

Abstract
It is well established that contact order and folding rates are correlated for small proteins. The folding rates of stefins A and B differ by nearly two orders of magnitude despite sharing an identical native fold and hence contact order. We break down the determinants of this behavior and demonstrate that the modulation of contact order effects can be accounted for by the combined contributions of a framework-like mechanism, characterized by intrinsic helix stabilities, together with nonnative helical backbone conformation and nonnative hydrophobic interactions within the folding transition state. These contributions result in the formation of nonnative interactions in the transition state as evidenced by the opposing effects on folding rate and stability of these proteins.
Introduction
For proteins that fold with two-state kinetics, the correlation between folding rate and contact order (the average separation of residues in physical contact in the native state divided by the sequence length) suggests that folding rates are determined by the topology of the native fold and thus independent of the stability of the final folded state (1). Structures of folding transition states are then predicted to be similar in small proteins with equivalent native topologies and defined by the spatial probability of forming native interactions (1). The introduction of contact order extends the nucleation-condensation view of two-state protein folding (2,3) (where both secondary and tertiary contacts are formed simultaneously around a folding nucleus) by taking into account the effective concentration of the residues involved. This model differs from framework (4) and diffusion collision models (5), where it is the hierarchical formation and association of preformed secondary structures (framework model) and/or the association of folding microdomains (diffusion-collision model) that controls the rate of folding. In these latter two models, folding rates are dependent on the stabilities of individual elements of secondary structure, whether or not this leads to an observable population of stable kinetic intermediates.
The cysteine proteinase inhibitors (6) stefins A and B are well-studied examples of proteins that fold with two-state kinetics (7–9). Both proteins comprise 98 residues and share a high level of sequence identity (54%), with no insertions or deletions within the alignment (Fig. 1 A). They have a characteristic hot dog fold in which an α-helix is surrounded by a 5-stranded, antiparallel β-sheet with a topology β1αβ2β3β4β5,(Fig. 1, B and C) (10–12). Despite the similarity in fold and contact order, stefin B folds 50 times faster than stefin A (7–9). Clearly in this case, contact order is not the sole determinant of folding rate.
Figure 1.
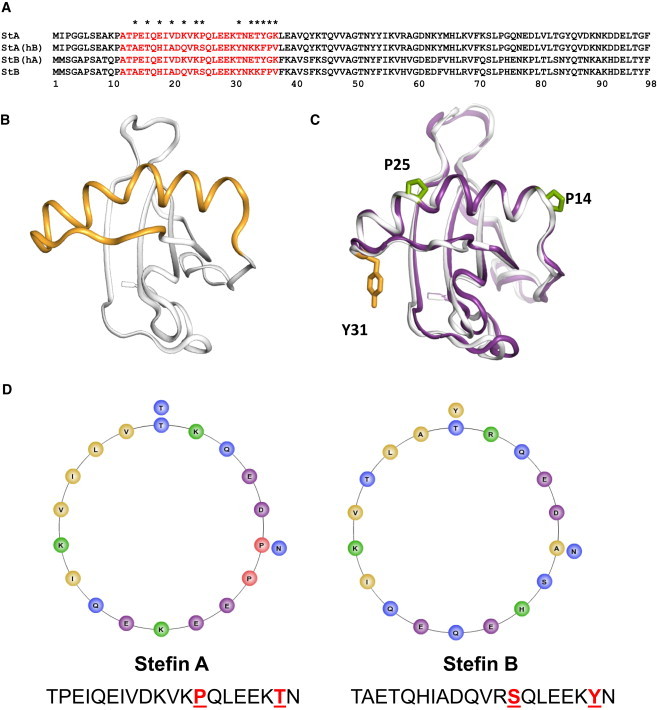
Structural features of stefin constructs. (A) Sequence alignments of stefin A, stefin B, stefin A (helixB), and stefin B (helixA). Regions defining the chimeras are highlighted and nonidentical residues marked with an asterisk. (B) Ribbon representation (pyMOL, Delano Scientific, 2002) of the structure of stefin A (pdb code 1NB5J (10)) showing the regions of the protein that have been exchanged to produce the chimeras. (C) Ribbon representations (pyMOL, Delano Scientific, 2002) of the superimposed structures of stefin A (darker color) and stefin B (white) (pdb codes 1NB5J (10) and 1STF (12)). The c-alpha atoms of the α-helices (residues 13–32) were superimposed using LSQKAB, showing a high similarity (rmsd = 1.07 Å). In (C) the positions of P25 in stefin A and Y31 in stefin B are shown as sticks. Residue P14 is also shown to pinpoint the N-terminal of the helix. (D) Helical wheel projections of the helices (residues 13–32) of stefin A and stefin B, where different shades represent hydrophobic, polar, positively charged, negatively charged, and proline residues, respectively. The corresponding amino acid sequences are shown beneath and the residues mutated in this study underlined. Projections were generated using the web-based program: (www-nmr.cabm.rutgers.edu/bioinformatics/Proteomic_tools/Helical_wheel/).
