

Abstract
Long interspersed nuclear element-1 is a highly abundant mammalian retrotransposon that comprises 17% of the human genome. L1 retrotransposition requires the protein encoded by open reading frame-1 (ORF1p), which binds single-stranded RNA with high affinity and functions as a nucleic acid chaperone. ORF1p has been shown to adopt a homo-trimeric, asymmetric dumbbell-shaped structure. However, its atomic-level structure and mechanism of RNA binding remains poorly understood. Here, we report the results of a site-directed spin labeling electron paramagnetic resonance (SDSL-EPR) study of 27 residues within the RNA binding region of the full-length protein. The EPR data are compatible with the large RNA binding lobe of ORF1p containing a RNA recognition motif (RRM) domain and a carboxyl-terminal domain (CTD) that are predicted from crystallographic and NMR studies of smaller fragments of the protein. Interestingly, the EPR data indicate that residues in strands β3 and β4 of the RRM are structurally unstable, compatible with the previously observed sensitivity of this region to proteolysis. Affinity measurements and RNA-dependent EPR spectral changes map the RNA binding site on ORF1p to residues located in strands β3 and β4 of the RRM domain and to helix α1 of the CTD. Complementary in vivo studies also identify residues within the RRM domain that are required for retrotransposition. We propose that in the context of the full-length trimeric protein these distinct surfaces are positioned adjacent to one another providing a continuous surface that may interact with nucleic acids.
Keywords: retrotransposon, LINE-1, L1, nucleic acid chaperone, RNA-binding, electron paramagnetic resonance spectroscopy, RRM, SDSL-EPR
Introduction
The mammalian long interspersed nuclear element-1 (LINE-1 or L1) is an active retrotransposon that has generated an astounding 28% of the human genome (reviews see Refs.1–7). De novo insertion or unequal homologous recombination between L1 elements in germinal chromosomes has caused a number of diseases and has been implicated in brain development, the regulation of gene expression, and X-chromosome inactivation. Although the majority of L1s are defective due to truncation at the 5′ terminus, it has recently become clear that hundreds of elements remain active in the human population, generating a significant and underappreciated amount of structural and phenotypic variation.8–11 L1 replicates via a target primed reverse transcription (TPRT) mechanism in which the RNA copy of the element is reverse transcribed at the site of genomic insertion.12 Two L1 encoded proteins are required for retrotransposition: open reading frame-1 (ORF1p), which binds single stranded nucleic acids,13,14 and ORF2p, which has endonuclease and reverse transcriptase functions.12,15,16
L1 retrotransposition requires ORF1p, which has at least two functions.17 First, it binds to L1 RNA during the cytoplasmic phase of the replication cycle and may prevent its degradation by cellular nucleases and the RNA interference pathway.18 Second, ORF1p functions as a nucleic acid chaperone; the isolated protein accelerates strand annealing, promotes melting of mismatched duplexes, and facilitates strand exchange.14 Thus, ORF1p directly participates in the TPRT reaction by facilitating nucleic acid strand transfers and by disrupting RNA structures that would impede the progress of ORF2p. The structure of ORF1p from the mouse L1 retrotransposon has been studied in the greatest detail. It forms a trimeric asymmetric dumbbell-shaped structure in which the polypeptide chains are arranged in parallel.19 Based on proteolysis sensitivity, ORF1p was proposed to contain three domains20: (1) a coiled coil domain, (2) a middle domain (M-domain), and (3) a carboxyl-terminal domain (CTD). The coiled-coil domain allows the protein to trimerize—presumably forming the dumbbell handle—whereas the M-domain and CTD form the larger lobe that binds RNA.
The structure of the larger lobe responsible for binding RNA is controversial. In the absence of RNA, a ∼50 amino acid long segment located between the M-domain and CTD of mouse ORF1p was found to be highly sensitive to proteolysis [referred to as the “linker,” Fig. 1(A)], and proposed to be structurally disordered.20 Thus, the full-length trimeric protein may adopt a “hydra-like” structure in which each CTD is connected to the body of the protein via a structurally disordered linker. However, a recent crystal structure of a monomeric fragment from the homologous human ORF1 protein, which corresponds to residues from the M-domain and linker, suggests that the linker is ordered, forming part of an autonomously folded, RNA recognition motif (RRM) type structure.23
Figure 1.
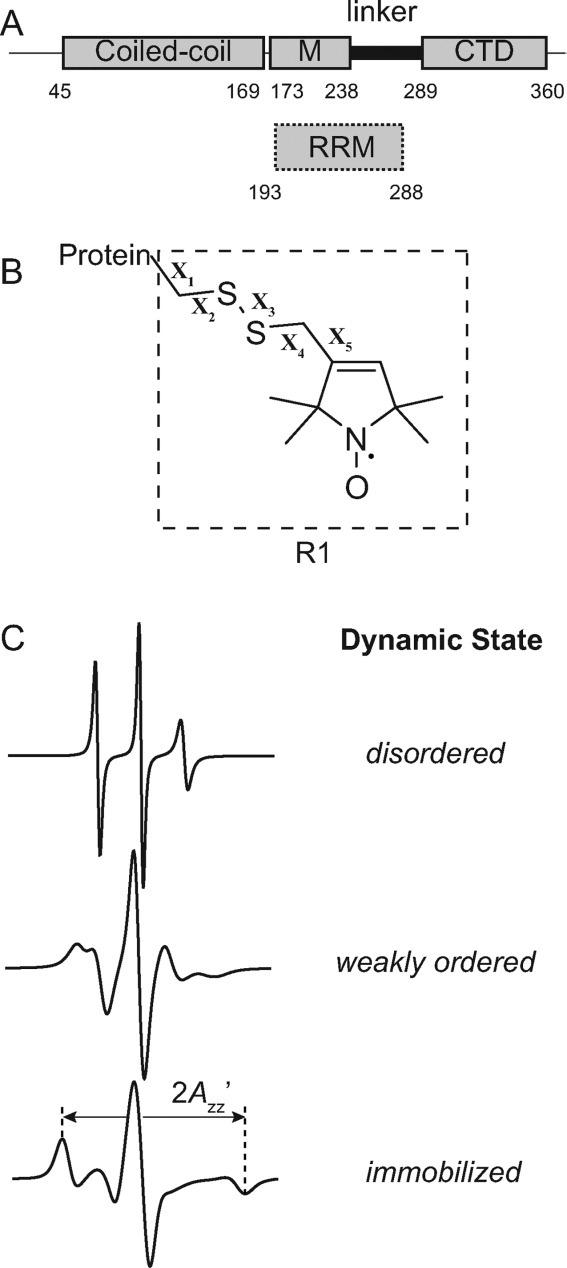
Schematic of ORF1p, the R1 spin label, and representative EPR spectra. (A) The top image displays a schematic of the ORF1p protein from the mouse L1 retrotransposon. Regions of the protein previously shown to be partially resistant to trypsin digestion are indicated by rectangles and correspond to a coiled-coil region, a middle region (M) and a C-terminal domain (CTD). The segment connecting the CTD and middle region is susceptible to trypsin proteolysis and is referred to as the linker. The lower image indicates where the presumed RNA recognition motif (RRM) binding domain is located based on the recently determined crystal structure of this domain from the human ORF1p protein (hORF1p). The amino acid number defining the beginning and end of each element is indicated. (B) Structure of the R1 side chain showing the dihedral angle designations (X1–X5). (C) Simulated EPR spectra corresponding to three fundamental dynamic modes of the R1 side chain in proteins: disordered (top trace), weakly ordered (middle trace), and immobilized (lower trace). Simulations were carried out using the NLSL.MOMD program21 available at (http://www.acert.cornell.edu/index_files/acert_ftp_links.php). In each case, the order parameter (S) and correlation time (τ) for the motion are given in the format {S,τ}. Details of spectral simulations are presented by Columbus et al.22
To better define the structure and dynamics of the RNA-binding region within ORF1p, site-directed spin labeling (SDSL) was used to study the linker region by electron paramagnetic resonance (EPR) spectroscopy. SDSL has emerged as a useful approach for investigating protein structure and dynamics by EPR spectroscopy.24–27 The technique is not limited by the size of the protein and has the potential to reveal both local backbone dynamics and conformational dynamics. In SDSL, a unique cysteine is introduced into a protein by site-directed mutagenesis, and then subsequently modified with a sulfhydryl-specific reagent to generate a covalently linked nitroxide side chain. The most commonly used spin label is designated R1 [Fig. 1(B)], which was used in this study.
