual RMSD increase in some regions, particularly the X-loop, A-loop, and α3-α4 loop (Fig. 5, Supporting Information Fig. S8 and Movie S9). These changes are not transient, as they can be seen in the long simulations and in the comparison of average structures [Fig. 3(B)], and are cumulative. The conformational changes observed within the 100 ps time scale are still far from those observed in the long simulations, especially in the helical sub-domain region, meaning that much conformational rearrangement still occurs long after hydrolysis, and that these processes are slow. Surprisingly, the α3-α4 loop, which is relatively distant from the binding site, responds to ATP hydrolysis quite rapidly. More generally, some of the more structured and core regions of the NBD present low conformational change in response to hydrolysis, and we propose they could function as transmission rigid regions to other particular regions of the protein. To sum up the mechanistic events, the binding pocket suffers conformational rearrangements right after hydrolysis, which are afterwards transmitted to specific regions of the NBD dimer, gradually and cumulatively, probably through regions that are almost structurally unaffected by the hydrolysis.
Figure 5.
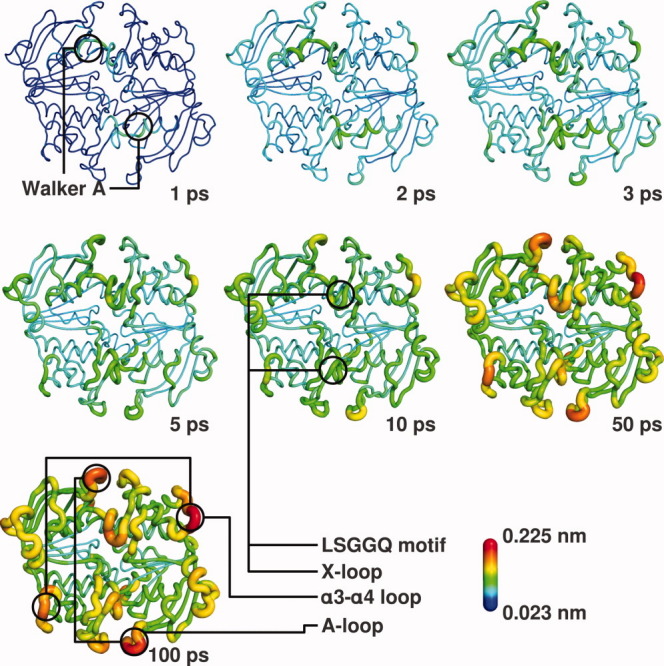
Mapping of the RMSD in the 100 ps following ATP hydrolysis on the NBD structure. The RMSD values at specific times (1, 2, 3, 5, 10, 50, and 100 ps) after ATP hydrolysis (see Supporting Information Fig. S8) are mapped on the average (over the 100 replicates) perturbed HlyB structure at that time, in a color scheme according to the presented scale. Cartoon sausage thickness is also related to RMSD values for additional clarity. ADP and IP are in stick representation. Some important motifs of the structure are highlighted and labeled. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Concluding Remarks
ABC transporters have been largely studied during the past decades but there is still much to know about their mechanism of transport. In this work, we intended to bring light to some of the most controversial issues in structural studies of ABC transporters through the simulations of the NBD dimer of a known exporter, HlyB. Although we could not exclude the possibility of NBD dimer opening as a result of ATP hydrolysis, we did not observe it within the simulated time scale of any of the replicate simulations. Nonetheless, we observed differences in the contact surface between the monomers after hydrolysis, which could be a possible precursor of a full or partial opening event. Nonetheless, more simulation and structural studies of other known ABC transporters are essential to a complete understanding of this process. We have also observed an outward movement of some regions of the helical sub-domain after ATP hydrolysis, showing that this is the region that experiences most conformational changes in response to hydrolysis. Still, we did not observe an entire rotation of the helical sub-domain. Even though, like in our previous studies on MJ0796,25 the A-loop and the α3-α4 loop regions show significant changes, in the present simulations, the X-loop stands out as the region that is most affected by ATP hydrolysis, shown by average structures comparison, fluctuation analysis, and even in the nonequilibrium analysis of the fast events after the hydrolysis. This motif, and particularly Glu601, may be the cornerstone for the coupling between ATP-hydrolysis and substrate transport in ABC exporters, because it is not present in importers. We suggest that mutation experiments may be carried out on Glu601 (or on equivalent residues in other exporters) by mutating it to a glutamine or alanine, to assess the effect of a change of charge and size of this particular residue on the transport process of ABC exporters.
