

Abstract
Aromatic amino acids of membrane proteins are enriched at the lipid-water interface. The role of tryptophan on the folding and stability of an integral membrane protein is investigated with ultraviolet resonance Raman and fluorescence spectroscopy. We investigate a model system, the β-barrel outer membrane protein A (OmpA), and focus on interfacial tryptophan residues oriented toward the lipid bilayer (trp-7, trp-170, or trp-15) or the interior of the β-barrel pore (trp-102). OmpA mutants with a single tryptophan residue at a nonnative position 170 (Trp-170) or a native position 7 (Trp-7) exhibit the greatest stability, with Gibbs free energies of unfolding in the absence of denaturant of 9.4 and 6.7 kcal/mol, respectively. These mutants are more stable than the tryptophan-free OmpA mutant, which exhibits a free energy of unfolding of 2.6 kcal/mol. Ultraviolet resonance Raman spectra of Trp-170 and Trp-7 reveal evolution of a hydrogen bond in a nonpolar environment during the folding reaction, evidenced by systematic shifts in hydrophobicity and hydrogen bond markers. These observations suggest that the hydrogen bond acceptor is the lipid acyl carbonyl group, and this interaction contributes significantly to membrane protein stabilization. Other spectral changes are observed for a tryptophan residue at position 15, and these modifications are attributed to development of a tryptophan-lipid cation-π interaction that is more stabilizing than an intraprotein hydrogen bond by ∼2 kcal/mol. As expected, there is no evidence for lipid-protein interactions for the tryptophan residue oriented toward the interior of the β-barrel pore. These results highlight the significance of lipid-protein interactions, and indicate that the bilayer provides more than a hydrophobic environment for membrane protein folding. Instead, a paradigm of lipid-assisted membrane protein folding and stabilization must be adopted.
Introduction
Membrane protein folding is an important area of research because this process is relevant to numerous topics, including protein misfolding diseases, drug discovery, and pathogenesis. Recent experimental and computational studies have focused on the thermodynamics, kinetics, and mechanisms of protein folding in the complex milieu of lipid bilayers (1–6). Hydrophobic interactions between protein side chains and the bilayer provide a key driving force for the insertion of transmembrane segments into the bilayer (7,8). Additionally, the energetic cost of burying nonhydrogen-bonded peptide backbone units is sufficiently high to require that membrane proteins adopt secondary structure in the interior of the hydrophobic bilayer (1,9,10). These and other thermodynamic considerations form the basis of current models for membrane protein folding in which the reaction is decomposed into thermodynamically isolated events, such as partitioning, folding, insertion, and association (1–3).
In contrast to the principles that focus on thermodynamics and hydrophobic effect, there is a lack of knowledge of specific protein-lipid interactions. For example, the role of aromatic side chains on the stability of membrane proteins has been explored; however, the prevalence or strengths of protein-lipid interactions, such as dipole-dipole, hydrogen-bonding, and cation-π interactions, which give rise to the enrichment of aromatic residues at the interfacial region, are not known. One reason for this lack of progress is the paucity of tools capable of providing molecular-level detail of lipid-protein interactions during the folding process. As an alternative, emphasis has been placed on the macroscopic properties of the lipid bilayer. It is now well known that bilayer properties, such as thickness, fluidity, charge density, and lateral stress, greatly influence membrane protein folding and stability (11). Although these and other findings have advanced our knowledge of membrane proteins, it is clear that a molecular understanding of protein-lipid interactions is crucial to develop a comprehensive picture of membrane protein folding.
We and others have previously aimed to obtain molecular insight into the role of anchoring aromatic amino acids at the bilayer-water interface (12–15). Tryptophan is a particularly important residue because it has the largest driving force for the bilayer interface as revealed by the White-Wimley hydrophobicity scale (1). The versatile molecular properties of tryptophan are the key to its importance in membrane protein folding and stability: tryptophan exhibits the largest nonpolar surface area, is the most polarizable residue, possesses an indole N-H moiety that is capable of hydrogen bond donation, and displays the greatest electrostatic potential for cation-π interactions (16,17). This diverse range of physical properties renders tryptophan an ideal amphiphilic residue for stabilizing the interfacial region of a membrane protein.