The potential contribution from a framework-like mechanism can be inferred from previous work using chimeras of stefins A and B, in which residues corresponding to the α-helix (residues 12–37) are replaced with the equivalent sequence from the counterpart protein (stefin A (helixB) and stefin B (helixA)) (Fig. 1 B) (9). Although the helix represents only 25% of the protein chain, its sole presence appears to dictate the rate of folding and therefore the stability of the transition state. In other words, we observe that the stefin B (helixA) chimera, which is mostly stefin B, folds at a similar rate to stefin A. Conversely, the stefin A (helixB) chimera folds at a similar rate to stefin B, indicating that the transition state of this chimera is stabilized relative to that of wild-type stefin A by the insertion of the helix of stefin B (9) (Table 1, Fig. 2 A).
Table 1.
Thermodynamic parameters for stefins and derived peptides
| Polypeptide | kfo (s−1) | ΔGF/Uo (kcal mol−1) |
|---|---|---|
| Stefin A peptide | – | +3.2 ± 0.2∗ |
| Stefin B peptide | – | +2.3 ± 0.2∗ |
| Stefin A | 14 ± 1.0 | −8.7 ± 0.3 |
| Stefin B (Y31) | 620 ± 120 | −4.5 ± 0.5 |
| Stefin A (helix B) | 310 ± 50 | −5.5 ± 0.3 |
| Stefin B (helixA) | 8.2 ± 2.0 | −4.7 ± 0.3 |
| Stefin A (P25S) | 26 ± 3 | −7.6 ± 0.2 |
| Stefin A (T31Y) | 15 ± 1.3 | −6.8 ± 0.2 |
| Stefin B (E31) | 66 ± 2.0 | −6.7 ± 0.6† |
| Stefin B (Y31T) | 55 ± 4.0 | −5.5 ± 0.3 |
Folding rate constants in water (kfo (s−1)) and stability of the folded state (ΔGF/Uo) were calculated from the fit of kinetic refolding and unfolding transitions for all proteins except where indicated. All errors quoted are standard errors determined from least squares fit to the data, based on a 95% confidence limit. Values for Stefin A, stefin B (Y31), stefin A (helix A), and stefin B (helix B) were reported previously (9). Buffer conditions were 0.01 M sodium phosphate, 0.2 M NaCl, pH 7.0, and 25°C, the same for all samples and maintained the same as in (9).
∗†Stability measurements derived from equilibrium helix induction curves and equilibrium denaturation data, respectively.
Figure 2.