The EPR spectrum of an R1 spin labeled protein reflects the motion of the nitroxide on the nanosecond timescale. When R1 is introduced into structured elements of the protein, interactions between the spin label side chain and the local protein environment restrict nitroxide motion, and the resulting EPR spectrum can reflect a weakly ordered or immobilized state of R1.28,29 In contrast, when R1 is located at a site within a sequence that is dynamically disordered on the nanosecond timescale, the narrow lines of the EPR spectrum reflect essentially fast isotropic motion.30,31 Hence, by scanning R1 through the linker region of ORF1p, the distinctive features of the EPR spectra can be used to qualitatively assess whether or not this region is ordered in the context of the full-length protein and determine the structural topology. In addition, regions of the protein where local structural changes occur upon RNA binding can be identified.
Simulated, single component EPR spectra corresponding to representative examples of disordered, weakly ordered, and immobilized states of R1 are shown in Figure 1(C). A disordered state of R1 suggests that the substituted residue resides in an unstructured part of the protein, whereas a weakly ordered or immobilized state indicates that the native residue is solvent exposed or buried in the hydrophobic core, respectively. However, the EPR spectra of R1 spin labeled proteins are often multicomponent (i.e., linear combinations of these single component spectra) and, as will be shown, such spectra were observed for nearly all spin labeled ORF1p mutants studied. Even in this case, the topology of the native residue can still be predicted from the qualitative features present in the EPR spectrum, and further details regarding the qualitative level of analysis of nitroxide EPR spectra used in the present communication are provided by Crane et al.32 and Kusnetzow et al.33
The EPR data reported here are consistent with the large RNA binding lobe of mouse ORF1p containing a RRM domain and a CTD. Interestingly, the data indicate that the C-terminal end of the RRM is structurally unstable, compatible with the sensitivity of this region to proteolysis. RNA-dependent EPR spectral changes, affinity measurements, and an in vivo retrotransposition assay suggest that residues located in strands β3 and β4 of the RRM domain and helix α1 of the CTD are important for RNA binding. From these data and the previously determined structures of the human RRM and mouse CTD proteins, we propose a model for ORF1p RNA binding.
Results
RNA binding properties of ORF1p mutants
A total of 27 single cysteine mutants were generated to introduce R1 at selected sites in the RNA binding region of the full-length protein. Of the selected sites, 21 reside in the protease sensitive segment of the putative RRM domain, and six are located in the CTD [Fig. 1(A)].
The RNA binding affinity of each cysteine mutant was quantitatively determined using a double-filter nitrocellulose assay. For this study, a 60-mer RNA molecule, comprising nucleotides 4330–4390 of the mouse Tf-5 L1 element, was used because it was previously shown to bind ORF1p with high affinity.34 To quantitatively interpret the binding data, the stoichiometry of the wild-type protein-RNA complex was determined by performing the assay with approximately equimolar concentrations of each component [Fig. 2(A)]. These data indicate that 1.2 ORF1p trimers interact with the 60-mer, which is consistent with a previous study that concluded that each trimeric subunit engages ∼17 nucleotides of RNA.35
Figure 2.
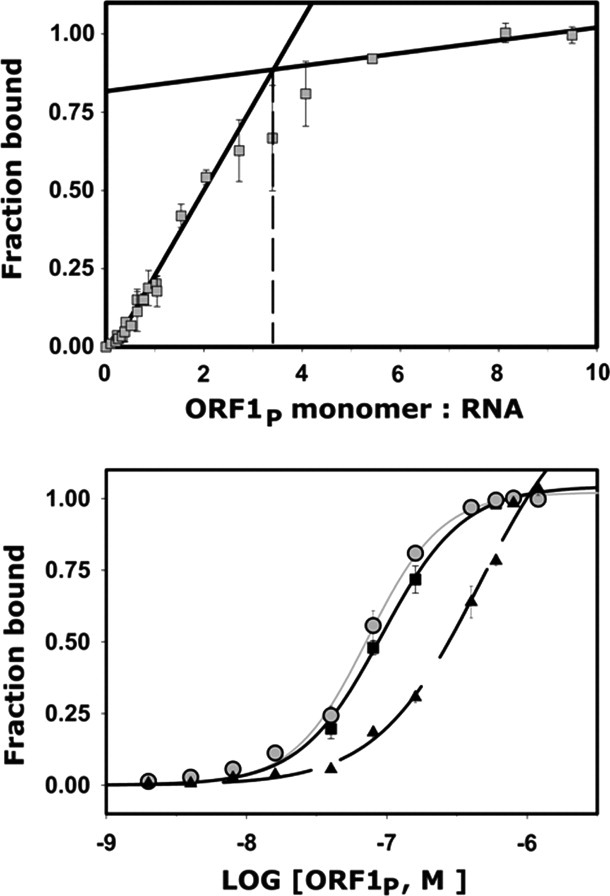
Representative RNA binding data from the full length and mutant ORF1p proteins. (A) Binding assay used to determine the stoichiometry of the complex between full-length ORF1p and a 60-mer L1 RNA molecule. The results of a double filter nitrocellulose binding assay using near equal molar concentrations of each component is shown. The black lines plot the linear regression for data points in the linear and saturation phases of the binding curve. Extrapolation (dotted lines) revealed ∼1.1 ORF1p trimers bind to a single RNA molecule. (B) Representative RNA binding assays of wild-type and mutant ORF1p proteins. The strength of RNA binding to a 60-mer L1 RNA was tested. Shown is data for: wild-type ORF1p (black squares, black line), and Arg256Cys/Cys174Arg (gray circles, gray lines), and Asn262Cys/Cys174Arg (black triangles, black dashed line) mutants. The data were fit to a binding model that assumed 1:1 stoichiometry and yielded KD values of 92 ± 9 nM, 47 ± 16 nM, and >1500 nM, respectively. A complete list of the binding data is presented in Table I.
For the wild-type protein, fitting of the binding isotherm data yielded a dissociation constant (KD) of 92 ± 9 nM (Table I). Representative quantitative binding data for the wild-type protein and two mutants are shown in Figure 2(B), the dissociation constants are given in Table I, and the effects on binding are summarized in Figure 3. In general, the cysteine mutants bind the 60-mer RNA with similar affinities to the wild-type protein (i.e., ≤ threefold increase in the KD). However, three mutants are significantly impaired in their binding ability (i.e., Arg251Cys, Arg256Cys, and Arg284Cys), as these proteins exhibit at least a 15-fold reduction in affinity compared with the wild-type protein and fail to saturate the RNA molecule at high concentrations. Thus, these residues are likely to be important for RNA binding (see Discussion). A NMR chemical-shift perturbation study of the isolated CTD also implicated several residues in RNA binding.20 Therefore, selected residues on the surface of the CTD were individually mutated to alanine, and the ability of each mutant to bind RNA was assessed by the aforementioned assay. All of the alanine mutants bind RNA with affinities that are within threefold of the wild-type protein (Table I and Fig. 3).
Table I.