The model protein used in the current study is outer membrane protein OmpA. OmpA is one of the most abundant proteins in the outer membrane of Gram-negative bacteria, and consists of 325 residues that form an eight-stranded transmembrane β-barrel and a soluble C-terminal domain that is located in the periplasm (see Fig. 1). The relative ease with which wild-type and mutant OmpA can be overexpressed in Escherichia coli combined with the reversible and spontaneous nature in which the protein folds into synthetic bilayers make OmpA an ideal candidate for studies of membrane protein folding (18–22). Prior biophysical studies have included investigations of the photophysics, thermodynamics, and kinetics associated with specific tryptophan residues (23–26). Experiments with OmpA mutants that contain single tryptophan residues are especially informative because these studies reveal site-specific information without significant modification of native structure. These residues may be placed in any one of the five native positions or in a nonnative tryptophan location as we have pursued here, to provide a comprehensive view of the aromatic belt region of the protein.
Figure 1.
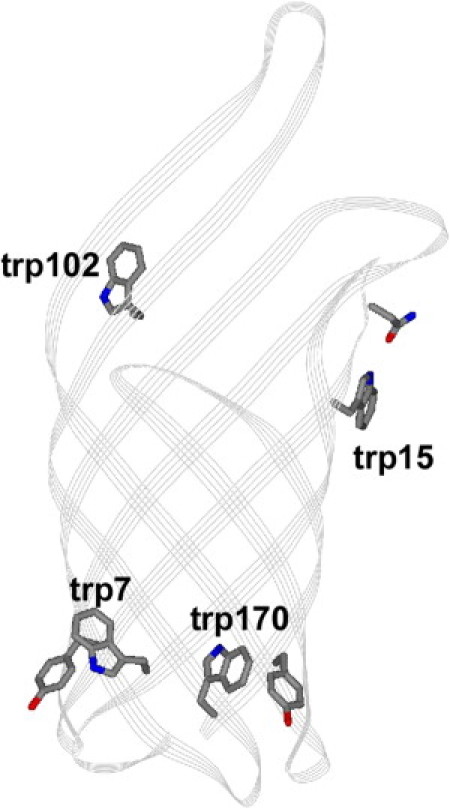
Structure of transmembrane portion of OmpA (PDB 1QJP). The native phenylalanine side chain at position 170 has been replaced with an indole group of a tryptophan residue. Trp-7, trp-15, and trp-170 are located exterior to the barrel; trp-102 is located in the interior of the barrel. Other amino acids that are suggested by the crystal structure to interact with these tryptophan residues are also shown.
The choice of ultraviolet (UV) resonance Raman (UVRR) for further studies of the anchoring tryptophan residues of OmpA is motivated by the need for structural information. This vibrational tool has been successfully applied to soluble and membrane-associated proteins and peptides, as well as to topics in protein folding and aggregation (27–31). Tryptophan is the most widely studied amino acid by UVRR, and correlations to structure/environment have been established for tryptophan normal modes (32–35). In contrast, fluorescence spectra are dependent on electronic properties associated with the chromophore and do not provide detailed structural information. Instead, fluorescence spectroscopy is a complementary method that reveals protein stability and local environment with high sensitivity. Here, we describe stabilizing molecular interactions between tryptophan residues and the bilayer-water interface during a folding reaction using UVRR and fluorescence spectroscopy. These results highlight the role of the lipid in membrane protein folding reactions, and emphasize that lipid-assisted membrane protein folding models are appropriate for this complex in vitro process.
Materials and Methods
Vesicle and sample preparation
Small unilamellar vesicles were prepared using a previously published procedure (26). Briefly, lipid solutions (Avanti Polar Lipids, Alabaster, AL) of 5 mg/mL 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, gel-to-liquid crystalline phase transition temperature Tc = 23°C), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, Tc = 41°C), or 7:3 mol ratio of DMPC/DMPE (DMPE is 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine, Tc = 50°C) in 20 mM potassium phosphate (KPi) buffer, pH 7.3, were sonicated above their respective phase transition temperatures to produce small unilamellar vesicles with diameters less than 50 nm. Vesicle solutions containing DMPC, DPPC, or DMPC/DMPE mixtures were equilibrated at temperatures above Tc up to 12 h before use. These lipids were selected because they allowed for study of folded OmpA (DMPC), an adsorbed intermediate (DPPC), or strength of protein-lipid cation-π interaction (DMPE).
OmpA mutants were generated as previously described (26). Native OmpA has five tryptophan residues at positions 7, 15, 57, 102, and 143. Five mutants are the subject of this report. Three mutants contain single tryptophan residues at positions 7, 15, or 102 and the remaining native tryptophan residues were replaced with phenylalanine. The fourth mutant contains a single tryptophan residue at a nonnative location, 170 (native residue is phe-170), with the five native tryptophan residues replaced with phenylalanine. The final mutant is tryptophan-free, and has phenylalanine in place of all tryptophan residues.