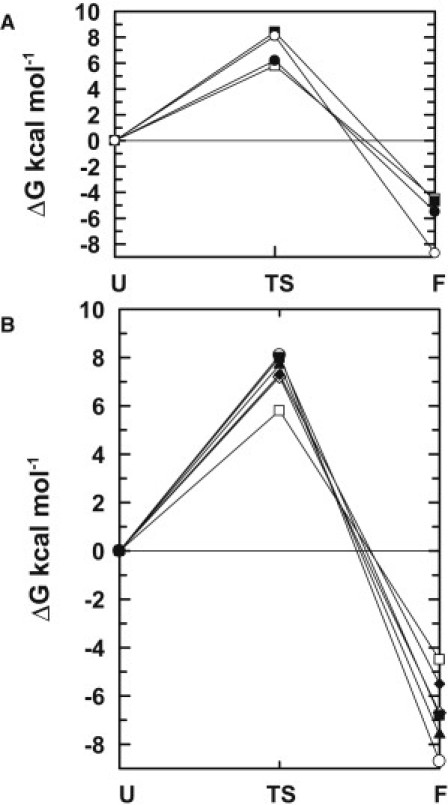
Free-energy profiles for the folding of stefin A (open circles) and Y31 stefin B (open squares). In (A) they are compared with their chimeras stefin A (helix B) (solid circles) and stefin B (helix A) (solid squares). In (B), they are shown with respect to the different single site mutants with E31 stefin B (open diamonds), Y31T stefin B (solid diamonds), T31Y stefin A (solid squares), and P25S stefin A (solid triangles). The order of stabilities in the folded state generally opposes that seen in the transition state. The activation energy for folding was derived from the rate constants (Table 1 (9),) using a preexponential factor of 107. All ΔGs are relative to the unfolded state (U).
Further scrutiny of the data does however lead to the conclusion that the transition state is stabilized by nonnative interactions, something that is not entirely consistent with a simple framework-like folding assembly or with a contact order driven model. Whereas the helix of stefin B is stabilizing and that of stefin A is destabilizing to the folding transition state of the stefins, the effect on the folded state is variable such that the effect on the stability of the transition state opposes that of the folded protein. This results in a crossover in the free-energy profiles for the folding of these proteins (Fig. 2 A) (9) and indicates that the transition state is stabilized by nonnative interactions. In other words, any contribution from framework-like behavior is not transferred to the final folded state.
Here, we elucidate the factors governing this behavior by analysis of the sequences comprising the α-helices of stefins A and B. We quantify the stabilities of the isolated helices and thereby determine the potential contribution from framework-like behavior to the folding mechanism. We then investigate the roles of a helix-breaking proline residue (P25) that is present in stefin A but absent in stefin B, a human polymorphism in stefin B (Y/E31) that is located at the C-terminal end of the helix and the equivalent residue (T31) in stefin A. We show how different effects combine to modulate contact order as the main determinant of folding rates in the stefin proteins.
Materials and Methods
Protein and peptide preparation
All site-directed mutants were made using the Quik-change method (Agilent, Wokingham, UK). For all proteins, expression and purification was performed as described (13,14) using the recombinant stefin A and stefin B constructs (15,16) bearing the C3S mutation. The structural integrity of all mutants was verified using 1D 1H-NMR. Stefin B isoforms Y/E31 show no structural differences as evidenced by the absence of significant chemical shift change in the 1H15N-hsqc spectra. Peptides corresponding to the isolated helices of stefins A and B (residues 12–33) were purchased from Peptide Protein Research, Fareham, UK. To exclude contributions from nonnative helical structure, peptides are three residues shorter than the insert used to form the chimeras, which include part of the loop region (residues 34–37).
Equilibrium denaturation
The 30 μM protein samples were incubated for 30 min at 25°C in 0.01 M sodium phosphate, 0.2 M NaCl, pH 7.0 at varying concentrations of guanidine hydrochloride (GuHCl). Circular dichroism (CD) spectra were measured using a JASCO (Great Dunmow, UK) J-810 spectropolarimeter and ellipticity at 222 nm plotted as a function of GuHCl. Data were fitted to a two-state model assuming a linear free-energy relationship (17) using Grafit (Erithracus Software, East Grinstead, UK).