RNA Binding Affinities of Full-Length Mouse ORF1p
| Proteina | KD (nM)b | Fold increasec | Locationd |
|---|---|---|---|
| Wild-type ORF1p | 92 ± 9 | — | |
| S250C/C174R | 151 ± 21 | 1.6 | RRM (β2-β3 loop) |
| R251C/C174R | >1500 | >15 | RRM (β2-β3 loop) |
| I255C/C174R | 80 ± 5 | 0.9 | RRM (β3) |
| R256C/C174R | >1500 | >15 | RRM (β3) |
| N259C/C174R | 49 ± 8 | 0.5 | RRM (β3-α2 loop) |
| N262C/C174R | 47 ± 16 | 0.5 | RRM (α2) |
| K263C/C174R | 71 ± 5 | 0.8 | RRM (α2) |
| D264C/C174R | 40 ± 5 | 0.4 | RRM (α2) |
| I266C/C174R | 139 ± 31 | 1.5 | RRM (α2) |
| L267C/C174R | 562 ± 104 | 6.1 | RRM (α2) |
| K268C/C174R | 38 ± 18 | 0.4 | RRM (α2) |
| A269C/C174R | 523 ± 127 | 5.7 | RRM (α2) |
| R271C/C174R | 151 ± 21 | 1.6 | RRM (α2) |
| K273C/C174R | 357 ± 140 | 3.9 | RRM (α2) |
| Q275C/C174R | 68 ± 16 | 0.7 | RRM (α2-β3′ loop) |
| V276C/C174R | 128 ± 31 | 1.4 | RRM (β3′) |
| Y278C/C174R | 202 ± 49 | 2.2 | RRM (β3′) |
| K279C/C174R | 86 ± 10 | 0.9 | RRM (β3′-β4 hairpin) |
| I283C/C174R | 135 ± 5 | 1.5 | RRM (β4) |
| R284C/C174R | >1500 | >15 | RRM (β4) |
| I285C/C174R | 127 ± 55 | 1.4 | RRM (β4) |
| F289A | 234 ± 11 | 2.5 | CTD (pre α1) |
| E292A | 84 ± 6 | 0.9 | CTD (α1) |
| M294A | 145 ± 7 | 1.6 | CTD (α1) |
| A296C/C174R | 275 ± 7 | 3 | CTD (α1) |
| R308A | 111 ± 4 | 1.2 | CTD (α1) |
| R315A | 93 ± 8 | 1 | CTD (β1) |
| L317A | 139 ± 34 | 1.5 | CTD (β1-β2 loop) |
| Y318C/C174R | 304 ± 111 | 3.3 | CTD (β1-β2 loop) |
| A320C/C174R | 121 ± 50 | 1.3 | CTD (β1-β2 loop) |
| K331C/C174R | 130 ± 14 | 1.4 | CTD (β3) |
| F333C/C174R | 127 ± 37 | 1.4 | CTD (β3) |
| E335A | 141 ± 38 | 1.9 | CTD (β3) |
| Y359C/C174R | 41 ± ND | 0.4 | Post CTD |
The wild-type protein is the ORF1p from the Tf5 type mouse L1 retrotransposon.
The dissociation constant (KD) is the average value determined from three double-filter binding experiments. The error is the standard deviation of these measurements, unless otherwise indicated (ND, not determined). The affinity of the protein for a 60 nt 32P-labeled RNA from the L1 retrotransposon RNA was measured. Accurate binding affinities cannot be determined for the mutants R251C, R256C, and R284C, but each must bind RNA >15-fold less tightly than the wild-type protein with a KD that exceeds ∼1500 nM.
The fold decrease is the RNA affinity of the mutant relative to the wild-type protein.
Figure 3.
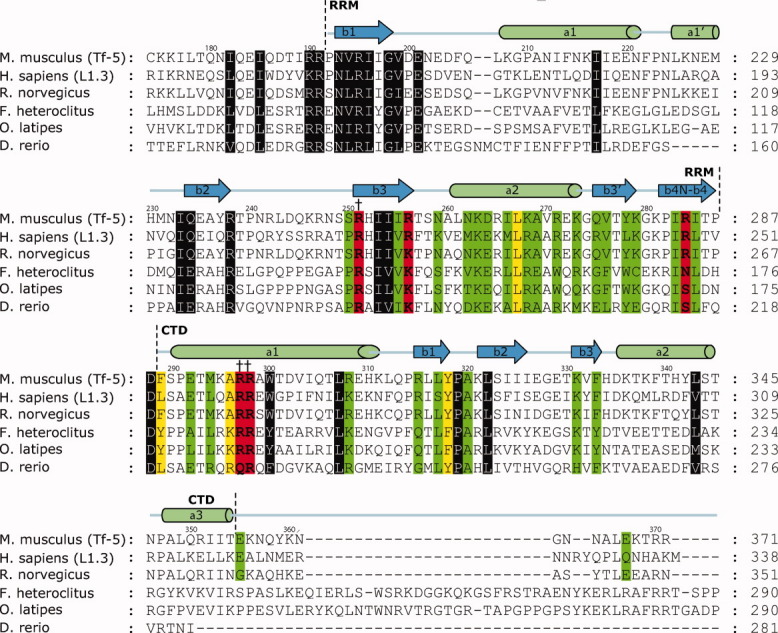
Primary sequence alignment of ORF1 proteins and a summary of the RNA binding and in vivo results. The alignment of amino acids of ORF1p contains the RRM identified by crystallography and the CTD identified by NMR. Amino acids that were mutated to cysteine or alanine residues and determined for their ability to bind a 60-mer L1 RNA molecule are color coded: green (KD < 3X wild type), yellow (3X wild type ≤ KD ≤ 6.1X wild type), and red (KD > 6.1X wild type). Residues that are highly conserved but were not mutated are colored gray. Mutants that disrupted in vivo retrotransposition are indicated by daggers. The ORF1 protein alignments were generated for mouse (from the Tf-5 retrotransposon, AAC53541.1), human (from the human L1.3 retrotransposon, AAB59367), rat (S21345), mumichog (AF055640), Zebrafish (CAD61093), and medaka (AAS83199) using CLUSTALW.36
EPR spectra of the RNA-free protein
Using the aforementioned cysteine mutants, the R1 spin label was introduced into ORF1p at selected sites located within the protease sensitive “linker” segment (239–288) and the CTD (289–360) [Fig. 1(A)]. EPR spectra of the RNA-free ORF1p mutants are shown as black traces in Figure 4. Qualitatively, the spectra are indicative of restricted nitroxide motion at nearly all sites studied, suggesting that both regions are structurally ordered in the full-length trimeric protein.
Figure 4.
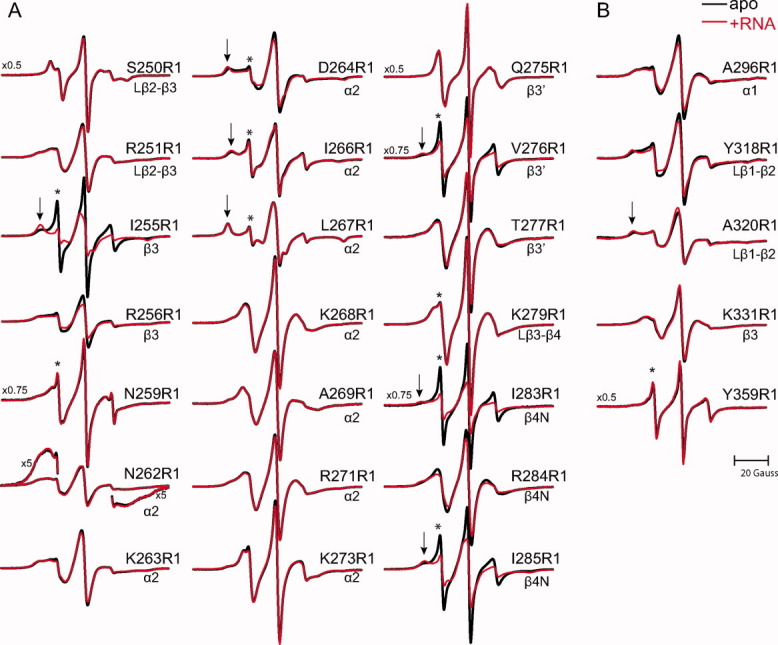
EPR spectra of apo and RNA bound mouse ORF1p. EPR spectra of ORF1p R1 mutants at sites in the RRM (panel A) or CTD (panel B) in the absence (black) and a presence of a 60-mer L1 RNA (red). Spectral features indicating highly dynamic components are designated by an asterisk; those indicating relatively immobilized states are designated by an arrow. The magnetic field scan width is 100 G. All spectra are normalized to the same concentration of nitroxide. Vertical scaling factors (left side of spectrum) were applied to select spectra for display purposes.