Unfolded protein samples were prepared by dilution of stock unfolded protein into 20 mM KPi buffer; residual urea concentration in the prepared samples was 0.3 to 0.8 M. Spectra of unfolded protein were collected immediately after preparation of the sample to avoid protein aggregation. OmpA mutants adsorbed on the vesicle were prepared by dilution of stock protein into buffered solution containing DPPC vesicles at 23°C, which is below its phase transition temperature; it was previously shown that OmpA does not insert into bilayers below the phase transition temperature (36). The sample was equilibrated at this temperature for 30 min before spectroscopic measurements were made. For the studies of OmpA folding into DMPC vesicles, the folding process was initiated by addition of stock protein into a buffered solution of equilibrated vesicles. Spectra were collected immediately (t = 0 min) or incubated at a temperature of 37°C until the appropriate time point. Each time point was analyzed with fresh sample. Protein samples and blanks containing DMPC/DMPE vesicle mixtures were equilibrated at 50°C for 3 h.
Protein conformation was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis and fluorescence spectroscopy (36). Adsorbed and unfolded protein has an apparent molecular mass of ∼35 kDa, whereas the folded protein has an apparent molecular mass of ∼30 kDa. This differential mobility was used in a gel-shift assay to determine the free energy of unfolding of the tryptophan-free OmpA mutant via gel densitometry (20,26).
Spectroscopic measurements
Absorption and fluorescence measurements, including generation of refolding curves, have been described previously (26). Fluorescence measurements were performed on a JY Horiba (Edison, NJ) FL-3 fluorometer with 290-nm excitation. Samples were maintained at temperatures of 30°C for solutions that contained DMPC vesicles, 23°C for solutions that contained DPPC vesicles, or above 50°C for solutions that contained DMPE vesicles.
The Raman instrumentation has been described previously (27,33). UVRR spectra were collected for the OmpA mutants at indicated time points following initiation of the folding reaction via mixing of protein + lipid solutions. The final protein concentration was ∼20 μM for all mutants. Each sample and blank UVRR spectrum was collected for a total of 5 min. Therefore, spectra collected at nominally t = 0 min reflect sample within the first ∼5 min following mixing. Similarly, spectra collected at t = 20, 40, 60, and 180 min reflect samples 20–25, 40–45, 60–65, and 180–185 min following mixing, respectively. For a given 5-min window of data collection, the individual 1-min spectra were analyzed and no significant changes were observed in any of the 5-min observation windows. Steady-state UVRR spectra of unfolded and adsorbed forms of the protein were also collected. UVRR spectra of blanks with and without urea were acquired, and the spectra presented here are difference spectra in which contributions from quartz capillary, buffer, and urea have been removed. Double-difference spectra of isolated tryptophan peaks in the Fermi doublet region are also presented. These double-difference spectra were generated by subtracting contribution of the tryptophan-free mutant from spectra of the tryptophan-containing mutants at equivalent time points in the folding reaction.
Results
The following convention is used throughout this report: tryptophan residues are referred to as trpX, and single-tryptophan OmpA mutants are referred to as TrpX, where X indicates the position for the single tryptophan in the mutant; the tryptophan-free OmpA mutant is denoted Trp-free. Vibrational modes in the UVRR spectra follow literature convention (32). Steady-state UVRR difference spectra of the five OmpA mutants folded in DMPC and unfolded in KPi buffer are shown in Fig. S1 of the Supporting Material. Strong bands arising from the 17 native tyrosine residues are observed at ∼831 and ∼853 cm−1 (Y1 + 2Y16a), ∼1179 cm−1 (Y9a), ∼1209 cm−1 (Y7a), and ∼1611 cm−1 (Y8a, with strong overlap of the trp mode W1). Signal from these tyrosine residues show minimal variation for the unfolded, adsorbed, and folded protein (Fig. S2), and will not be discussed in the current report. The vibrational modes for the single tryptophan residue are distinguishable from the tyrosine modes, and appear at ∼760 cm−1 (W18), ∼879 cm−1 (W17), ∼1238 cm−1 (W10), ∼1341/1360 cm−1 (W7), ∼1549 cm−1 (W3), and ∼1611 cm−1 (W1 with strong overlap with Y8a).