Materials and Methods
Preparation of HlyB NBD dimer starting structure: E631Q mutant and E631 wild type
The 2.7 Å resolution crystal structure of E631Q mutant HlyB NBD dimer with two bound ATP molecules (PDB code: 2FGK)37 was chosen for this work. One of the dimer structures present in the unit cell was removed (chains C and D) and only the water molecules with less than 50% relative accessibility to the solvent, calculated with the program ASC,40 were kept. The E631Q mutant protein was reverted to the E631 wild-type protein, by in silico mutation of the glutamine to a glutamate. It is known that the wild-type dimer has Mg2+ ions bound to ATP, whereas this mutant has Na+ ions bound instead.33 Because there were no ions modeled in the structure that was chosen, the appropriate type of ions were then added to each structure, with their atomic positions taken from a structural water (atom number 7703) and from a Mg2+ position of another HlyB dimmer structure (after structural alignment with our structure; PDB code: 1XEF).10 We determined the protonation states of each individual group of the protein at pH 7.0 with continuum electrostatic calculations using MEAD (version 1.1.8)41,42 and Monte Carlo sampling of protonation states using PETIT (version 1.3).43 The methodology was described in detail elsewhere.43,44 From the results (data not shown), all glutamate and aspartate amino acids were found deprotonated, all lysines and arginines protonated, and all histidines neutral. The C terminals were found deprotonated and the N terminals protonated. The protonation states of ATP, ADP, and IP were the same as used in Oliveira et al.,25 meaning that we have chosen the single protonated state for IP 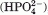 , and protonation states for ATP and ADP that correspond to an overall charge of −3 and −2, respectively.
, and protonation states for ATP and ADP that correspond to an overall charge of −3 and −2, respectively.
General setup of the molecular dynamics simulations
For the molecular mechanics description of our systems, we used the 43A1 GROMOS96 force field45,46 and the SPC water model.47 ATP, ADP, and IP parameters were the same as in Oliveira et al.25 For the molecular dynamics simulations, the GROMACS 3.3.1 package48,49 was used. The simulations were performed using periodic boundary conditions at 300 K and 1 atm, with an integration time step of 0.002 ps. The temperature and pressure were kept constant with Berendsen coupling baths,50 with separate temperature coupling for solutes and solvent. In the production simulations, the pressure coupling constant was 0.5 ps and the heat coupling constant was 0.1 ps. The van der Waals interactions were considered up to 14 Å and the electrostatic interactions beyond 9 Å were treated with a smooth particle mesh Ewald method.51,52 The neighbor lists were updated every 10 steps. All bonds were constrained to their equilibrium lengths with the LINCS algorithm,53 except the water molecules, that were kept rigid with the SETTLE algorithm.54
Molecular dynamics of E631 wild-type prehydrolysis system
The E631 wild-type starting structure was solvated in a rhombic dodecahedral box, considering a minimum distance of 9 Å between the protein and the box walls, ending up with a total of 14,532 water molecules. We performed a steepest descent energy minimization with position restraints of 1000 kJ mol−1 nm−1, consisting of 2000 steps with restraints on all heavy atoms and 2000 steps with restraints on all Cα atoms. From the resulting conformation, we performed 5 molecular dynamics simulations, initiated with different sets of random velocities following a Maxwell–Boltzmann distribution. The simulations started by 200-ps long restrained simulations as previously described,25 followed by 100-ns long unrestrained simulations, summing up to a total of 500 ns of production simulation of the prehydrolysis system (or ATP system; see Supporting Information Fig. S10A). Some E631Q mutant control simulations were performed to assess the effect of the reversion of the mutation on the structure, just as previously described,25 in a total of five simulations of 10 ns each.
ATP hydrolysis using the slow growth method and posthydrolysis molecular dynamics
For the modeling of the ATP hydrolysis, we used a coupling parameter approach55 (which is used in the slow growth method56) for the conversion of ATP to ADP and IP. Note that, with this procedure, we had no intention of calculating the free energy associated with the hydrolysis process or study the process itself. Our intention was to have a protocol that modeled a smooth conversion of the nucleotides with the objective of analyzing the structural effects on the protein. From each 5-ns equilibrated conformation of the prehydrolysis system simulations (see Supporting Information Fig. S10A), we used the slow growth method to convert ATP into ADP and IP. Specifically, the IP molecule atom positions were created from the Pγ and the four oxygen atoms bound to it. Afterward, the IP tetrahedral configuration, centered on phosphor, was rotated so that the oxygen between Pβ and Pγ stayed in the diametric opposite side of Pγ. Then, a proton atom was added to one of the IP oxygens as previously mentioned. The slow-growth conversion process was done during 5 ps of molecular dynamics, with the same conditions as described for the regular molecular dynamics, except for an integration time step of 0.0005 ps and an update of the neighbor lists every five steps. During this process, the force-field parameters of our hybrid system with ATP and IP are coupled to a lambda (λ) parameter. While the λ parameter changes continuously from 0 to 1, the IP nonbonded parameters are being “turned on” and the ATP gamma phosphate bonded and nonbonded interactions are being “turned off.” After this, we performed 100-ns long regular molecular dynamics simulations, summing up to a total of 500 ns of production simulation of the posthydrolysis system (or ADP-IP system; see Supporting Information Fig. S10A).
