

Abstract
Protein–protein interactions play an essential role in the functioning of cell. The importance of charged residues and their diverse role in protein–protein interactions have been well studied using experimental and computational methods. Often, charged residues located in protein interaction interfaces are conserved across the families of homologous proteins and protein complexes. However, on a large scale, it has been recently shown that charged residues are significantly less conserved than other residue types in protein interaction interfaces. The goal of this work is to understand the role of charged residues in the protein interaction interfaces through their conservation patterns. Here, we propose a simple approach where the structural conservation of the charged residue pairs is analyzed among the pairs of homologous binary complexes. Specifically, we determine a large set of homologous interactions using an interaction interface similarity measure and catalog the basic types of conservation patterns among the charged residue pairs. We find an unexpected conservation pattern, which we call the correlated reappearance, occurring among the pairs of homologous interfaces more frequently than the fully conserved pairs of charged residues. Furthermore, the analysis of the conservation patterns across different superkingdoms as well as structural classes of proteins has revealed that the correlated reappearance of charged residues is by far the most prevalent conservation pattern, often occurring more frequently than the unconserved charged residues. We discuss a possible role that the new conservation pattern may play in the long-range electrostatic steering effect.
Keywords: charged residue conservation, protein interaction interfaces, electrostatic steering, protein–protein interactions, interface similarity
Introduction
With the appearance of high-throughput methods for detecting protein–protein interactions, it became clear that proteins in a cell seldom function in isolation. Instead, they form molecular complexes, interacting with each other as well as with RNA and DNA molecules.1 A detailed characterization of protein–protein interactions can provide crucial information about the function and evolution of protein complexes and can be helpful in medicine, for example, when studying interactions that underlie infectious diseases.2,3
Although cataloging all the structural and physico-chemical components that are critical in determining the protein interface is still an open question, the important role that the charged residues play in protein–protein interactions has been well documented.4–11 In some interactions, charged residues are shown to be instrumental in defining binding specificity, while sometimes contributing little binding energy to the interactions themselves.5,12 In other cases, charged residues were found to promote the high affinity binding.13,14 They are also the main players in “electrostatic steering,” a long-range mechanism, in which electrostatic forces can steer a ligand protein into a binding site on the receptor protein, which drastically increase the association rate.15,16 Often, the charged residues important for protein–protein interactions are conserved across families of evolutionary related proteins and protein complexes.17–19 However, on a large scale, the charged residues appear to be significantly less conserved in protein interaction interfaces than, for example, hydrophobic and aromatic residues that are enriched in the clusters of conserved residues found in the interfaces.20 This seemingly counterintuitive result suggests that further more detailed study is required to gain the insights on the conservation of charged residues in the protein interfaces.
The main goal of this work is to further understand the role of charged residues in protein interaction interfaces by studying the conservation patterns they form across homologous interactions. Studying protein–protein interactions in the evolutionary-related protein complexes has allowed scientists to learn about the features that are conserved in protein interfaces and binding sites. For example, when analyzing the relationship between sequence similarity and protein binding orientation, it has been shown that the geometry of interactions is often conserved between similar pairs of proteins.21 Another large-scale study of protein interactions revealed that homologous proteins frequently have their binding sites in similar locations of protein surfaces to interact with other, often unrelated, proteins.22 Here, by determining several types of conservation patterns and characterizing the pairs of charged residues that are in close contact with each other using these patterns, we have identified an intriguing phenomenon that could shed light on the role of charged residues in protein–protein interactions and the mechanisms that underlie their evolutionary conservation.
The article is organized as follows. In the Results section, we report statistics on the conservation pattern occurrences in the interaction interfaces as well as provide a detailed analysis of the types of conservation using case studies. We discuss the obtained results, propose a new hypothesis on the diverse role of charged residues in forming protein interaction interfaces, and outline the directions for future research. Finally, in the Materials and Methods section, we determine a set of homologous interactions using an interaction interface similarity measure, define a set of conservation patterns among charged residue pairs, and describe a protocol for obtaining statistics on these patterns on a large scale.
Results
We first introduce several concepts that are used throughout the article. A protein–protein interaction is defined as triple (S1, S2, O), where S1 and S2 are the two interacting subunits (a subunit can be a protein or protein domain) and O is their relative orientation. Residue r1 of S1 is in contact with residue r2 of S2 if r1 has at least one atom within 6 Å of an atom of r2. A protein binding site of an interacting subunit is defined as the set of all its residues that are in contact with any residues of another subunit. Given a protein–protein interaction, its interaction interface is defined as triple (B1, B2, C), where B1 and B2 are protein binding sites of the interacting subunits and C is a set of all pairs of contact residues. Two protein–protein interactions with similar interaction interfaces are called homologous if a subunit in the first interaction shares homology with a subunit in the second interaction and the remaining two subunits also share homology between each other. The corresponding similar interfaces are also called homologous.