Refolding kinetics
Apparent rate constants for the refolding of P25S and T31Y stefin A were measured using intrinsic fluorescence on a SX-17MV stopped-flow machine (Applied Photophysics, Leatherhead, UK) at 25°C in 0.01 M sodium phosphate, 0.2 M NaCl, pH 7.0. Refolding rates of the E31 stefin B isoform and the Y31T mutant (for which there is no change in intrinsic fluorescence) were obtained in the same buffers using CD spectroscopy. Folding rates for stefin B E31 were collected using Synchrotron radiation CD at 222 nm (Synchrotron Radiation Source, Daresbury, UK; Beamline CD-12) coupled to a custom-made stopped-flow device equipped with a commercially available data acquisition system (Applied Photophysics). Stefin B Y31T rates were measured using a Pi-Star stopped-flow machine in CD mode (Applied Photophysics). The GuHCl dependence of the rate constants was fitted according to (17): kobs = kfo exp (−mkf [GuHCl]), where kobs is the observed rate constant and kfo is the rate constant of folding in water. The data were fitted using Grafit (Erithracus). We observed no change in intrinsic fluorescence upon unfolding of E31 stefin B (i.e., Y31 is solely responsible for reporting this change in Y31 stefin B). We found previously that folding rates of stefin A, stefin B (Y31), and the chimeric proteins, stefin A (helixB) and stefin B (helixA), are not probe dependent (9), which justifies our use of CD spectropolarimetry to measure the refolding kinetics of E31 and T31 stefin B.
We have chosen to fit our data to a model in which the free energies of the mutant unfolded states are unchanged. The alternative view where changes are seen in the unfolded state free energy would favor a model where point mutations have consistent effects. This is not what is seen in our data (for example, T31Y stefin A versus Y31T stefin B).
Peptide stability and helix propensity
Peptide samples in 0.01 M sodium phosphate, 0.2 M sodium sulfate, pH 7.0 were incubated at room temperature for 30 min at varying trifluoroethanol (TFE) concentrations. CD spectra were recorded using a JASCO J-810 spectropolarimeter. Helix-induction curves were fitted using Grafit (Erithracus) according to a two-state model (18). The percentage helicity attained at the endpoint of each titration was calculated as described elsewhere (19).
Results and Discussion
Contribution of framework-like behavior
The formation of a well-populated, isolated helix is not apparent as an intermediate in the folding kinetics of the stefins. However, the stability of the helix may determine how productively it behaves as a template on which the rate-limiting processes build. If the observed kinetics is due to the presence of such a high energy, helix-folded intermediate, then this should be reflected in the behavior of isolated peptides corresponding to the helical regions of the amino acid sequences.
The helix induction curves obtained for peptides corresponding to the helical regions of stefins A and B are sigmoidal in shape indicating a cooperative transition to a helical structure at high concentrations of 2,2,2 trifluoroethanol (TFE) (Fig. 3 A). The curves are independent of peptide concentration over a range of 0.2–1.6 mM and a clear isodichroic point at 203 nm is also observed (Fig. 3 B) thus validating the analysis of these data using a simple two-state helix-coil transition model. It is well established that TFE stabilizes α-helical conformations of peptides (e.g., 20,21), although the mechanism by which this occurs is still a matter of debate (22). Despite these disagreements, it has been shown experimentally that a linear free-energy relationship with the concentration of TFE can be assumed to determine the stability of helices in water (18). This analysis yields a stability for the isolated helix of stefin B, which is greater than that of stefin A by ΔΔG = 0.9 ± 0.4 kcal mol−1 (Table 1), a value that also defines the energetic contribution from framework-like behavior to the folding transition state. Using the relationship Δkf = e−ΔΔG/RT, this could result in a fourfold acceleration in the folding rate of stefin B relative to stefin A or up to ninefold when taking experimental error into consideration. Although substantial, this is not sufficient to explain the observed 50-fold rate enhancement (where ΔΔGTS ∼2.3 kcal mol−1 (Fig. 2 and Table 1) (9)).
Figure 3.