Interestingly, nearly all of the spectra are multicomponent, that is, reflect multiple nitroxide dynamic states in slow exchange (>100 ns). Such spectra may arise from multiple rotamers of the spin label side chain that engage in different interactions with the protein, or multiple protein conformational states.37 In the remainder of this section, the individual dynamic components (i.e., disordered, weakly ordered, and relatively immobilized) observed in the EPR spectra are presented as well as a short description of their structural and dynamic implications.
A relatively immobilized state is the predominant component in the spectra of Asp264R1, Ile266R1, Leu267R1, and Ala320R1 (denoted by an arrow in Fig. 4), characterized by well-resolved hyperfine extrema with splittings [2Azz′, as defined in Fig. 1(C)] ranging from 64 to 69 Gauss. Such restricted motions generally occur for R1 side chains at sites buried in the protein where the nitroxide has multiple interactions with nearby residues.28,29 The spectra of Ile255R1, Val276R1, Ile283R1, and Ile285R1 contain similar immobile components [Fig. 4(A)], although representing only 20–40% of the total population (Figs. 4 and 5). The EPR data are generally consistent with the structure of the human ORF1p RRM domain, as residues Ile255, Ile266, Leu267, Val276, Ile283, and Ile285 in the mouse protein are predicted to be buried within the hydrophobic core based on primary sequence homology. However, the side chain of Asp264 is predicted to be solvent exposed in the mouse protein, so the immobilized component observed in the Asp264R1 might result from a subtle difference between the human and mouse RRM structures, or because Asp264 engages in interdomain interactions in the context of the full-length trimeric protein (see Discussion).
Figure 5.
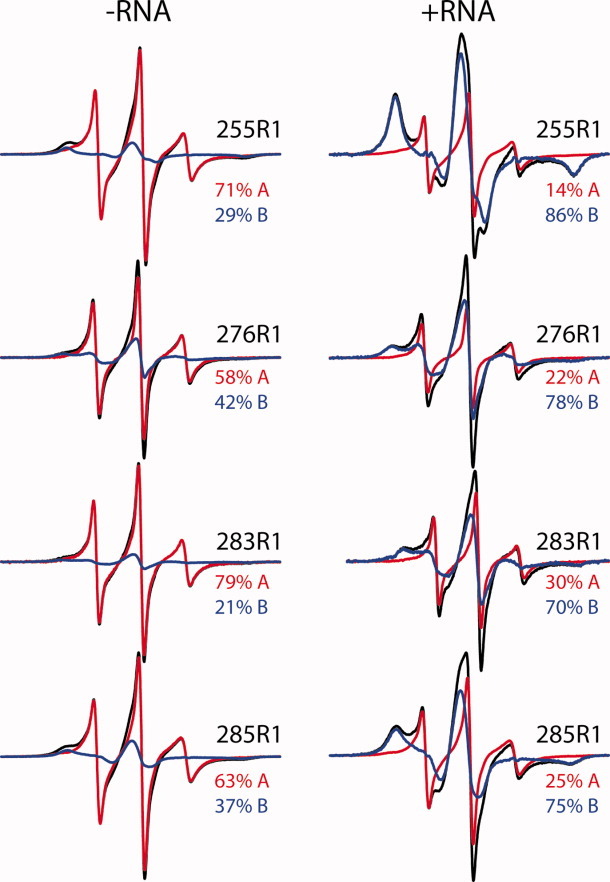
Individual dynamic components of Ile255R1, Val276R1, Ile283R1, and Ile285R1 spectra. EPR spectra of the indicated ORF1p mutants with (right panel) or without RNA (left panel) shown as black traces. For each mutant in the presence or absence of RNA, the EPR spectrum is modeled as a weighted sum of the same two dynamic components (red, blue traces), with the percentage of each component present given on the right. In the presence of RNA, an increase in the relatively immobilized component and corresponding decrease in the highly dynamic component is evident.
The spectrum of Asn262R1 is distinct from the other spectra in Figure 4 because it exhibits spectral broadening indicative of magnetic dipolar interactions between nitroxides that are closer than ∼15 Å. Although spectral broadening can also arise from motional restrictions that due to dipolar interaction is unique and is manifested in the spectrum by an atypical ratio of spectral intensities38 and spectral “wings” that extend beyond the range characteristic for motional broadening39; both effects are evident in the spectrum of Asn262R1 [Fig. 4(A)]. The spectra of Arg251R1 and Arg256R1 may also reflect weak dipolar broadening, but confirmation would require further analysis. The dipolar broadening observed in the Asn262R1 spectrum suggests that the nitroxides are in close proximity in the context of the full-length trimer, and thus the corresponding residues likely reside at an inter-subunit interface. In the human RRM structure, the residues homologous to 256 through 263 are located within a segment that connects strand β3 to helix α2, and the EPR data suggest that these segments may be packed close to one another in the trimeric structure (see Discussion).
A weakly ordered dynamic state is the dominant component in the spectra of Lys268R1, Ala269R1, Arg271R1, Lys273R1, Thr277R1, Lys279R1, Arg284R1, and 331R1. Variation among the spectra of weakly ordered states is likely due to differences in local backbone motions at the site of attachment. Such dynamic states reflect moderate restrictions on nitroxide motion and typically occur at solvent-exposed sites.40 The EPR spectrum of Gln275R1 [Fig. 4(A)] also exhibits a weakly ordered motion, although it is distinct from the aforementioned weakly ordered states as evidenced by the extremely narrow central resonance relative to the other spectra. Similar EPR spectra have been observed for R1 in other proteins and are believed to arise from an unusual anisotropic motion41,42 that could have contributions from restricted backbone motions. Thus, the EPR data suggest that the corresponding residues reside at solvent-exposed regions of the protein, which is consistent with the locations of homologous residues within the structure of the human RRM protein.
In addition to structural insights, the EPR data offer insights into the local nanosecond backbone motions present in the trimeric protein. Indeed, a spectral component corresponding to a highly mobile disordered state (denoted by an asterisk in Fig. 4) was observed in the spectra of Ile255R1, Asn259R1, Asp264R1, Ile266R1, Leu267R1, Val276R1, Lys279R1, Ile283R1, Ile285R1, and Tyr359R1. This component reflects fast, isotropic motion (<2 ns) and likely results from large amplitude backbone fluctuations at the site of attachment. Although this component represents a relatively minor population in most of these spectra (<5%), it represents a substantial population (ca. 50–80%, Fig. 5) of Ile255R1, V276R1, Ile283R1, Ile285R1, and nearly 100% of Tyr359R1. Although a highly dynamic state was expected for Tyr359R1 because the corresponding residue resides in an unstructured portion of the CTD,20 it was not expected in the spectra of Ile255R1, V276R1, Ile283R1, or Ile285R1 because the corresponding residues are buried in the human RRM structure (see Discussion).