Similar Interfaces Extracted from Homologous Protein–Protein Interactions
The data collection protocol applied to a comprehensive set of all protein–protein interactions extracted from PIBASE resulted in two sets of homologous interactions, one at each redundancy level. We determined 2668 pairs of similar interfaces at the 100% redundancy level, whose interacting subunits are classified into 581 SCOP families and 361 SCOP superfamilies, and 372 pairs at the 95% redundancy level, with interacting subunits classified into 178 SCOP families and 137 SCOP superfamilies. When applying the definition of a charged residue pair to the interfaces at each redundancy level, we found that 843 and 90 interface pairs did not have a single pair of residues for either interface. The remaining 1825 and 282 interface pairs had 3357 and 481 charged residue pairs, correspondingly.
Charged Residue Pairs and Their Conservation Patterns
In this work, we aimed to (i) determine whether or not the charged residue pairs were structurally conserved across the interfaces of homologous interactions and (ii) identify the type and degree of the conservation. To do that, we proposed several types of conservation patterns among the pairs of charged residues based on the patterns of their conservation across the homologous interfaces and then calculated the basic occurrence statistics for each type. For this work, we restricted ourselves by considering only the charged residue pairs that were in close contact. A positively charged residue (Arginine or Lysine) and a negatively charged residue (Aspartic or Glutamic acid) are in close contact if they have at least one pair of atoms within 3 Å. We will be referring to “a pair of charged residues in close contact” simply as a “pair of charged residues” for the remainder of the article. In the set of 282 nonredundant interface pairs, there were on average 6.2 residue pairs in close contact (both charged and uncharged) per interface, including an average of approximately two pairs of charged residues.
We observed four basic types of conservation patterns in charged residue pairs. The type of each conservation pattern was determined by first aligning two interfaces using the subunit–subunit alignment from the iiRMSD protocol that produces the smallest iiRMSD value. Then, we analyzed the colocalization of the charged residue pairs across the two interfaces according to the alignment. The two pairs of residues, each pair from a different interface, were called colocalized if in the structurally aligned interfaces, the Cα–Cα distance between each two aligned residues was less than 3 Å. The conservation pattern was called Type 1, referred to as unconserved charged residues (Fig. 1) when the pairs of charged residues were found in only one of the two interfaces. Type 2 patterns, referred to as conserved charged residues, were defined by the occurrence of any two pairs of charged residues, one from each interface, that were colocalized with the corresponding charges being conserved. The situation in which two pairs of charged residues were colocalized, but the corresponding charges in one interface were swapped between the interacting binding sites compared to another interface, was designated as a Type 3 pattern, referred to as swapped charged residues. Finally, the conservation pattern was termed Type 4, referred to as charged residues of correlated reappearance, when for any two charged residue pairs, one per each interface, when each charged residue pair was not colocalized with any other charged residue pairs. Although many pairs of interfaces were found to have only a single conservation pattern, there were cases of several patterns occurring in the same pair of interfaces (see Charged Residue Conservation Patterns Across Homologous Interfaces in Intraspecies and Interspecies Interactions: Case Studies section for examples). To avoid contribution to multiple conservation patterns by a single charged residue, the conservation patterns, from Types 1 to 4, were determined by consecutively excluding those pairs of charged residues that had already contributed to a conservation pattern.
Figure 1.

The flow chart of the analysis protocol. (1) Extracting structures of binary protein–protein interaction complexes from PIBASE and assigning SCOP superfamily IDs to each interacting subunit. (2) Obtaining a nonredundant dataset of interaction interfaces. (3) Determining pairs of similar homologous interfaces by using iiRMSD similarity measure and the assigned SCOP superfamily IDs. (4) Defining charged residue contacts and their conservation patterns. (5) Calculating the frequency of occurrence for each conservation pattern.
We next determined a conservation pattern for each set of two pairs of charged residues. Based on how conserved the charged residues are in the interaction interfaces, one can expect two basic scenarios for the distribution of conservation patterns. On one hand, if the positions of charged residues were highly conserved across the homologous interactions, one would expect most interface pairs containing charged residues to be of Type 2. On the other hand, in the absence of such conservation of the charged residue pairs and under a simple equiprobable-substitution model, one would expect to see  times more occurrences of the patterns of Type 1 than of Type 2, where Pk is the probability for two pairs of corresponding residues to share a conservation pattern of Type k. Similarly, one would expect
times more occurrences of the patterns of Type 1 than of Type 2, where Pk is the probability for two pairs of corresponding residues to share a conservation pattern of Type k. Similarly, one would expect 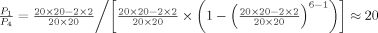 times more occurrences of Type 1 than of Type 4 (recall that for each interface there are on average approximately six pairs of residues in close contact, each of which can potentially be a pair of charged residues). Strikingly, we found that there were almost as many interface pairs of Type 4 as of Type 1 (Table I). In addition, the number of conservation patterns of Types 1 and 4 prevailed over the conservation patterns of Type 2. We also found that the homologous complexes whose interfaces contain the conservation patterns of Type 2 shared significant structural and sequence similarity: each set of two pairs of charged residues classified as Type 2 was obtained from a pair of complexes whose subunits were exclusively classified to the same SCOP family. This was not a common feature for all conservation patterns: the interface pairs associated with other types of conservation patterns were oftentimes obtained from the complexes whose subunits did not belong to the same SCOP families, although they did belong to the same SCOP superfamilies. Finally, the distributions of the conservation patterns across the five types, four types for charged residues and one for uncharged residues, were consistent when comparing the datasets at both redundancy levels (Table I).