Differences in tryptophan mode frequencies and intensities are observed for the unfolded, adsorbed, and folded species. The W17 and W7 peaks are hydrogen bonding and hydrophobicity markers, respectively (33,37,38). The W17 mode appears near 875–880 cm−1, with the lower frequency corresponding to strong hydrogen-bonded N-H. The W7 mode is a sensitive marker for tryptophan environment hydrophobicity; this mode is in Fermi resonance with a combination band and appears as part of a doublet (38). The ratio of the intensities of the Fermi doublet, RFD (I∼1360/I∼1340), increases when tryptophan is in a nonpolar environment, and the RFD values reported here are consistent with those of model compounds in different solvents under resonance excitation (33). A summary of the UVRR data along with corresponding fluorescence maxima are presented in Table 1 and Table S1.
Table 1.
UVRR and fluorescence data for OmpA mutants Trp-7, Trp-15, Trp-102, and Trp-170
| Mutant | Condition | W17 | RFD | λmax |
|---|---|---|---|---|
| Trp-7 | Unfolded | 881 | 1.0 | 353 |
| Adsorbed | 879 | 1.1 | 344 | |
| DMPC, t = 0 min | 881 | 1.3 | 339 | |
| DMPC, t = 180 min | 877 | 1.7 | 327 | |
| Trp-15 | Unfolded | 881 | 1.1 | 350 |
| Adsorbed | 881 | 1.3 | 345 | |
| DMPC, t = 0 min | 879 | 1.1 | 344 | |
| DMPC, t = 180 min | 881 | 1.2 | 335 | |
| Trp-102 | Unfolded | 879 | 1.3 | 352 |
| Adsorbed | 879 | 1.1 | 347 | |
| DMPC, t = 0 min | 879 | 1.3 | 337 | |
| DMPC, t = 180 min | 877 | 1.5 | 330 | |
| Trp-170 | Unfolded | 879 | 1.3 | 353 |
| Adsorbed | 879 | 1.4 | 340 | |
| DMPC, t = 0 min | 879 | 1.5 | 337 | |
| DMPC, t = 180 min | 875 | 1.9 | 331 |
Data for mutants unfolded in buffer, adsorbed on DPPC vesicles, and during a folding reaction into DMPC vesicles immediately and 180 min following mixing of protein with vesicles. W17 is in cm−1, RFD is the ratio of the W7 Fermi doublet, I1360/I1340, and fluorescence maximum (λmax) is in nm.
The W3 mode is a UVRR conformational marker whose frequency reflects the dihedral torsion angle, χ2,1, about the C2-C3-Cβ-Cα linkage (39). Minor differences are observed in the W3 frequency, but these changes are not systematic and within the experimental error of ±2 cm−1. The W3 mode frequencies and corresponding torsion angles obtained for the folded protein are 1549 cm−1 (90°), 1551 cm−1 (96°), 1552 cm−1 (100°), and 1549 cm−1 (90°) for Trp-7, Trp-15, Trp-102, and Trp-170, respectively. The values obtained from the crystal structure are 83°, 45°, and 93° for Trp-7, Trp-15, and Trp-102, respectively. Trp-170 in Fig. 1 is modeled with the same torsion angle as the native phe-170 residue, 88°.
UVRR spectra of OmpA mutants
Trp-15
Key portions of the UVRR spectra of Trp-15 are shown in Fig. 2. The 700–910 cm−1 region that includes the W18 (∼760 cm−1) and W17 (∼881 cm−1) peaks, as well as the W7 region 1310–1400 cm−1 of the UVRR spectra of Trp-15 unfolded in KPi buffer, adsorbed on DPPC, and during the folding reaction in DMPC are displayed. A shoulder on the high-frequency edge of W18 near 778 cm−1 appears at ∼20 min following initiation of the folding reaction. The two bands in this 730–800 cm−1 region are fit with Gaussian profiles. The intensity of the high-frequency peak increases until it reaches ∼30% of the intensity of the W18 peak when the protein is fully folded at 180 min. This high-frequency shoulder persists in the spectra of Trp-15 folded in 7:3 DMPC/DMPE vesicles, and is eliminated when OmpA is folded in the neutral detergent octyl glucoside (Fig. S3).
Figure 2.