EPR spectra of the RNA-bound protein
Through changes in the EPR spectra, the ORF1p R1 mutants have the potential to reveal regions of the protein involved in RNA binding. To map the regions of the protein affected by RNA binding, the ORF1p R1 mutants were studied in the presence of a 10-fold molar excess of the 60-mer RNA molecule; the resulting EPR spectra are shown as red traces for the RRM [Fig. 4(A)] and CTD [Fig. 4(B)]. Several spectra show a noticeable change upon the addition of RNA, including the RRM mutants Ile255R1, Arg256R1, Asp264R1, Val276R1, Ile283R1, Arg284R1, Ile285R1, and the CTD mutants Ala296R1 and Tyr318R1.
The most striking RNA-dependent changes are observed in the spectra of Ile255R1, Val276R1, Ile283R1, and Ile285R1. For these mutants when RNA is present, the fraction of the relatively immobilized component (designated by an arrow) increases at the expense of the highly dynamic component (Fig. 5). This indicates that these residues reside in conformationally heterogeneous regions of the protein. Indeed, the homologous residues are buried in the human RRM structure, so a direct interaction of the spin label with the RNA is unlikely.
In contrast to the dramatic changes observed for the aforementioned mutants, subtle changes are observed in the spectra of Arg256R1, Asp264R1, Arg284R1, and the CTD mutants Ala296R1 and Tyr318R1. In the presence of RNA, the effective hyperfine splitting [2Azz′, Fig. 1(C)] of the relatively immobilized state slightly increases in the mutants Asp264R1 and Tyr318R1, indicating that the nitroxide motion is further restricted in the RNA bound state.
Interestingly, even though mutation of Arg256 or Arg284 drastically reduces the binding affinity of the protein (Table I), the spectral changes observed for the 256R1 and 284R1 mutants suggest that these mutants are still capable of RNA binding, although the affinity is presumably reduced.
Identification of ORF1p amino acids required for retrotransposition
Mouse retrotransposons harboring an Arg297Ala/Arg298Ala double mutation in the CTD of ORF1p are impaired in their ability to retrotranspose in a cell culture assay.13 To test whether residues within the RRM domain are also essential for retrotransposition, the ability of Arg251Ala and Arg284Ala ORF1p mutants to promote retrotransposition was assessed as these mutants show diminished RNA binding activity in vitro (Table I). Retrotransposon activity was determined using an autonomous retrotransposition assay in HeLa cells as described previously.13,15 The Arg251Ala mutant, which alters the surface formed by the β2-β3 loop of the RRM, is significantly impaired in its ability to promote retrotransposition (10% of wild-type protein), whereas the Arg284Ala mutant, located in strand β4, retains ∼70% activity. These results provide the first experimental evidence that RNA interactions involving the RRM domain are required for retrotransposition in vivo.
Discussion
ORF1p is required for the retrotransposition of the highly successful mammalian L1 element.1–5 Previous studies have led to different views of its global structure. A protease digestion and NMR study of mouse ORF1p suggests that the C-terminal, RNA binding region contains a CTD that is tethered to the body the protein by a ∼50 amino acid protease sensitive “linker” polypeptide segment [residues 239–288, Fig. 1(A)].20 Thus, in the absence of RNA, ORF1p might adopt a “hydra-like” structure in which three CTD modules are connected by disordered “linkers” that project from an elongated trimeric coiled-coil domain. However, a recent crystal structure of a monomeric polypeptide fragment from human ORF1p revealed that amino acids homologous to the protease sensitive linker segment in mouse ORF1p are structured, adopting a noncanonical RRM-type fold.23
In this study, we used SDSL-EPR to investigate mouse ORF1p to better understand its structure and to delineate how it interacts with RNA to promote retrotransposition. SDSL-EPR is a powerful method for investigating protein structure, exploring protein dynamics, and detecting conformational changes in large systems that are refractory to NMR or crystallographic methods. The data suggest that the linker region in mouse ORF1p is structured in the context of the intact trimeric protein and are compatible with it adopting a RRM-type structure. In addition, the data provide new insights into the conformational dynamics of the trimer, its quaternary structure, and mechanism of RNA binding.
Overall, the EPR data suggest that the linker region of mouse ORF1p adopts a similar structure to the RRM fold identified in the human ORF1 protein. For example, the spectra of Ile255R1, Ile266R1, Leu267R1, Val276R1, Ile283R1, and Ile285R1 [Fig. 4(A)] each exhibit a relatively immobilized component, suggesting that the corresponding residues either interact with the tertiary structure or are buried in the ORF1p structure. The homologous residues in the structure of the human RRM domain form part of the nonpolar core [Fig. 6(A)], thus the EPR data are consistent with the mouse protein containing an RRM domain. Additional evidence for structural similarity comes from the spectra of Lys268R1, Arg271R1, Gln275R1, Thr277R1, Lys279R1, and Arg284R1 [Fig. 4(A)]. The dominant dynamic state of R1 reflected by these spectra show that the corresponding residues are solvent-exposed, consistent with the locations of the homologous residues in the crystal structure of the human RRM domain [Fig. 6(A)].
Figure 6.
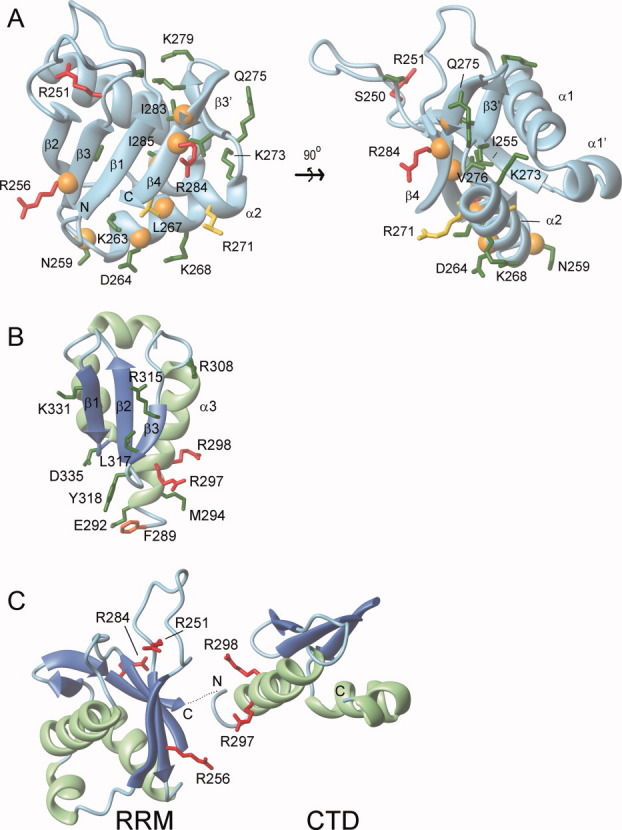
RNA binding model for the RRM and CTD. Residues important for RNA binding within the (A) RRM domain and (B) CTD. Amino acids within ORF1p that were mutated to cysteine or alanine residues and determined for their ability to bind a 60-mer L1 RNA molecule are color coded: green (KD < 3X wild type), yellow (3X wild type ≤ KD ≤ 6X wild type), and red (KD > 6X wild type). An orange sphere positioned at the alpha carbon atom indicates R1 labeled cysteine mutants that exhibited RNA-dependent changes in their EPR spectra. (C) Proposed primary interaction surface with RNA. The CTD and RRM domain are positioned adjacent to one to form a continuous surface that interacts with single stranded RNA. It is unclear whether this surface is formed by the RRM and CTD from a single monomer or from different monomers within the trimer. The orientation of the RNA molecule on this surface is also unknown. In panels B and C, ribbon diagrams of the RRM domain and CTD are shown with the sheet and helices colored blue and green, respectively. The secondary structure topology is labeled accordingly for the RRM (PDB 2w7a)23 and the CTD (PDB 2jrb).20
The EPR spectra of R1 at sites in the CTD [Fig. 4(B)] indicate that this region of the protein is also structured in the context of the full length protein. Indeed, the spectrum of Tyr359R1 indicates that this residue is located in an unstructured region of the protein, whereas the dominant components in the spectra of Ala296R1, Tyr318R1, and Ala320R1 indicate that the corresponding residues reside at tertiary interaction sites. These data are consistent with the previously determined CTD solution structure, suggesting that this structure is retained in the context of the full-length ORF1p trimer.