times more occurrences of Type 1 than of Type 4 (recall that for each interface there are on average approximately six pairs of residues in close contact, each of which can potentially be a pair of charged residues). Strikingly, we found that there were almost as many interface pairs of Type 4 as of Type 1 (Table I). In addition, the number of conservation patterns of Types 1 and 4 prevailed over the conservation patterns of Type 2. We also found that the homologous complexes whose interfaces contain the conservation patterns of Type 2 shared significant structural and sequence similarity: each set of two pairs of charged residues classified as Type 2 was obtained from a pair of complexes whose subunits were exclusively classified to the same SCOP family. This was not a common feature for all conservation patterns: the interface pairs associated with other types of conservation patterns were oftentimes obtained from the complexes whose subunits did not belong to the same SCOP families, although they did belong to the same SCOP superfamilies. Finally, the distributions of the conservation patterns across the five types, four types for charged residues and one for uncharged residues, were consistent when comparing the datasets at both redundancy levels (Table I).
Table I.
Statistical Analysis of Conservation Patterns for Charged Residue Pairs in Homologous Interfaces
| Number of interface pairs | Number of charged residue pairs | % of total charged residue pairs | ||||
|---|---|---|---|---|---|---|
| Type | (A) | (B) | (A) | (B) | (A) | (B) |
| No charge | 843 | 90 | 0 | 0 | 0.00 | 0.00 |
| 1. Unconserved | 934 | 143 | 1571 | 222 | 46.80 | 46.20 |
| 2. Conserved | 891 | 139 | 560 | 50 | 16.68 | 10.40 |
| 3. Swapped | 2 | 1 | 0.06 | 0.21 | ||
| 4.Correlated reappearance | 1224 | 208 | 36.46 | 43.20 | ||
| Total | 2668 | 372 | 3357 | 481 | ||
The analysis was done for two redundancy levels: 100% (A) and 95% (B).
Protein–protein interactions constituting a complex are often associated with one or several of its functions. Therefore, we wanted to see if specific conservation patterns were associated with some specific functions of protein complexes. There are several challenges in studying the relationship between the interaction-mediated functions and the conservation patterns of the charged residues. First, a large-scale PDB-wide functional annotation, such as Gene Ontology (GO) functional annotation,23 often associates a function with the entire PDB structure, which can possibly have more than one interaction. Second, as multiple conservation patterns can occur in a single pair of interaction interfaces, same function can be potentially associated with more than one conservation pattern. In this work, we inquired whether or not the fact that a pair of similar interfaces shared the same function depended on the type of conservation pattern. To address this question, we compared the sets of GO biological process terms at the first hierarchy level across the interface pairs with residue pairs from three main conservation Types: 1, 2 and 4, where each GO term was extracted from the GO annotation of a PDB structure containing the interaction. Specifically, for each two pairs of charged residues, we determined if the sets of GO terms between the corresponding protein–protein interactions (1) were identical or (2) had a nonempty overlap. As a result, at the 95% redundancy level, there were 40% of protein–protein interaction pairs containing residue pairs of Type 1 with identical sets of GO annotations and 70% with at least one GO term in common. The conserved residue pairs of Type 2 accounted for a similar amount of 38% and 76% of protein–protein interactions, and residue pairs of Type 4 accounted for 22% and 79% of protein–protein interactions. Finally, we found that the sets of interactions associated with each conservation type shared similar sets of GO annotation terms (data not shown). The latter fact suggested that the conservation patterns of charged residues are not likely to be associated with any specific biological function.
Conservation Patterns of Charged Residues are Similar Across Different Superkingdoms and Structural Classes of Proteins
To determine if the conservation of charged residues is intrinsically different within individual domains of life, we analyzed the conservation patterns of charged residues across the Archaea, Bacteria, and Eucarya superkingdoms, as well as Viruses (V) (Table II and Supporting Information Table S1). Although there were only a few pairs of interactions involving viral proteins, the analysis across the three superkingdoms revealed that the correlated reappearance (Type 4) is the most frequent conservation pattern among pairs of charged residues. Moreover, a pair of interfaces formed by proteins from the same superkingdom is more likely to be of Type 4 than of Type 1. The correlated reappearance was also the most likely pattern when comparing the interfaces of protein complexes from two different kingdoms; the only exception was the set of interface pairs where one protein interface was from an Archaea species and another one was from a Eucarya species, in which conserved pairs of charged residues (Type 2) were most frequently found.