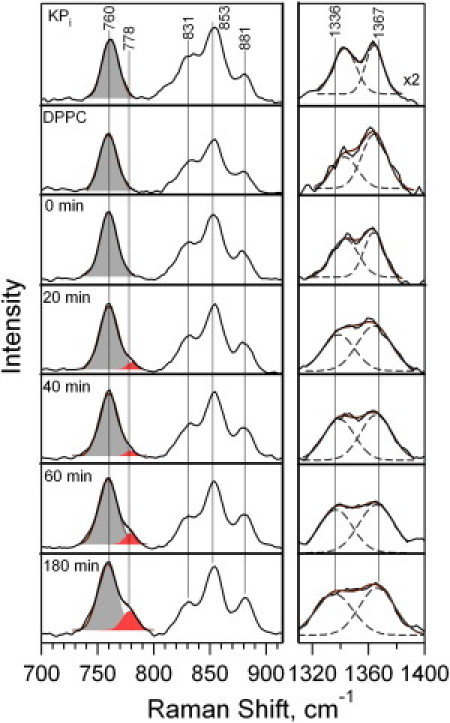
UVRR spectra of Trp-15 unfolded in KPi buffer, adsorbed on DPPC vesicles, and during a folding reaction into DMPC vesicles at t = 0 to 180 min following mixing of protein with vesicles. Gaussian decompositions are shown in the W18 (solid bands, left) and W7 (dashed lines, right) regions. Sum of Gaussian bands is shown as solid overlay. W7 region is shown as double difference spectra in which contribution from tyrosine peaks has been subtracted. Other tryptophan (W17, 881 cm−1) and tyrosine (831 and 853 cm−1) peaks are labeled.
The W7 RFD value for Trp-15 was 1.1 to 1.2 for protein during the folding reaction, and ∼1.3 for the adsorbed protein. Although the relative intensities of the W7 doublet were generally constant during folding, the positions and shapes of the peaks evolved systematically. The separation between the doublet peaks increased, and the peaks became broader during the folding process. The changes are shown by the Gaussian decompositions in Fig. 2. Unfolded Trp-15 exhibits W7 frequencies of 1343 cm−1 and 1363 cm−1, with full-width at half-maximum values of ∼18 cm−1. In the folded protein at t = 180 min, these peaks shifted to 1336 cm−1 (−7 cm−1 relative to unfolded) and 1367 cm−1 (+3 cm−1 relative to unfolded), with full-width at half-maximum values of ∼30 cm−1 (+12 cm−1 relative to unfolded) for both peaks. These changes appear at t = 20 min during the folding reaction.
The position of the W3 peak at 1551 cm−1 suggests that the dihedral angle for trp-15 obtained via UVRR is significantly different from that of the x-ray crystal structure. Trp-15 was modeled with the appropriate χ2,1 of 96° from UVRR spectra, and shown in Fig. 3. The structure of trp-15 from the crystal structure (with dihedral angle 45°) is shown for comparison.
Figure 3.
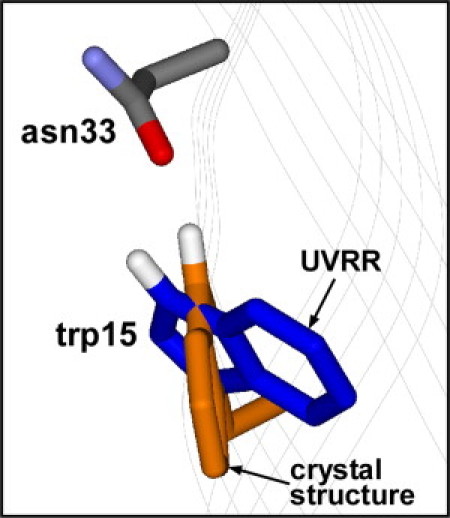
Structure of trp-15 folded in DMPC bilayer based on UVRR spectra. Crystal structure (PDB 1QJP) of trp-15 and possible hydrogen-bonding partner, Asn-33, are shown for comparison.
UVRR spectra of OmpA mutants
Trp-7
UVRR spectra or Trp-7 unfolded in buffer, adsorbed on DPPC vesicles, and at indicated time points following initiation of the folding reaction in DMPC are shown in Fig. S4. Spectral regions near W17 and W7 are shown in Figs. 4 and 5, respectively. A systematic downshift in W17 frequency from 881 to 877 cm−1 for trp-7 is observed. Fig. 5 shows the W7 Fermi doublet region in the absence of the tyrosine signal for Trp-7. The RFD value for Trp-7 increases significantly from 1.0 to 1.7 during the folding reaction, and is 1.1 for the adsorbed species.
Figure 4.
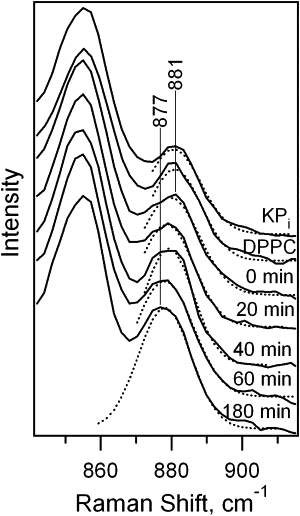
UVRR spectra (W17 region) of Trp-7 unfolded in KPi buffer, adsorbed on DPPC vesicles, and folding into DMPC vesicles at t = 0 to 180 min following mixing of protein with vesicles. Results from Gaussian decomposition are shown as the dotted curves.