Information about the trimeric structure of ORF1p is revealed in the EPR spectra by dipolar broadening. Such broadening is evident in the EPR spectrum of Asn262R1 [Fig. 4(A)] (and possibly others in the segment comprising residues 256–263), showing that the residues are in proximity (≤15 Å) in the quaternary structure. In the RRM crystal structure, the homologous residues form a β/α motif that is in close proximity to the N-terminus of the domain [Fig. 6(A)]. As the coiled coil domain terminates at this point, the EPR data indicate that the RRMs are positioned such that these motifs are located close to one another in the ORF1p trimer. This view is further supported by the EPR spectrum of Asp264R1 [Fig. 4(A)], which reflects an immobilized state characteristic of a buried R1 residue rather than a solvent-exposed one as in the monomeric human RRM structure [Fig. 6(A)]. The immobilization of Asp264R1 may result from an interaction with the coiled-coil domain, another RRM domain, or both.
Although the EPR data indicate that the linker region of mouse ORF1p adopts a similar structure to the human RRM domain, the data also reveal the presence of a small population of an unfolded state. This apparent structural equilibrium is evident in the multicomponent spectra of Ile255R1, Val276R1, Ile283R1, and Ile285R1 [Figs. 4(A) and 5]. As discussed above, the immobilized states indicate that these residues are located within the hydrophobic core in the ordered state, whereas the highly dynamic states are characteristic of an unfolded conformation. A spectral component corresponding to a highly dynamic state is not observed for R1 at buried sites in α-helices or β-sheets unless the structure has only marginal stability.28,29,43 Thus, the simplest interpretation of the ORF1p data is that these residues are located in a structurally unstable portion of the protein that undergoes exchange between a folded and unfolded conformation, providing an explanation for its previously observed protease susceptibility. If this instability extends toward the C-terminus of the protein that contains the junction point between the RRM and CTD, then the CTD could undergo segmental motions that rearrange its positioning relative to the body of the protein, as previously postulated in the “hydra-model” of ORF1p structure.
Regions within ORF1p that are affected by RNA binding are also revealed in the EPR spectra. The four mutants that exhibit the most dramatic spectral changes in the presence of RNA are Ile255R1, Val276R1, Ile283R1, and Ile285R1 (Fig. 5). Interestingly, the population of the highly dynamic state in all four spectra is reduced in the presence of RNA, indicating that the bound nucleic acid shifts the equilibrium to favor the more locally structured state. More subtle RNA-dependent changes were observed in the spectra of Arg256R1, Asp264R1, Arg284R1, Ala296R1, and Tyr318R1 (Fig. 4). Although the mutation of either β-sheet surface residue Arg256 or Arg284 dramatically reduces binding affinity (Table I), the spectral changes observed in both R1 mutants are consistent with these residues directly interacting with the RNA. On the other hand, residues Ala296 and Tyr318 interact with elements of the tertiary structure in the CTD [Fig. 6(B)], so the spectral changes observed in the presence of RNA could reflect subtle changes in the tertiary packing of the CTD upon RNA binding. As described above, the EPR data indicate that Asp264 is buried in the ORF1p trimer, so the spectral changes observed for Asp264R1 are likely the result of subtle changes in packing associated with RNA binding.
The EPR and biochemical data are compatible with the RRM interacting with RNA. Mutation of residues Arg256 and Arg284 located on the surface of the RRM β-sheet significantly affects RNA binding (Table I). These side chains may directly interact with the bound RNA, which would in part explain why the Arg284Ala mutant has reduced retrotransposition activity (70% of wild-type). Residues in the β2-β3 loop adjacent to the β-sheet may also play an important role in RNA binding. This is evident from the binding behavior of the Arg251Cys mutant, which interacts with RNA weakly, and our finding that retrotransposons harboring an Arg251Ala mutation retrotranspose only 10% as efficiently as the wild-type L1 element. In the structure of the human RRM, Arg251 appears to play a structural role by forming a salt-bridge with Glu201 of the β1-α1 loop. This interaction may function to properly position the β2-β3 loop to engage the RNA molecule along with the RRM sheet. This is consistent with the recent finding that a monomeric fragment from human ORF1p containing a triple Arg206Ala/Arg210Ala/Arg211Ala mutation binds RNA weakly, as these residues are located in the β2-β3 loop such that their side chains project towards the sheet.23 The notion that the sheet is the primary contact surface is also supported by numerous structures of RRM-nucleic acid complexes, which have shown that this surface is commonly used for nucleic acid recognition.44
In the full-length trimeric protein, helix α1 of the CTD may form a second RNA contact surface. The CTD is comprised of three α helices that are packed against one face of a three stranded β-sheet [Fig. 6(B)]. Previous studies have shown that RNA binding is partially mediated by Arg297 and Arg298 located in helix α1.14,15 Our systematic mutagenesis of residues surrounding this segment reveals that only mutation of residues Phe289, Ala296, and Tyr318 cause modest reductions in binding affinity (i.e., 2.5- to 3.3-fold KD increase compared with wild-type). Thus, all of the important side chains either project from one face of the helix (Phe289, Ala296, Arg297, and Arg298) or are positioned just proximal to this site (Tyr318). It therefore seems likely that only this small surface on the CTD forms significant interactions with the RNA in the context of the full-length protein. Studies of the human protein support this assertion, as a monomeric polypeptide containing its RRM and CTD bearing a Tyr318Ala/Lys321Ala (mouse numbering) mutation in the β sheet of the CTD retains wild-type RNA binding activity when assayed by gel filtration chromatography.23
In summary, our mutagenesis data has identified two distinct surfaces on the RRM and CTD domains that are required for productive interactions with RNA. However, RNA binding is mediated by a trimer of polypeptides that are positioned adjacent to one another at the end of the dumbbell-shaped structure.35 Presumably, the RNA binding surfaces are positioned adjacent to one another in the context of the full-length trimer, thereby providing a continuous surface for interaction with single stranded RNA. At present, it is not clear whether or not this continuous surface is formed by the RRM and CTD from a single polypeptide chain, but recent studies of a monomeric polypeptide of hORF1p containing both the CTD and RRM indicate that domains on a single polypeptide chain can work together to bind RNA.23 The EPR and mutagenesis data provide insights into how this might occur. Figure 6(C) shows a view of the RRM and CTD that positions the C-terminal end of the RRM adjacent to the N-terminal end of the CTD. The two domains have been rotated so as to juxtapose the amino acids in each protein that are important for binding. In this orientation, the single strand nucleic acid is contacted by the β-sheet and the β2-β3 loop of the RRM on one side as well as by a more limited interface on the α1 helix of the CTD. Therefore, the two surfaces from both the RRM and CTD could work together to form a pocket and capture the RNA. Additional structural studies of the free and bound states of ORF1p are needed to test the validity of this model.