Table II.
Distribution of charged residues among the pairs of interfaces (95% redundancy level) across the Archaea (A), Bacteria (B), Eucarya (E) superkingdoms as well as Viruses (V)
| A | B | E | V | |
|---|---|---|---|---|
| (A) | ||||
| A | 7 | 16 | 11 | 2 |
| B | 116 | 85 | 0 | |
| E | 133 | 0 | ||
| V | 2 | |||
| (B) | ||||
| A | 0 | 1 | 0 | 0 |
| B | 34 | 7 | 0 | |
| E | 48 | 0 | ||
| V | 0 | |||
| (C) | ||||
| A | 0 | 15 | 12 | 1 |
| B | 52 | 82 | 0 | |
| E | 60 | 0 | ||
| V | 0 | |||
| (D) | ||||
| A | 4 | 1 | 9 | 1 |
| B | 21 | 3 | 0 | |
| E | 10 | 0 | ||
| V | 1 | |||
| (E) | ||||
| A | 14 | 4 | 4 | 0 |
| B | 68 | 34 | 0 | |
| E | 83 | 0 | ||
| V | 1 | |||
(A) Distribution of homologous interaction pairs considered for the analysis. (B) Distribution of interaction pairs where neither of interactions has a charged residue pair. (C) Distribution of unconserved charged residue pairs (Type 1). (D) Distribution of conserved charged residue pairs (Type 2). (E). Distribution of correlated reappearance charged residue pairs (Type 4). For tables (A and B), position (X,Y) in a table corresponds to the number of interaction pairs from an interface formed by two proteins from kingdom X and an interface formed by two proteins from kingdom Y. For tables (C–E), position (X,Y) in a table corresponds to the number of charged residue pairs of the given type between a pair of residues from an interface formed by two proteins from kingdom X and a residue pair from another interface formed by two proteins from kingdom Y. Note that a single interface can contribute to more than one class of charged residue pairs.
We next studied whether the conservation patterns of charged residues depended on the structural properties of the proteins forming corresponding interaction interfaces (Table III and Supporting Information Table S2). Specifically, we analyzed the distribution of conservation types for five protein SCOP classes of proteins, a–d, and g (other SCOP classes, e, f and h–k, contributed only to a few interactions and were excluded from the analysis). We found that correlated reappearance is the prevailing conservation pattern for the charged residues irrespective of the structural class of proteins forming the interface. Interestingly, interfaces formed exclusively by proteins consisting of the segregated alpha and beta regions (SCOP class d) were found to have a larger proportion of fully conserved charged residue pairs than other interfaces.
Table III.
Distribution of charged residues among the pairs of interfaces (95% redundancy level) across five SCOP classes
| a | b | c | d | g | |
|---|---|---|---|---|---|
| (A) | |||||
| a | 74 | 14 | 12 | 10 | 6 |
| b | 24 | 83 | 12 | 0 | |
| c | 56 | 13 | 0 | ||
| d | 42 | 2 | |||
| g | 16 | ||||
| (B) | |||||
| a | 33 | 2 | 1 | 3 | 1 |
| b | 4 | 15 | 4 | 0 | |
| c | 8 | 2 | 0 | ||
| d | 6 | 1 | |||
| g | 8 | ||||
| (C) | |||||
| a | 31 | 8 | 9 | 3 | 6 |
| b | 20 | 61 | 1 | 0 | |
| c | 43 | 6 | 0 | ||
| d | 24 | 2 | |||
| g | 7 | ||||
| (D) | |||||
| a | 5 | 3 | 2 | 2 | 0 |
| b | 0 | 11 | 3 | 0 | |
| c | 2 | 3 | 1 | ||
| d | 17 | 0 | |||
| g | 1 | ||||
| (E) | |||||
| a | 30 | 5 | 4 | 6 | 0 |
| b | 4 | 64 | 6 | 0 | |
| c | 36 | 9 | 1 | ||
| d | 38 | 0 | |||
| g | 0 | ||||
(A) Distribution of homologous interaction pairs considered for the analysis. (B) Distribution of interaction pairs where neither interaction has a charged residue pair. (C) Distribution of unconserved charged residue pairs. (D) Distribution of conserved charged residue pairs. (E) Distribution of correlated reappearance charged residue pairs. For tables (A) and (B), position (X,Y) in a table corresponds to the number of charged residue pairs of the given type between a pair of residues from an interface formed by a protein from SCOP class X and a protein from SCOP class Y. For tables (C–E), position (X,Y) in a table corresponds to the number of two pairs of residues, each pair from a distinct interface formed by a protein from SCOP class X and a protein from SCOP class Y.