Figure 5.
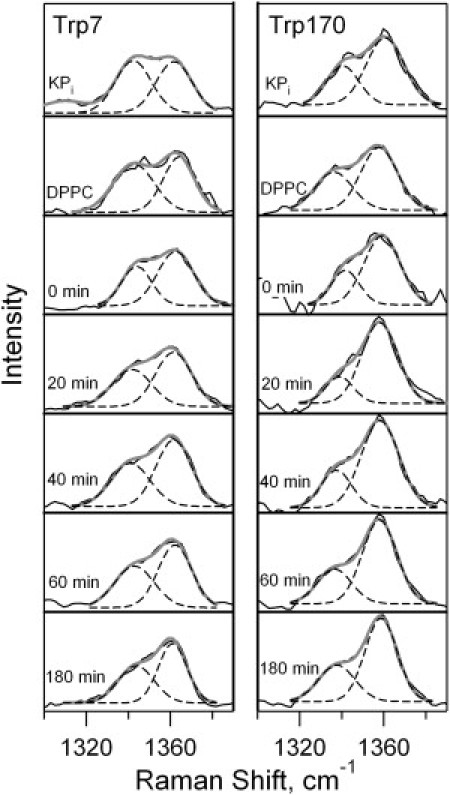
Fermi doublet regions in the UVRR double difference spectra of Trp-7 (left) and Trp-170 (right) unfolded in KPi buffer, adsorbed on DPPC vesicles, and during a folding reaction into DMPC vesicles at t = 0 to 180 min following mixing of protein with vesicles. Gaussian decompositions are shown as dashed lines, and the sum of the Gaussian peaks is shown as the solid overlay.
UVRR spectra of OmpA mutants
Trp-170 and Trp-102
UVRR and fluorescence spectra were obtained for the previously uncharacterized nonnative OmpA mutant, Trp-170. Fig. 5 illustrates the increase in RFD value from 1.3 to 1.9 during the folding reaction. The W17 peak for Trp-170 evolves in a similar manner to the Trp-7 mutant, and shifts −4 cm−1 during the folding reaction.
Results for Trp-102 are included in Table 1 and Table S1. Unfolded and folded protein exhibit RFD values between 1.3 and 1.5, whereas the adsorbed species has a value of 1.1. The hydrogen-bonding indicator, W17, shifts one pixel in a systematic manner from 879 to 877 cm−1.
Free energies of unfolding
The refolding curves for Trp-170 and Trp-free (Fig. S5) reveal a Gibbs free energy of unfolding in the absence of denaturant of 9.4 ± 1.9 and 2.6 ± 0.4 kcal/mol, respectively. values for the other mutants Trp-7 (6.7 ± 1.0), Trp-15 (4.8 ± 0.7), and Trp-102 (4.7 ± 0.7), have been previously published (26). We have remeasured the value for wild-type OmpA and determined it to be 10.3 ± 1.5 kcal/mol.
Adsorbed intermediate
A blue shift in fluorescence is observed for both the adsorbed intermediate and folded OmpA relative to unfolded protein (Table 1). However, the magnitude of the blue shift between unfolded and adsorbed protein (max change of 13 nm) is different from the blue shift associated with protein insertion into the bilayer (max change of 26 nm). It is known that the adsorbed intermediate exhibits a similar amount of secondary structure as the folded protein (36). These data suggest that the tryptophan residues of the structured, adsorbed intermediate are partially buried in a nonpolar, protein environment. The adsorbed intermediate will be discussed in greater depth in a future article.
Discussion
Protein-lipid interactions are crucial for the folding and stability of integral membrane proteins. Properties such as lipid chain length, extent of saturation, headgroup size and charge, and vesicle curvature alter the stability and folding rates of OmpA (20,40,41) as well as those of α-helical membrane proteins (42–44). These and other effects of lipid on membrane proteins are not surprising given the chemical and physical heterogeneity of the fluid lipid bilayer (5,45). The bilayer-water interfacial region of the liquid crystalline bilayer is especially complex, and the role of anchoring amino acid residues on membrane protein folding remains an active area of research. The unique role of tryptophan, evidenced by its localization to the bilayer-water interface (13,46–48), and large thermodynamic driving force are now widely accepted; however, the specific interactions between tryptophan and lipid have not been investigated until now.