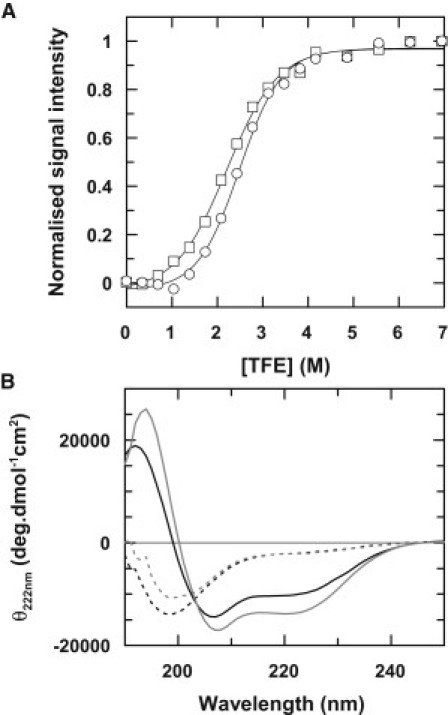
Stability and helical propensity of stefin A and stefin B peptides. (A) Helical induction curves for the isolated α-helices of stefin A (open circles) and stefin B (open squares). Data are normalized to the endpoints of each titration, which correspond to the maximum helical content attained for each peptide. (B) CD spectra are shown for stefin A (black) and stefin B (gray) peptides at 0 M (dashed lines) and 6 M (solid lines) TFE. The percentage helicity attained for the stefin A and stefin B peptides corresponding to the endpoints of the TFE titrations are 30% for stefin A and 40% for stefin B, which presumably reflects breakage of the stefin A helix at P25.
Effect of helix backbone conformation
Dramatic contributions from a nonnative backbone conformation to folding rates and stability have been reported for the SH3 domain, in which a single kink in the β-sheet that stabilizes the native protein destabilizes the transition state during folding (23). Using an analogous argument, it is plausible that the proline-free stefin B helix is able to form stabilizing contacts within the transition state, thus accelerating the folding rate while kinking of the stefin A helix by proline 25 (serine in stefin B) promotes contacts with the β-sheet and stabilizes the folded state relative to stefin B (24,25, Fig. 1 C). In accordance with these arguments, the crystal structures of both proteins show curvature in the helices, and the percentage helicity we observe for the stefin A and stefin B peptides corresponding to the endpoints of the TFE titrations is 30% for stefin A and 40% for stefin B (Fig. 3 B). This suggests that the helix of stefin A curves naturally lowering the overall CD measurement, whereas stefin B is able to form a more stable and extensive helix.
Should the change in the folding rates of the stefins arise as a result of such a model, then substitution of the proline residue by serine in stefin A (P25S) would be predicted to accelerate the folding reaction (by stabilization of the transition state) while destabilizing the final folded state. We measured an ∼twofold rate enhancement coupled with a reduced stability of the native state by 1.1 ± 0.4 kcal mol−1 for this mutant (Figs. 4, 2 B, and Table 1). The data show that the nonnative backbone conformation arising from the P25S mutation has only a modest contribution to transition state stabilization.
Figure 4.
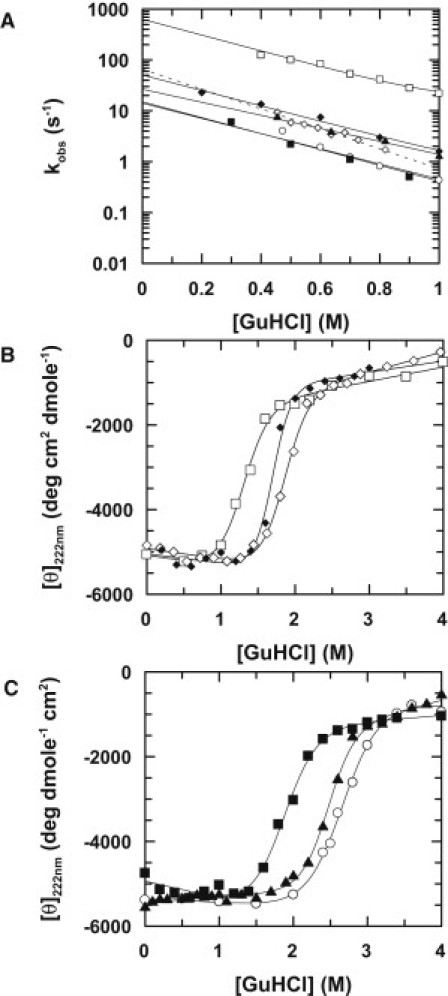
Effects of point mutations on the folding transition state of stefins. (A) Observed folding rate constants for Y31 stefin B (open squares), E31 stefin B (open diamonds), and Y31T stefin B (solid diamonds), stefin A (open circles), P25S stefin A (solid triangles), and T31Y stefin A (solid squares). Linear fits of the data are represented by continuous lines (see Methods). (B) Equilibrium denaturation monitored using CD spectroscopy of stefin B polymorphs, Y31 (open squares) and E31 (open diamonds) as well as the mutant Y31T (solid diamonds). (C) Equilibrium denaturation monitored using CD spectroscopy of stefin A wild-type (open circles) and its mutants P25S (solid triangles) and T31Y (solid squares). Mean molar residue ellipticity is plotted against [GuHCl] (see Methods) and lines represent data-fitting to a two-state transition.