Charged Residue Conservation Patterns Across Homologous Interfaces in Intraspecies and Interspecies Interactions: Case Studies
We performed a more detailed analysis of the conservation patterns by considering several case studies (Fig. 2). We first selected five pairs of homologous interfaces that exhibited different types of conservation patterns. Specifically, we were interested in comparing the conservation patterns for (i) pairs of highly similar interfaces, those sharing the same SCOP families [Fig. 2(A–C)] and (ii) more distantly related interfaces, those formed by proteins sharing the same SCOP superfamilies but not the same families [Fig. 2(D,E)]. First, we found that the conserved residues pairs (Type 2) occurred almost exclusively among the highly similar interfaces [Fig. 2(A)], while other conservation types, Types 3 and 4, were found in both highly similar and distantly related interfaces [Fig. 2(B–E)]. In addition, the analysis revealed some interfaces with several groups of charged residue pairs, each of which corresponded to a different conservation pattern [Fig. 2(B,D)], including the only two detected cases of swapped charges (Type 3). All other interfaces had residue pairs only of a single conservation pattern [Fig. 2(A,C,E)].
Figure 2.

Case studies of the conservation of charged residue pairs in homologous interfaces. For each case study, the following are shown: (i) the overall structural alignment of the interacting complexes, (ii) the structural alignment of the interaction interfaces, and (ii) the superposed binding sites with the shown positively charged (blue and cyan) and negatively charged (red and magenta) residues delineated. A: Conserved charged residue pairs. B and D: Swapped charged residue pairs. C and E: Charged residue pairs of correlated reappearance. In the first three cases (A–C), each pair of homologous subunits that forms the interaction is from the same SCOP family, while in the last two cases (D and E), at least one pair of the interfaces is from two different families, although within the same SCOP superfamily.
We also investigated the conservation patterns in protein–protein interactions between host and pathogen organisms. Although the number of host–pathogen interactions (HPIs) was limited in our dataset, detailed examination of the HPI case studies can provide insight into intrinsic differences between the intraspecies and interspecies protein–protein interactions. In our next case study, the first complex (PDB ID: 1PVH) was an intraspecies interaction between a human cell-surface signaling receptor gp130, which is known to interact with a variety of cytokines and other proteins, and leukemia inhibitory factor (LIF). With this interaction, we found one pair of charged residues in contact (Glu141 and Arg15, correspondingly).24 The second complex was a homologous HPI occurring between the same receptor gp130 and Kaposi's sarcoma-associated herpesvirus interleukin-6 protein, vIL6 (PDB ID: 1I1R), which has been suggested to mimic the human intraspecies interaction.25 Interestingly, when structurally aligned with LIF, vIL6 did not have a charged residue corresponding to Arg15 (Supporting Information Fig. S1); moreover, Glu141 of gp130 did not even participate in the gp130–vIL6 interaction.24 The absence of the charged contact residue in the HPI interface suggests that it may play a secondary role in forming interaction interfaces between gp130 and its partners.
Discussion
In this work, we performed a large-scale analysis of the conservation patterns for the charged residue pairs detected in the interfaces of homologous interactions. To do so, we first defined two structural interface similarity measures and then selected the most accurate one based on an analysis of how well each similarity measure distinguishes between the pairs of similar and dissimilar interfaces. Using the selected similarity measure, we defined a concept of a homologous interface and extracted pairs of homologous interfaces from a structural database of protein–protein interactions. Based on the preliminary data, we then introduced four basic types of the conservation patterns, characterized all charged residue pairs by type, and proceeded with a statistical analysis of occurrence frequency for each type in the pairs of homologous interfaces.
The main result of this work is the identification of an unexpectedly high number of conservation patterns of a new type, which we call the correlated reappearance patterns. Specifically, for a pair of interfaces where one homologous interface has a charged residue pair, the other interface “loses” a charge residue pair in the same location but “gains” it in a different region of the interface. Together with unconserved charged residue pairs, they were the most abundant conservation patterns. These observations were further analyzed based on a set of five case studies, where we found that a pair of homologous interaction interfaces may exhibit more than one conservation pattern. The analysis of the conservation patterns of charged residue across different superkingdoms as well as structural classes of proteins has revealed that the correlated reappearance type is by far the most dominating conservation pattern, often occurring more frequently than the unconserved charged residues. We have also determined an interesting, but rather rare, phenomenon in which “charge swapping” is demonstrated among the colocalized pairs of charged residues, which is a good example of correlated mutations occurring in the interface. These findings suggest expanding the principles of structural conservation of interaction residues.
The obtained picture is consistent with both the fact that the conservation of charged residues is frequently observed in the binding sites of homologous proteins as well as the fact that on average, the charged residue pairs are not as conserved as other residue types. The latter is due to the definitions of residue conservation that the current methods use: either sequentially, through a conserved position in a sequence alignment or structurally through a conserved location on a consensus surface obtained by a structural superposition. We hypothesize that to form a protein-protein interaction, it is often not a specific location of the charged residue pairs that needs to be conserved, but rather their mere presence in the protein interface. This hypothesis could perhaps be associated with the long-range steering mechanisms mediated by electrostatic interactions by the charged residues. We suggest it is the recognition of a receptor binding site by a ligand protein binding site, rather than a precise orientation of the two binding sites, that is the primary role for such pairs of charged residues. We are planning to obtain further insights into the intrinsic mechanisms behind the electrostatic steering by evaluating the overall charge distribution across the interface surfaces and associating the global and local charge distribution patterns with the magnitude of correlated reappearance events among the pairs of charged residues.