Materials and Methods
Generation of expression constructs and site directed mutagenesis of ORF1p
Expression plasmids encoding wild-type full-length ORF1p of the Tf5 type L1 element were generated by PCR using the previously described mouse L1-TF5 ORF1p expression plasmid as a template45 and cloned into the pCOLD I vector (Takara Bio). Single cysteine and alanine amino acid mutations of ORF1p were produced using the Quikchange site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the pCOLD I ORF1p construct as the template. All cysteine mutants contain a Cys174Arg mutation that reduces spurious nitroxide labeling. The Cys174Arg mutation does not affect RNA binding consistent with the location of this residue in the coiled-coil region of the protein and its poor conservation in mammalian ORF1 proteins (data not shown). Arginine was introduced at this site because this residue is present in the human protein based on primary sequence homology. The identity of the ORF1p cysteine mutants and alanine mutants were confirmed by DNA sequencing. Mutant L1 T-fc retrotransposon constructs were generated by making single point mutations within ORF1 by site-directed mutagenesis.13 The resulting constructs were also verified for fidelity by DNA sequencing.
Sample preparation of ORF1 proteins
Because previously described production methods yielded only small quantities of soluble ORF1p,34,46 we developed a new cold-shock vector based expression system that produces ∼5 mg of >95% pure ORF1p per liter of Escherichia coli culture. Expression vectors producing His252Cys, Ile253Cys, and Arg281Cys (each in the same Cys174Arg background) were also constructed, but the overexpressed proteins were insoluble (data not shown). Wild-type and mutant versions of ORF1p were over expressed in Rosetta-2 cells (Novagen-EMD Biosciences, Madison, WI). One-liter cultures were grown at 37°C to an OD600 nm ∼ 0.4, and then placed in an ice bath for 30 min. The cultures were then induced with 1 mM IPTG at 15°C for 24 h. The cells were then harvested at 8000 rpm (JA-10), resuspended in lysis buffer: 50 mM Tris-HCl pH 7.3, 500 mM NaCl, 6 μg/mL (final concentration) RNaseA (Fisher BioReagents, Fair Lawn, NJ), 750 μg/mL lysozyme (Sigma, St. Louis, MO), and Protease Inhibitor Cocktail Set 2 (Calbiochem-EMD Biocsciences, Madison, WI). The resuspended cells were sonicated on ice and then incubated with 0.5% Triton X-100 and 0.03% PEI. Afterward, the whole cell lysate was centrifuged at 15,000 rpm for 30 min (JA-20 rotor). ORF1p was then precipitated out of the supernatant by the addition of ammonium sulfate (50%, w/v), and then subsequently centrifuged at 15,000 rpm for 30 min. The pellet was resuspended in Resuspension Buffer: 50 mM phosphate, pH = 8.0, 500 mM NaCl, 1M urea, 10 mM imidazole, and 10% glycerol. The resuspension was added to Ni-NTA beads (Qiagen, Valencia, CA) and purified using the manufacturers' instructions with two exceptions: Wash Buffer is 50 mM phosphate, pH = 8.0, 500 mM NaCl, 10 mM imidazole, and 10% glycerol as well as Elution Buffer is 50 mM phosphate, pH = 8.0, 500 mM NaCl, 250 mM imidazole, and 10% glycerol. The eluted protein samples were then diluted to 50 mM NaCl and applied to a SP-Sepharose Fast Flow XK-16 column using Buffer A (50 mM phosphate pH 6.0, 2.5 mM EDTA, 5 mM dithiothreitol (DTT), and 10% glycerol (vol/vol)) and eluted using a gradient of 1M NaCl (0–100%) in buffer B (50 mM phosphate pH 6.0, 2.5 mM EDTA, 5 mM DTT, 10% glycerol (vol/vol), 1M NaCl). Pooled protein fractions were then concentrated to ∼1 mL.
RNA sample preparation
The following RNA substrate that contain nucleotides 4330–4390 of the L1 retrotransposon was created by in vitro transcription: a 60-mer (5′ GGC AAC AAU UAC UUU UCC UUA AUA UCU CUU AAC AUC AAU GGU CUC AAC UCG CCA AUA AAA 3′).47 Using complementary DNA oligonucleotides (Integrated DNA Technologies, Coralville, IA) with a T7 promoter and labeled [α-32P]-UTP with NTPs (Amersham Pharmacia Biosciences, Piscataway, NJ), radiolabeled RNA products were in vitro transcribed, run on a 10–15% acrylamide—8M urea gel, and then cut out of the gel. The gel fragments were then incubated over night in water and ethanol precipitated the following day. Large scale milligram production of the unlabeled 60-mer RNA was also produced by in vitro transcription. Unlabeled RNA was purified using a 6% acrylamide—8M urea tube gel electrophoresis apparatus in combination with an Akta FPLC system (Amersham Pharmacia Biosciences, Piscataway, NJ).
Nitrocellulose double-filter binding assay
The double-filter binding assay was conducted in a similar manner to previously published work.20,34 Wild-type ORF1p, the cysteine and alanine mutant ORF1p constructs were incubated with 1 nM of 60 nt 32P-labeled RNA (for binding affinity measurements) and 20 nM of 60-mer 32P-labeled RNA (for binding stoichiometry measurements) for 60 min on ice in binding buffer (20 mM Hepes pH 7.5, 250 mM NaCl, 1 mM DTT, 5 mM EDTA). Using a 96-well dot blot system (Schleicher & Schuell, Keene, NH), 25 μL reactions were filtered through nitrocellulose and DE81 (preincubated in binding buffer), and washed three times with 25-μL ice-cold binding buffer. The nitrocellulose and DE81 were allowed to dry and subsequently exposed to a K-screen (Amersham Pharmacia Biosciences, Piscataway, NJ). Radioactive RNA was then quantified by imaging analysis using Quantity One software (Amersham Pharmacia Biosciences, Piscataway, NJ) and exported to Sigma Plot 2000 (SPSS, Chicago, IL) for analysis. The dissociation constant (KD) and Hill coefficient (h) were calculated by plotting the log of the total concentration of protein (log[Ptotal]) as a function of the fraction of the RNA that was bound (y). Data was fit to a cooperative binding model using the equation y = (ymax)/(1 + e∧([Ptotal] − KD)/[Ptotal]/h), where h is the Hill coefficient.
Cell culture and autonomous retrotransposition
Mouse L1 T-fc and ORF1p mutant L1 T-fc retrotransposon activity were determined using an autonomous retrotransposition assay with an antisense intron-containing neomycin resistance cassette, as described previously.13,15 Briefly, HeLa cells were grown at 37°C in high glucose Dubecco's modified Eagle's medium (DMEM) lacking pyruvate. Cells were passaged by standard methods. HeLa cells were then seeded and grown to 70% confluency in DMEM. Cells were transfected with the lipofectamine transfection reagent (Invitrogen). Each transfection consisted of 1 mL of Opti-mem (Invitrogen) (1 mg of DNA and 7 mL of lipofectamine reagent). Twelve to fourteen days after transfection, cells were trypsinized, pooled, and counted with a hemocytometer. Dilutions were plated in DMEM containing G418. After 14 days, the G418 resistant cells were fixed to plates and stained with 0.4% Giemsa for visualization. The number of G418 resistant colonies were scored and activity for mutants was assessed compared with wild-type activity.
Spin labeling and EPR spectroscopy
Before spin labeling, each ORF1p mutant was exchanged into Labeling Buffer [20 mM MES, pH = 6.8; 250 mM NaCl; 10% (vol/vol) glycerol] using two tandem 5 mL HiTrap HP desalting columns (GE Healthcare). Immediately upon the completion buffer exchange, a fivefold molar excess of 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate (a gift of Kálmán Hideg, University of Pécs, Hungary)48 and urea (to a final concentration of 1M) were added to each mutant, and then incubated with constant agitation at 10°C overnight. Samples were then desalted to remove unreacted nitroxide reagent by the aforementioned desalting procedure, and then concentrated using an Amicon Ultra-15 concentrator fitted with a 30 K molecular weight cutoff membrane (Millipore).