Effect of tyrosine/glutamate polymorphism in stefin B
At this point, we conclude that the identity of the helix controls folding, but that its stability in isolation and a principal source of its distortion in the native fold can only partially account for the variation in folding rates between stefin A and stefin B. This leaves the remaining 10 changes in helix sequence as the main source of these differences (Fig. 1). However, from previous work we know that a single polymorphism in the human stefin B gene at residue 31 (tyrosine to glutamate) changes the distribution between monomeric and dimeric forms of stefin B and profoundly influences the rates of amyloidogenesis (13,16,26,27). This suggests that there is an underlying effect on the stability of the folded state and the dynamics of the folding mechanism. Our measurements indicate a folding rate for the E31 isoform of stefin B that is ∼10-fold slower than Y31 coupled to an increase in folded state stability of 2.2 ± 1.1 kcal mol−1 (Table 1 and Figs. 4 and 2 B). These effects are substantial and reflect the same trends observed when comparing stefin B (Y31) to stefin A. Of importance, the Y31T stefin B mutant, in which Y31 is replaced by the equivalent residue in the stefin A sequence produces identical results (Table 1 and Figs. 4 and 2 B), indicating that it is the nature of the tyrosine residue at this position, not glutamate that gives rise to the observed changes.
Residue 31 is a surface-exposed residue located at the C-terminus of the stefin B helix (Fig. 1 C). We can discount the idea that differences in stability arise from different helix-capping abilities since tyrosine, glutamate, or threonine residues are not of the appropriate polarity. A far more likely source of the increase in folding rate is the greater hydrophobicity of tyrosine relative to threonine or glutamate. The importance of hydrophobic interactions in protein folding reactions is well documented, where the exclusion of hydrophobic surface from solvent provides the dominant driving force for the early collapse of the polypeptide chain (28). Hydrophobic clustering can be a feature of denatured and transition states but to increase the folding rate, the stabilization of the latter must be greater. Hence, we infer from our data that the greatest degree of burial of the tyrosine residue in stefin B is found within the transition state for folding and that destabilization of the folded state is attributable to increased solvation of the tyrosine side chain relative to the denatured state, a phenomenon sometimes referred to as the reverse hydrophobic effect (29). We reference our free-energy diagrams to the unfolded state to provide a clear visual comparison of the changes in folding rate that we observe (Fig. 2). We determined whether Y31 in stefin B is sufficient to build a cluster with the remaining stefin A sequence, as seen for the stefin A (helix B) chimera (Fig. 2 A). Although we do observe a comparable reverse hydrophobic effect in stefin A (the folded state of the T31Y mutant is destabilized by 1.9 ± 0.5 kcal mol−1), the refolding rate of stefin A T31Y is indistinguishable from that of the wild-type protein (Figs. 4 and 2 B). This indicates that the transition state stabilization observed during refolding of Y31 stefin B results from interaction between multiple side chains originally substituted in the stefin chimeras (residues 12 to 37, with 11 combinatorial possibilities) (9).
Surface hydrophobic residues have been reported previously to stabilize folding transition states with respect to the native state (30–34), and we propose here that this is a major source for the observed crossover in the free-energy plots of the stefins and their helix-substituted chimeras (Fig. 2). We conclude that hydrophobic clustering, in which Y31 plays a central role, is a major effect in this system.