Nonequilibrium analysis: the subtraction technique method
The long simulations allow us to compare the ATP and ADP-IP systems in equilibrium. Nonetheless, it may be of interest to study the fast events that occur in the short time scale (few picoseconds) after the hydrolysis, during which the ADP-IP system is still in nonequilibrium. However, the small response to ATP hydrolysis occurring at that short time scale could be easily masked by the commensurate fluctuations in the system. To circumvent this problem, we used the subtraction technique,57,58 as we have done previously in another system experiencing conformational changes.59 This approach requires a large number of short simulations to be reliable, so we performed 10 molecular dynamics simulations of the prehydrolysis state, each of them 10 ns long (five of which were prolonged to get the 100-ns long simulations mentioned above; see Supporting Information Fig. S10B). Then, from the 5-ns equilibrated part of each trajectory, we sampled conformations at each 500 ps, covering the whole range of the equilibrated trajectory, to a total of 100 conformations. Each of these conformations served as a starting point for a simulation of the posthydrolysis state, consisting of 5 ps of slow growth molecular dynamics followed by 95 ps of molecular dynamics, as described above. The response of the system to the perturbation (ATP hydrolysis) can then be calculated as the difference of a given property measured in the perturbed and unperturbed states at the same simulation time, averaged over the large number of simulations, giving us the time evolution of the response.
Supplementary material
References
- 1.Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- 2.Higgins CF, Hiles ID, Whalley K, Jamieson DJ. Nucleotide binding by membrane components of bacterial periplasmic binding protein-dependent transport systems. EMBO J. 1985;4:1033–1039. doi: 10.1002/j.1460-2075.1985.tb03735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mimmack ML, Gallagher MP, Pearce SR, Hyde SC, Booth IR, Higgins CF. Energy coupling to periplasmic binding protein-dependent transport systems: stoichiometry of ATP hydrolysis during transport in vivo. Proc Natl Acad Sci USA. 1989;86:8257–8261. doi: 10.1073/pnas.86.21.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottesman MM, Ambudkar SV. Overview: ABC transporters and human disease. J Bioenerg Biomembr. 2001;33:453–458. doi: 10.1023/a:1012866803188. [DOI] [PubMed] [Google Scholar]
- 5.Lee VT, Schneewind O. Protein secretion and the pathogenesis of bacterial infections. Genes Dev. 2001;15:1725–1752. doi: 10.1101/gad.896801. [DOI] [PubMed] [Google Scholar]
- 6.Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deeley RG, Westlake C, Cole SPC. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006;86:849–899. doi: 10.1152/physrev.00035.2005. [DOI] [PubMed] [Google Scholar]
- 8.Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152:211–229. doi: 10.1016/s0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 9. The PyMOL Molecular Graphics System, Version 1.2r1, Schro¨dinger, LLC.
- 10.Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollenstein K, Dawson RJP, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Jones PM, O'Mara ML, George AM. ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem Sci. 2009;34:520–531. doi: 10.1016/j.tibs.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 14.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 15.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol Cell. 2009;33:528–536. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou P, Bortolus M, McHaourab HS. Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J Mol Biol. 2009;393:586–597. doi: 10.1016/j.jmb.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchaklian AH, Klug CS. Characterization of the LSGGQ and H motifs from the Escherichia coli lipid A transporter MsbA. Biochemistry. 2006;45:12539–12546. doi: 10.1021/bi060830a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newstead S, Fowler PW, Bilton P, Carpenter EP, Sadler PJ, Campopiano DJ, Sansom MSP, Iwata S. Insights into how nucleotide-binding domains power ABC transport. Structure. 2009;17:1213–1222. doi: 10.1016/j.str.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen P-C, Tajkhorshid E. Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys J. 2008;95:5100–5110. doi: 10.1529/biophysj.108.139444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones PM, George AM. Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins. 2009;75:387–396. doi: 10.1002/prot.22250. [DOI] [PubMed] [Google Scholar]
- 24.Oliveira ASF, Baptista AM, Soares CM. Conformational changes induced by ATP-hydrolysis in an ABC transporter: A molecular dynamics study of the Sav1866 exporter. Proteins. 2011;79:1977–1990. doi: 10.1002/prot.23023. [DOI] [PubMed] [Google Scholar]
- 25.Oliveira ASF, Baptista AM, Soares CM. Insights into the molecular mechanism of an ABC transporter: conformational changes in the NBD dimer of MJ0796. J Phys Chem B. 2010;114:5486–5496. doi: 10.1021/jp905735y. [DOI] [PubMed] [Google Scholar]