The tryptophan residues discussed here were selected for study based on their relative locations; residues trp-7 and trp-170 are on opposite sides of the bilayer relative to trp-15 and trp-102 (see Fig. 1). These mutants allow us to test a simple model of membrane protein folding. For example, a traditional scheme of membrane insertion in which the β-barrel is partially formed before insertion (49) assumes that trp-15 and trp-102 traverse the bilayer, whereas trp-7 and trp-170 do not (36). The local environment of these tryptophan residues is also significant. Trp-7 residue is close to several aromatic residues, including Phe-170. Pairwise aromatic interactions are important stabilizing forces (24), and we studied the Phe-to-Trp mutant Trp-170 to gain additional insight into this aromatic cluster. Trp-15 may also be important for protein stability because in the crystal structure, it participates in an intramolecular hydrogen bond with Asn-33. Trp-102 is located in a loop that is likely not membrane-bound, and is the only tryptophan residue to face the interior of the β-barrel and not the lipids. The solvent-accessible surface areas for the indole groups of trp-7, trp-170, trp-15, and trp-102 are 100%, 100%, 82%, and 76%, respectively, and therefore, the indole moieties are exposed and interact strongly with the lipid (trp-7, trp-170, trp-15) or other protein (trp-102) environment. In the case of trp-170, the indole ring was modeled in the same plane as the native phenylalanine residue, but can be oriented with χ2,1 at +88° or −88°, giving high solvent-accessible surface area values of 100% or 82%. We do not discuss the other two native tryptophan residues, trp-57 and trp-143, because the stabilities of the Trp-57 and Trp-143 mutants (26) are comparable to that of Trp-free OmpA, suggesting that these particular tryptophan residues do not provide significant stabilization of the protein.
Protein stability
Thermodynamic measurements of these and similar mutants have previously been reported (24,26). These prior studies indicated that among the five native tryptophan residues, trp-7 makes the greatest contribution to the overall stability of OmpA. Mutation of trp-7 to alanine destabilized OmpA by the greatest amount (3.6 kcal/mol) relative to other tryptophan residues (24). In fact, residues in this aromatic pocket, such as trp-7 and tyr-43, provide significant stability. Our results indicate that addition of a nonnative trp residue in this aromatic pocket, trp-170, is also highly stabilizing. The OmpA mutant with a single tryptophan residue at position 170 (Trp-170 = 9.4 kcal/mol) is nearly as stable as wild-type OmpA, which has five native tryptophan residues (wild-type OmpA = 10.3 kcal/mol). As a comparison, OmpA with a single tryptophan residue at position 7 is destabilized 3.6 kcal/mol relative to wild-type while other mutants, Trp-15 and Trp-102, are destabilized by ∼5.5 kcal/mol. The free energy of unfolding of Trp-free is 2.6 kcal/mol. The observed variation of free energies suggests that the stabilizing interactions are sensitive to local environment, and that molecular interactions of tryptophan with lipid and/or protein strongly influence the energetic contribution to membrane protein stability. This finding further emphasizes the need to apply structure-sensitive spectroscopic methods to obtain a more detailed picture of the molecular interactions of membrane proteins.
Site-specific molecular interactions
UVRR is an ideal technique for studies of protein dynamics because of its selectivity, sensitivity, and structure-specific vibrational markers. The investigation of Trp-7 and Trp-170 reveals the molecular basis and local environmental factors that give rise to the stabilizing nature of the tryptophan residues in these locations. The simultaneous rise in RFD and downshift of the W17 frequency as the protein folds and inserts into the membrane is consistent with the N-H moiety of the indole group participating in a hydrogen-bond in a nonpolar environment (33,37). Hydrogen bonds in nonpolar environments have been shown to be more favorable by up to 1.2 kcal/mol than in polar environments (50–53), and the current finding that both trp-7 and trp-170 form these favorable interactions provide molecular evidence of the stabilizing nature of these interactions in membranes.