Conclusions
Stefins A and B present us with an example of the interplay of different energetic contributions to the folding transition state. Elements of contact order, framework-like behavior, nonnative backbone conformation, and also nonnative hydrophobic clustering play their roles, side-by-side, to lead to the correct formation of what are relatively simple folds.
Acknowledgments
We thank the Council for the Central Laboratory of the Research Councils Daresbury Laboratory for the use of the synchrotron radiation circular dichroism beamline, CD-12.1, and associated equipment and resources. We are very grateful to Prof. Sheena Radford for the use of her Pi-Star stopped-flow machine in CD mode. We especially thank Prof. Tony Clarke for insightful discussions.
This work was supported by the Biotechnology and Biological Sciences Research Council (UK) (BB/C504035/1), the Royal Society (516002.K5631), and the Slovenian Research Agency (P1-0140, led by Boris Turk and P1-0048).
Footnotes
Clare Jelinska's present address is Medical Research Council Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford, UK.
Manca Kenig's present address is Lek Pharmaceuticals, Ljubljana, Slovenia.
References
- 1.Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 2.Fersht A.R. Optimization of rates of protein folding: the nucleation-condensation mechanism and its implications. Proc. Natl. Acad. Sci. USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abkevich V.I., Gutin A.M., Shakhnovich E.I. Specific nucleus as the transition state for protein folding: evidence from the lattice model. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 4.Kim P.S., Baldwin R.L. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 1982;51:459–489. doi: 10.1146/annurev.bi.51.070182.002331. [DOI] [PubMed] [Google Scholar]
- 5.Karplus M., Weaver D.L. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci. 1994;3:650–668. doi: 10.1002/pro.5560030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turk V., Stoka V., Turk D. Cystatins: biochemical and structural properties, and medical relevance. Front. Biosci. 2008;13:5406–5420. doi: 10.2741/3089. [DOI] [PubMed] [Google Scholar]
- 7.Zerovnik E., Virden R., Waltho J.P. On the mechanism of human stefin B folding: I. Comparison to homologous stefin A. Influence of pH and trifluoroethanol on the fast and slow folding phases. Proteins. 1998;32:296–303. [PubMed] [Google Scholar]
- 8.Zerovnik E., Jerala R., Waltho J.P. On the mechanism of human stefin B folding: II. Folding from GuHCl unfolded, TFE denatured, acid denatured, and acid intermediate states. Proteins. 1998;32:304–313. doi: 10.1002/(sici)1097-0134(19980815)32:3<304::aid-prot6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 9.Kenig M., Jenko-Kokalj S., Zerovnik E. Folding and amyloid-fibril formation for a series of human stefins' chimeras: any correlation? Proteins. 2006;62:918–927. doi: 10.1002/prot.20812. [DOI] [PubMed] [Google Scholar]
- 10.Jenko S., Dolenc I., Turk D. Crystal structure of Stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J. Mol. Biol. 2003;326:875–885. doi: 10.1016/s0022-2836(02)01432-8. [DOI] [PubMed] [Google Scholar]
- 11.Martin J.R., Craven C.J., Waltho J.P. The three-dimensional solution structure of human stefin A. J. Mol. Biol. 1995;246:331–343. doi: 10.1006/jmbi.1994.0088. [DOI] [PubMed] [Google Scholar]
- 12.Stubbs M.T., Laber B., Turk V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. EMBO J. 1990;9:1939–1947. doi: 10.1002/j.1460-2075.1990.tb08321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan G.J., Giannini S., Staniforth R.A. Exclusion of the native alpha-helix from the amyloid fibrils of a mixed alpha/beta protein. J. Mol. Biol. 2008;375:487–498. doi: 10.1016/j.jmb.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Staniforth R.A., Giannini S., Waltho J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001;20:4774–4781. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jerala R., Trstenjak M., Turk V. Cloning a synthetic gene for human stefin B and its expression in E. coli. FEBS Lett. 1988;239:41–44. doi: 10.1016/0014-5793(88)80541-6. [DOI] [PubMed] [Google Scholar]