Our future steps will include expanding the conservation analysis to other types of interactions that are known to be crucial in forming protein interaction interfaces, for example, disulfide and hydrogen bonds. We will also determine and compare the conservation patterns of the other residue types, such as aromatic residues, that are known to be more conserved in the interaction interfaces than the charged residues. Finally, we are planning to study the evolutionary changes in the conservation patterns themselves by comparing the patterns across all members of homologous families.
Materials and Methods
Our approach to study the conservation of charge residues in the interfaces of homologous structures on a large scale consists of four steps (Fig. 1). First, we introduce two structural measures of the interface similarity. One measure relies on aligning the entire structures of the interacting subunits, while the other relies on structural superposition of the interacting interfaces only. Second, we analyze the ability of each similarity measure to separate similar and nonsimilar interfaces and select the most accurate measure to define a nonredundant set of homologous interactions. Third, we define four types of conservation patterns among the charged residue pairs occurring in homologous interaction interfaces. Finally, every two pairs of charged residues, one from each interface, are classified into one conservation pattern type, and the occurrence frequency for each type is calculated.
Structural Interface Similarity Measures: iiRMSD Versus siRMSD
Although a widely used SCOP classification of similar and evolutionary-related protein and domain structures can be readily used to determine homologous subunit pairs, determining whether two interaction interfaces are similar is not straightforward. In this work, we consider two structural interface similarity measures, iiRMSD and siRMSD. By evaluating the performance of the two similarity measures on two datasets of similar and dissimilar interface pairs obtained from the 3D Complex database,26 we can select the measure that better separates the similar and dissimilar interfaces. The first similarity measure is called the interaction interface RMSD (iiRMSD) and is similar to the L_RMSD measure, a commonly used measure in the critical assessment of prediction of interactions.27 It is defined using the overall structures of the two interacting subunits. Given two protein–protein interactions, one between subunits A1 and A2 and another between subunits B1 and B2, iiRMSD is calculated in four steps:
structurally align one subunit Ai with another subunit Bj (i, j∈{1, 2}) using MultiProt software28;
superpose the remaining two subunits according to the above alignment;
calculate an average of Cα-based RMSD between the binding site residues for each pair of the superposed subunits; and
select iiRMSD to be the smallest of the Cα-based RMSD averages over four possible superposition scenarios in step 1: A1-B1, A1-B2, A2-B1, or A2-B2.
The second similarity measure is called the superposed interface RMSD (siRMSD) and is defined exclusively using the structures of the interaction interfaces. To calculate siRMSD, the two interfaces are first structurally aligned using the same MultiProt software followed by the calculation of the Cα-based RMSD between the aligned residues of the interaction interfaces.
Next, we compare the two similarity measures and select the one that distinguishes between similar and dissimilar interfaces more accurately. To achieve that, we first construct a reliable dataset of similar and dissimilar interface pairs and then estimate the difference between the distributions of similarities generated for the sets of similar and dissimilar interface pairs by each of the two measures. Our dataset of protein interfaces is obtained from 3D Complex, a nonredundant database of protein complexes that are grouped into hierarchically organized classes based on their similarity in sequence, structure, or topology.26 The hierarchical classification system in 3D Complex consists of 12 levels. In this work, the pairs of interfaces are extracted from the complexes at the third, quaternary structures (QS), level. At this level, the complexes grouped into the same class are structurally similar, as they share (i) the number of chains, (ii) pattern of contacts, (iii) the total number of distinct genes used to generate the subunits in each protein complex, and (iv) structural similarity between all corresponding pairs of subunits. We first select 5924 pairs of similar complexes from 4005 QS classes. We consider classes that have at least two protein complexes, randomly selecting one or more pairs of complexes from each class. We then select two structurally similar binary interactions, one from each complex, and extract the interaction interface from each binary interaction. The obtained dataset consists of 5924 pairs of similar interfaces. Second, we generate a set of 4491 pairs of structurally unrelated protein complexes. These complexes serve as the source of dissimilar binary interaction pairs. Each pair of binary interactions is selected by randomly picking two complexes from two distinct QS classes, such that the pairs of binary interactions extracted from these complexes are formed by four different subunits (different homologous chain IDs for all four subunits). As a result of the selection protocol, 4491 pairs of dissimilar interfaces are selected and added to the dataset.