- 26.Senior AE, al-Shawi MK, Urbatsch IL. The catalytic cycle of P-glycoprotein. FEBS Lett. 1995;377:285–289. doi: 10.1016/0014-5793(95)01345-8. [DOI] [PubMed] [Google Scholar]
- 27.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 28.Sauna ZE, Kim I-W, Nandigama K, Kopp S, Chiba P, Ambudkar SV. Catalytic cycle of ATP hydrolysis by P-glycoproteevidence for formation of the E·S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry. 2007;46:13787–13799. doi: 10.1021/bi701385t. [DOI] [PubMed] [Google Scholar]
- 29.van Gunsteren WF, Bakowies D, Baron R, Chandrasekhar I, Christen M, Daura X, Gee P, Geerke DP, Glättli A, Hünenberger PH, Kastenholz MA, Oostenbrink C, Schenk M, Trzesniak D, van der Vegt NFA, Yu HB. Biomolecular modeling: goals, problems, perspectives. Angew Chem Int Ed Engl. 2006;45:4064–4092. doi: 10.1002/anie.200502655. [DOI] [PubMed] [Google Scholar]
- 30.Jones PM, George AM. Nucleotide-dependent allostery within the ABC transporter ATP-binding cassette: a computational study of the MJ0796 dimer. J Biol Chem. 2007;282:22793–22803. doi: 10.1074/jbc.M700809200. [DOI] [PubMed] [Google Scholar]
- 31.Holland IB, Schmitt L, Young J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review) Mol Membr Biol. 2005;22:29–39. doi: 10.1080/09687860500042013. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 33.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol. 2008;18:726–733. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matthes D, de Groot BL. Secondary structure propensities in peptide folding simulations: a systematic comparison of molecular mechanics interaction schemes. Biophys J. 2009;97:599–608. doi: 10.1016/j.bpj.2009.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lange OF, van der Spoel D, de Groot BL. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys J. 2010;99:647–655. doi: 10.1016/j.bpj.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaitseva J, Oswald C, Jumpertz T, Jenewein S, Wiedenmann A, Holland IB, Schmitt L. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 2006;25:3432–3443. doi: 10.1038/sj.emboj.7601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 2001;9:571–586. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 39.Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF. The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J Biol Chem. 2001;276:32313–32321. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]
- 40.Eisenhaber F, Argos P. Improved strategy in analytic surface calculation for molecular systems: handling of singularities and computational efficiency. J Comput Chem. 1993;14:1272–1280. [Google Scholar]
- 41.Bashford D, Karplus M. pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 42.Bashford D, Gerwert K. Electrostatic calculations of the pKa values of ionizable groups in bacteriorhodopsin. J Mol Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- 43.Baptista AM, Soares CM. Some theoretical and computational aspects of the inclusion of proton isomerism in the protonation equilibrium of proteins. J Phys Chem B. 2001;105:293–309. [Google Scholar]
- 44.Teixeira V, Soares C, Baptista A. Studies of the reduction and protonation behavior of tetraheme cytochromes using atomic detail. J Biol Inorg Chem. 2002;7:200–216. doi: 10.1007/s007750100287. [DOI] [PubMed] [Google Scholar]
- 45.van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH, Kruger P, Mark AE, Scott WRP, Tironi IG. Biomolecular simulation: the GROMOS96 manual and user guide. Zurich, Switzerland: Verlag der Fachvereine Hochschulverlag AG an der ETH Zurich; 1996. [Google Scholar]
- 46.Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Kruger P, van Gunsteren WF. The GROMOS biomolecular simulation program package. J Phys Chem A. 1999;103:3596–3607. [Google Scholar]
- 47.Hermans J, Berendsen HJC, Gunsteren WFV, Postma JPM. A consistent empirical potential for water-protein interactions. Biopolymers. 1984;23:1513–1518. [Google Scholar]
- 48.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 49.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 50.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 51.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 52.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 53.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 54.Miyamoto S, Kollman PA. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 55.Mezei M, Beveridge DL. Free energy simulations. Ann NY Acad Sci. 1986;482:1–23. doi: 10.1111/j.1749-6632.1986.tb20933.x. [DOI] [PubMed] [Google Scholar]
- 56.Kollman P. Free energy calculations: applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 57.Ciccotti G, Jacucci G, McDonald IR. “Thought-experiments” by molecular dynamics. J Stat Phys. 1979;21:1–22. [Google Scholar]
- 58.Allen MP, Tildesley DJ. Computer simulation of liquids. USA: Oxford University Press; 1989. [Google Scholar]
- 59.Oliveira ASF, Teixeira VH, Baptista AM, Soares CM. Reorganization and conformational changes in the reduction of tetraheme cytochromes. Biophys J. 2005;89:3919–3930. doi: 10.1529/biophysj.105.065144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.