- 16.Rabzelj S., Turk V., Zerovnik E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with EPM1. Protein Sci. 2005;14:2713–2722. doi: 10.1110/ps.051609705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson S.E., Fersht A.R. Folding of chymotrypsin inhibitor 2. 2. Influence of proline isomerization on the folding kinetics and thermodynamic characterization of the transition state of folding. Biochemistry. 1991;30:10436–10443. doi: 10.1021/bi00107a011. [DOI] [PubMed] [Google Scholar]
- 18.Jasanoff A., Fersht A.R. Quantitative determination of helical propensities from trifluoroethanol titration curves. Biochemistry. 1994;33:2129–2135. doi: 10.1021/bi00174a020. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y.H., Yang J.T., Martinez H.M. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 1972;11:4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 20.Storrs R.W., Truckses D., Wemmer D.E. Helix propagation in trifluoroethanol solutions. Biopolymers. 1992;32:1695–1702. doi: 10.1002/bip.360321211. [DOI] [PubMed] [Google Scholar]
- 21.Segawa S., Fukuno T., Noda Y. Local structures in unfolded lysozyme and correlation with secondary structures in the native conformation: helix-forming or -breaking propensity of peptide segments. Biopolymers. 1991;31:497–509. doi: 10.1002/bip.360310505. [DOI] [PubMed] [Google Scholar]
- 22.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 23.Di Nardo A.A., Korzhnev D.M., Davidson A.R. Dramatic acceleration of protein folding by stabilization of a nonnative backbone conformation. Proc. Natl. Acad. Sci. USA. 2004;101:7954–7959. doi: 10.1073/pnas.0400550101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jerala R., Zerovnik E., Turk V. Structural basis for the difference in thermodynamic properties between the two cysteine proteinase inhibitors human stefins A and B. Protein Eng. 1994;7:977–984. doi: 10.1093/protein/7.8.977. [DOI] [PubMed] [Google Scholar]
- 25.Barlow D.J., Thornton J.M. Helix geometry in proteins. J. Mol. Biol. 1988;201:601–619. doi: 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
- 26.Zerovnik E., Pompe-Novak M., Turk V. Human stefin B readily forms amyloid fibrils in vitro. Biochim. Biophys. Acta. 2002;1594:1–5. doi: 10.1016/s0167-4838(01)00295-3. [DOI] [PubMed] [Google Scholar]
- 27.Jenko Kokalj S., Guncar G., Turk D. Essential role of proline isomerization in stefin B tetramer formation. J. Mol. Biol. 2007;366:1569–1579. doi: 10.1016/j.jmb.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 28.Dill K.A. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 29.Pakula A.A., Sauer R.T. Reverse hydrophobic effects relieved by amino-acid substitutions at a protein surface. Nature. 1990;344:363–364. doi: 10.1038/344363a0. [DOI] [PubMed] [Google Scholar]
- 30.Gu H., Doshi N., Baker D. Robustness of protein folding kinetics to surface hydrophobic substitutions. Protein Sci. 1999;8:2734–2741. doi: 10.1110/ps.8.12.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poso D., Sessions R.B., Clarke A.R. Progressive stabilization of intermediate and transition states in protein folding reactions by introducing surface hydrophobic residues. J. Biol. Chem. 2000;275:35723–35726. doi: 10.1074/jbc.M001747200. [DOI] [PubMed] [Google Scholar]
- 32.Viguera A.R., Vega C., Serrano L. Unspecific hydrophobic stabilization of folding transition states. Proc. Natl. Acad. Sci. USA. 2002;99:5349–5354. doi: 10.1073/pnas.072387799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machius M., Declerck N., Wiegand G. Kinetic stabilization of Bacillus licheniformis alpha-amylase through introduction of hydrophobic residues at the surface. J. Biol. Chem. 2003;278:11546–11553. doi: 10.1074/jbc.M212618200. [DOI] [PubMed] [Google Scholar]
- 34.Cranz-Mileva S., Friel C.T., Radford S.E. Helix stability and hydrophobicity in the folding mechanism of the bacterial immunity protein Im9. Protein Eng. Des. Sel. 2005;18:41–50. doi: 10.1093/protein/gzi002. [DOI] [PubMed] [Google Scholar]