To evaluate the ability of the iiRMSD and siRMSD similarity measures to distinguish between similar and dissimilar interfaces, we use the Bhattacharyya coefficient (BC)-based metric.29 Specifically, we compare the distributions of similarity values between the sets of similar and dissimilar interfaces generated by each measure for the entire datasets of similar interfaces and unrelated complexes. The BC-based metric is defined as  , where ρ(p,q) is the BC between two distributions p and q. The BC can be approximated using n-bin histograms as
, where ρ(p,q) is the BC between two distributions p and q. The BC can be approximated using n-bin histograms as 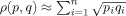 , where pi and qi are the normalized frequencies of the corresponding histograms.
, where pi and qi are the normalized frequencies of the corresponding histograms.
The analysis of the distributions for both similarity measures on similar and dissimilar sets using n = 50 bins (Fig. 3) reveals that the difference of mean values between the distributions of similar and dissimilar interfaces generated using iiRMSD measure (Δμ = 4.73) is larger than that one generated using siRMSD measure (Δμ = 1.11). Moreover, the comparison of two similarities using the BC-based metric has revealed the larger distance between the distributions generated by iiRMSD (dBC = 0.36) compared to those ones generated by siRMSD (dBC = 0.23). This suggests that the iiRMSD measure may better differentiate between similar and dissimilar interfaces than siRMSD. Therefore, the iiRMSD similarity measure is selected to define the comprehensive set of homologous protein–protein interactions.
Figure 3.

Histograms of iiRMSD and siRMSD value distributions calculated on the similar and dissimilar interface dataset obtained from 3D Complex. The two distributions of similarities for similar and dissimilar interfaces generated using the iiRMSD measure (A) show better separation than the corresponding distributions generated using siRMSD measure, which is further supported by our analysis using Bhattacharyya coefficient-based metric.
Collecting Protein Interaction Interface Data
To obtain a set of homologous protein–protein interactions, we apply a five-step protocol to a comprehensive dataset of protein–protein interaction structures extracted from PIBASE.30 First, we exclude the structures with resolution worse than 2.5 Å (the resolution information is obtained from PDB).31 Second, we define two levels of redundancy of the subunits participating in the interactions. At the first redundancy level, redundant subunits have 100% identical sequences as defined by ASTRAL SCOP 1.75.32 At the second level, redundant subunits share at least 95% sequence identity (seq_id). Third, for each subunit in a protein–protein interaction, we assigned a SCOP superfamily ID33; proteins or protein domains from the same SCOP superfamily are defined as evolutionary related, based on structural, functional, and sequence evidence. Fourth, all interactions are grouped based on the SCOP superfamily IDs of the participating subunits: the interactions within the same group share the same pairs of assigned SCOP superfamily IDs. Fifth, we calculate the iiRMSD of interfaces extracted from each pair of protein–protein interactions from the same group (Fig. 1). We define two interfaces to be similar if the iiRMSD measure between them is smaller than 8 Å. The threshold is selected based on our analysis of the iiRMSD values for similar and dissimilar interfaces and has allowed us to decrease the number of false positives, such as interfaces from two different binding modes formed by the same or homologous pairs of subunits.
Acknowledgments
The authors thank members of the Korkin and Shyu labs for helpful discussions and suggestions on the project.
Supplementary material
References
- 1.Alberts B. Essential cell biology: an introduction to the molecular biology of the cell. New York: Garland Pub; 1998. [Google Scholar]
- 2.Davis FP, Barkan DT, Eswar N, McKerrow JH, Sali A. Host pathogen protein interactions predicted by comparative modeling. Protein Sci. 2007;16:2585–2596. doi: 10.1110/ps.073228407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loewenstein Y, Raimondo D, Redfern OC, Watson J, Frishman D, Linial M, Orengo C, Thornton J, Tramontano A. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Shen BJ, Sebald W. A mixed-charge pair in human interleukin 4 dominates high-affinity interaction with the receptor alpha chain. Proc Natl Acad Sci USA. 1997;94:1657–1662. doi: 10.1073/pnas.94.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis SJ, Davies EA, Tucknott MG, Jones EY, van der Merwe PA. The role of charged residues mediating low affinity protein–protein recognition at the cell surface by CD2. Proc Natl Acad Sci USA. 1998;95:5490–5494. doi: 10.1073/pnas.95.10.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamburger ZA, Brown MS, Isberg RR, Bjorkman PJ. Crystal structure of invasa bacterial integrin-binding protein. Science. 1999;286:291–295. doi: 10.1126/science.286.5438.291. [DOI] [PubMed] [Google Scholar]