For EPR spectroscopy, 5-μL samples (typically 10–100 μM ORF1p in labeling buffer) were loaded into a glass capillary (0.6 mm i.d. × 0.8 mm o.d., VitroCom, NJ) sealed at one end. Samples of the ORF1p mutants in the presence of RNA were generated by adding a 10-fold molar excess of the 60-mer RNA to the protein solution. EPR spectra were recorded at room temperature on a Varian E-109 spectrometer operated at X-band (9 GHz) and fitted with a two-loop one-gap resonator using 2 mW incident microwave power and field modulation (100 kHz) amplitude optimized to the natural linewidth of each individual spectrum. For ORF1p R1 mutants exhibiting a sharp, highly dynamic component (e.g., 255R1), no detectable nitroxide signal was present in the filtrate, confirming that the sharp component was not simply a free nitroxide species.
EPR spectral component analysis
It is assumed that (1) the spectra of Ile255R1, Val276R1, Ile283R1, and Ile285R1 contain only two spectral components, and (2) the binding of each mutant RNA simply shifts the equilibrium between the two components without a concomitant change in the spectrum of either individual component. Note that this approach does not require that each component represent a single dynamic state. The individual dynamic components present in these spectra were identified by an interactive program written in LabVIEW called component analysis. Briefly, for each mutant, a suitable linear combination of the RNA-free and RNA bound spectrum results in a difference spectrum that represents the mobile or immobile component. If the spectral components are very distinct, as is the case for these four mutants, the scaling factors for the linear combination can easily be obtained interactively. The pure component spectra are normalized for the same number of spins, and then used to fit the original spectra as a weighted sum to obtain the percentage of each component.
References
- 1.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 2.Babushok DV, Kazazian HH., Jr. Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat. 2007;28:527–539. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- 3.Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? Bioessays. 2005;27:775–784. doi: 10.1002/bies.20257. [DOI] [PubMed] [Google Scholar]
- 4.Kazazian HH, Jr, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- 5.Ostertag EM, Kazazian HH. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 6.Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003;13:651–658. doi: 10.1016/j.gde.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 8.Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–1170. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewing AD, Kazazian HH., Jr. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20:1262–1270. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, Wheelan SJ, Ji H, Boeke JD, Burns KH. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141:1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin SL, Cruceanu M, Branciforte D, Wai-Lun Li P, Kwok SC, Hodges RS, Williams MC. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J Mol Biol. 2005;348:549–561. doi: 10.1016/j.jmb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Martin SL, Bushman FD. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol Cell Biol. 2001;21:467–475. doi: 10.1128/MCB.21.2.467-475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr. High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 16.Feng Q, Moran JV, Kazazian HH, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 17.Martin SL. The ORF1 protein encoded by LINE-1: structure and function during L1 retrotransposition. J Biomed Biotechnol. 2006:45621–45626. doi: 10.1155/JBB/2006/45621. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang N, Kazazian HH., Jr. L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- 19.Martin S, Branciforte D, Keller D, Bain DL. Trimeric structure for an essential protein in L1 retrotransposition. Proc Natl Acad Sci USA. 2003;100:13815–13820. doi: 10.1073/pnas.2336221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Januszyk K, Li PW, Villareal V, Branciforte D, Wu H, Xie Y, Feigon J, Loo JA, Martin SL, Clubb RT. Identification and solution structure of a highly conserved C-terminal domain within ORF1p required for retrotransposition of long interspersed nuclear element-1. J Biol Chem. 2007;282:24893–24904. doi: 10.1074/jbc.M702023200. [DOI] [PubMed] [Google Scholar]
- 21.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson A. 1996;120:155–189. [Google Scholar]
- 22.Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 23.Khazina E, Weichenrieder O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proc Natl Acad Sci USA. 2009;106:731–736. doi: 10.1073/pnas.0809964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 25.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 27.Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 28.Lietzow MA, Hubbell WL. Motion of spin label side chains in cellular retinol-binding protecorrelation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry. 2004;43:3137–3151. doi: 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- 29.Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 30.Columbus L, Hubbell WL. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4-58 bZip free and bound to DNA. Biochemistry. 2004;43:7273–7287. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- 31.Langen R, Cai K, Altenbach C, Khorana HG, Hubbell WL. Structural features of the C-terminal domain of bovine rhodopsa site-directed spin-labeling study. Biochemistry. 1999;38:7918–7924. doi: 10.1021/bi990010g. [DOI] [PubMed] [Google Scholar]
- 32.Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, Randall LL. Mapping of the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into the mechanism of ligand transfer during protein export. J Mol Biol. 2005;353:295–307. doi: 10.1016/j.jmb.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 33.Kusnetzow AK, Altenbach C, Hubbell WL. Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry. 2006;45:5538–5550. doi: 10.1021/bi060101v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolosha VO, Martin SL. High-affinity, non-sequence-specific RNA binding by the open reading frame 1 (ORF1) protein from long interspersed nuclear element 1 (LINE-1) J Biol Chem. 2003;278:8112–8117. doi: 10.1074/jbc.M210487200. [DOI] [PubMed] [Google Scholar]
- 35.Basame S, Wai-lun Li P, Howard G, Branciforte D, Keller D, Martin SL. Spatial assembly and RNA binding stoichiometry of a LINE-1 protein essential for retrotransposition. J Mol Biol. 2006;357:351–357. doi: 10.1016/j.jmb.2005.12.063. [DOI] [PubMed] [Google Scholar]
- 36.Thompson JD, Higgins DG, Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez CJ, Fleissner MR, Guo Z, Kusnetzow AK, Hubbell WL. Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 2009;18:1637–1652. doi: 10.1002/pro.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Likhtenshtein GI. Biophysical labeling methods in molecular biology. New York: Cambridge University Press; 1993. p. 305. [Google Scholar]
- 39.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 40.Fleissner MR, Cascio D, Hubbell WL. Structural origin of weakly ordered nitroxide motion in spin-labeled proteins. Protein Sci. 2009;18:893–908. doi: 10.1002/pro.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo Z, Cascio D, Hideg K, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: solvent-exposed sites in helix B of T4 lysozyme. Protein Sci. 2008;17:228–239. doi: 10.1110/ps.073174008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Eps N, Oldham WM, Hamm HE, Hubbell WL. Structural and dynamical changes in an alpha-subunit of a heterotrimeric G protein along the activation pathway. Proc Natl Acad Sci USA. 2006;103:16194–16199. doi: 10.1073/pnas.0607972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bridges MD, Hideg K, Hubbell WL. Resolving conformational and rotameric exchange in spin-labeled proteins using saturation recovery EPR. Appl Magn Reson. 2010;37:363–390. doi: 10.1007/s00723-009-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clery A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Martin SL, Li J, Weisz JA. Deletion analysis defines distinct functional domains for protein-protein and nucleic acid interactions in the ORF1 protein of mouse LINE-1. J Mol Biol. 2000;304:11–20. doi: 10.1006/jmbi.2000.4182. [DOI] [PubMed] [Google Scholar]
- 46.Kolosha VO, Martin SL. In vitro properties of the first ORF protein from mouse LINE-1 support its role in ribonucleoprotein particle formation during retrotransposition. Proc Natl Acad Sci USA. 1997;94:10155–10160. doi: 10.1073/pnas.94.19.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berliner LJ, Grunwald J, Hankovszky HO, Hideg K. A novel reversible thiol-specific spin label: papain active site labeling and inhibition. Anal Biochem. 1982;119:450–455. doi: 10.1016/0003-2697(82)90612-1. [DOI] [PubMed] [Google Scholar]