- 7.Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein–protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 8.Xu B, Stippec S, Robinson FL, Cobb MH. Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. J Biol Chem. 2001;276:26509–26515. doi: 10.1074/jbc.M102769200. [DOI] [PubMed] [Google Scholar]
- 9.Sinha N, Smith-Gill SJ. Electrostatics in protein binding and function. Curr Protein Pept Sci. 2002;3:601–614. doi: 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- 10.Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 11.Streb JW, Miano JM. Cross-species sequence analysis reveals multiple charged residue-rich domains that regulate nuclear/cytoplasmic partitioning and membrane localization of a kinase anchoring protein 12 (SSeCKS/Gravin) J Biol Chem. 2005;280:28007–28014. doi: 10.1074/jbc.M414017200. [DOI] [PubMed] [Google Scholar]
- 12.Slagle SP, Kozack RE, Subramaniam S. Role of electrostatics in antibody–antigen association: anti-hen egg lysozyme/lysozyme complex (HyHEL-5/HEL) J Biomol Struct Dyn. 1994;12:439–456. doi: 10.1080/07391102.1994.10508750. [DOI] [PubMed] [Google Scholar]
- 13.Nelson CA, Viner NJ, Young SP, Petzold SJ, Unanue ER. A negatively charged anchor residue promotes high affinity binding to the MHC class II molecule I-Ak. J Immunol. 1996;157:755–762. [PubMed] [Google Scholar]
- 14.Stenlund P, Lindberg MJ, Tibell LA. Structural requirements for high-affinity heparin binding: alanine scanning analysis of charged residues in the C-terminal domain of human extracellular superoxide dismutase. Biochemistry. 2002;41:3168–3175. doi: 10.1021/bi011454r. [DOI] [PubMed] [Google Scholar]
- 15.Wade RC, Gabdoulline RR, Ludemann SK, Lounnas V. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc Natl Acad Sci USA. 1998;95:5942–5949. doi: 10.1073/pnas.95.11.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber G. Kinetic studies of protein–protein interactions. Curr Opin Struct Biol. 2002;12:41–47. doi: 10.1016/s0959-440x(02)00287-7. [DOI] [PubMed] [Google Scholar]
- 17.Haberland J, Gerke V. Conserved charged residues in the leucine-rich repeat domain of the Ran GTPase activating protein are required for Ran binding and GTPase activation. Biochem J. 1999;343(Pt 3):653–662. [PMC free article] [PubMed] [Google Scholar]
- 18.Unkles SE, Rouch DA, Wang Y, Siddiqi MY, Glass AD, Kinghorn JR. Two perfectly conserved arginine residues are required for substrate binding in a high-affinity nitrate transporter. Proc Natl Acad Sci USA. 2004;101:17549–17554. doi: 10.1073/pnas.0405054101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawtin SR, Simms J, Conner M, Lawson Z, Parslow RA, Trim J, Sheppard A, Wheatley M. Charged extracellular residues, conserved throughout a G-protein-coupled receptor family, are required for ligand binding, receptor activation, and cell-surface expression. J Biol Chem. 2006;281:38478–38488. doi: 10.1074/jbc.M607639200. [DOI] [PubMed] [Google Scholar]
- 20.Guharoy M, Chakrabarti P. Conserved residue clusters at protein–protein interfaces and their use in binding site identification. BMC Bioinformatics. 2010;11:286. doi: 10.1186/1471-2105-11-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aloy P, Russell RB. InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics. 2003;19:161–162. doi: 10.1093/bioinformatics/19.1.161. [DOI] [PubMed] [Google Scholar]
- 22.Korkin D, Davis FP, Sali A. Localization of protein-binding sites within families of proteins. Protein Sci. 2005;14:2350–2360. doi: 10.1110/ps.051571905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulanger MJ, Bankovich AJ, Kortemme T, Baker D, Garcia KC. Convergent mechanisms for recognition of divergent cytokines by the shared signaling receptor gp130. Mol cell. 2003;12:577–589. doi: 10.1016/s1097-2765(03)00365-4. [DOI] [PubMed] [Google Scholar]
- 25.Chow D, He X, Snow AL, Rose-John S, Garcia KC. Structure of an extracellular gp130 cytokine receptor signaling complex. Science. 2001;291:2150–2155. doi: 10.1126/science.1058308. [DOI] [PubMed] [Google Scholar]
- 26.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janin J, Henrick K, Moult J, Eyck LT, Sternberg MJ, Vajda S, Vakser I, Wodak SJ. CAPRI: a Critical Assessment of PRedicted Interactions. Proteins. 2003;52:2–9. doi: 10.1002/prot.10381. [DOI] [PubMed] [Google Scholar]
- 28.Shatsky M, Nussinov R, Wolfson HJ. A method for simultaneous alignment of multiple protein structures. Proteins. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- 29.Comaniciu D, Ramesh V, Meer P. Kernel-based object tracking. IEEE Trans Pattern Anal Mach Intell. 2003;25:564–575. [Google Scholar]
- 30.Davis FP, Sali A. PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics. 2005;21:1901–1907. doi: 10.1093/bioinformatics/bti277. [DOI] [PubMed] [Google Scholar]
- 31.Berman HM. The Protein Data Bank: a historical perspective. Acta Crystallogr A. 2008;64:88–95. doi: 10.1107/S0108767307035623. [DOI] [PubMed] [Google Scholar]
- 32.Chandonia JM, Hon G, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. The ASTRAL Compendium in 2004. Nucleic Acids Res. 2004;32:D189–D192. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.