This role of indole to form hydrogen bonds has been demonstrated in membrane-associated peptides (28,46,54–56). However, the identity of the hydrogen bond acceptor is not known and has been proposed to be the acyl carbonyl group, phosphate oxygen, or lipid-embedded water molecules. Given that the large RFD values for these OmpA mutants (1.7–1.9) are most similar to that of cyclohexanone (RFD value of 1.7), the tryptophan residues at positions 7 and 170 in OmpA are likely closer to the hydrocarbon core than to the polar headgroups; tryptophan residues near polar headgroups would be expected to exhibit RFD values near 1.5, which is that of acetonitrile (33). The proximity of indole groups and analogs to the ester moiety has been demonstrated by experiments and simulations (13,47,57). We hypothesize that the hydrogen bond acceptor for trp-7 and trp-170 is the lipid acyl carbonyl group of DMPC, and this hydrogen bond in a nonpolar environment provides significant stability to the folded OmpA structure. Pairwise interactions in the aromatic cluster that contains trp-7 and trp-170 may also contribute to the stability of OmpA because the centroids of both of these residues are ∼5 Å from tyrosine centroids in a T-shaped arrangement (Fig. 1) (24,58,59). However, because phenylalanine can also provide significant aromatic interactions, we expect that the Trp-to-Phe mutants studied here primarily reflect differences in stability from hydrogen bonding because tryptophan is a hydrogen bond donor, whereas phenylalanine is not.
There was no evidence for strong hydrogen-bonding of trp-15. This lack of hydrogen-bonding is somewhat surprising given the close proximity of a hydrogen bond acceptor, Asn-33, in the crystal structure. However, the conformation of trp-15 based on UVRR analysis of the dihedral χ2,1 angle indicates that the crystal and solution structures of trp-15 are different. When the χ2,1 angle of trp-15 is modeled in the protein structure to match that of the UVRR data (96°) as shown in Fig. 3, the strength of the hydrogen bond likely decreases because of the less favorable geometry. The structural information provided by UVRR is more relevant given that OmpA is in the solution phase and folded in a bilayer, and environment differences between OmpA folded in detergent and vesicle have been previously reported (23).
A surprising result is the growth of a shoulder in the W18 region of the Trp-15 UVRR spectrum. The W18 mode is a delocalized in-plane breathing mode and is sensitive to changes in the π electron density that participates in cation-π interaction (32). In addition to this intense Raman-active mode, several indole hydrogen-out-of-plane (HOOP) normal modes are predicted to appear in the region between the W18 (∼760 cm−1) and W17 (∼880 cm−1) peaks; these HOOP modes consist of large out-of-plane displacements of the hydrogen atom (H2) bonded to C2 in the pyrrole ring and benzyl hydrogens (60,61). On- and off-resonance Raman spectra of model compounds such as N-acetyl-L-tryptophanamide support the presence of these additional modes between the W18 and W17 peaks (33). Alterations in relative intensities and frequencies of modes in this spectral region as well as others have been associated with cation-π interactions in proteins and model compounds (62,63).
The growth of the ∼778 cm−1 shoulder in Trp-15 is attributed to perturbations of the W18 and/or HOOP modes that reflect development of a cation-π interaction; this interpretation is based on a UVRR study of a model compound, normal mode calculations, and NMR analysis (64). Additional support for this interaction is provided by the observed broadening of the W7 Fermi doublet. Alterations in the separation between these peaks have previously been attributed to cation-π interactions, though a systematic trend has not been established (63,65). Participation of membrane protein tryptophan residues in cation-π interactions with the lipid headgroup has been proposed by others, and both quaternary and primary amine headgroups in phosphocholine (PC) and phosphoethanolamine (PE) lipids, respectively, are viable cation donors (47,56,57,66). The persistence of the 778 cm−1 shoulder in 100% PC and mixed PC/PE vesicles supports this interpretation. The disappearance of the shoulder when OmpA is folded in the detergentoctyl glucoside (OG) further supports the hypothesis that this novel spectral marker originates from protein-lipid, and not protein-protein, interactions. Finally, the relatively low RFD value of 1.2 when Trp-15 is folded in the bilayer supports the localization of this residue to the polar headgroup region where it can interact with the charged amine group of the PC moiety. Comparison of the free energies of unfolding of Trp-15 (4.8 kcal/mol) and Trp-free (2.6 kcal/mol) indicates that this tryptophan-π interaction provides ∼2 kcal/mol of stabilization. This intermolecular interaction is more favorable than an intramolecular hydrogen bond, and this finding reiterates the importance of specific lipid-protein interactions in the bilayer-water interface region for membrane proteins.
Trp-102 is the only tryptophan residue that is oriented toward the interior of the β-barrel pore and has several charged residues within a 7 Å radius of the indole ring. On the basis of its location and orientation, this residue likely does not interact with the bilayer. Nonetheless, we were motivated to study this mutant to determine the changes in local environment and structure during folding. We previously reported that trp-102 provides intraprotein stabilization because of its amphiphilic nature (26); substitution of trp-102 with Phe-102 decreases the free energy of unfolding by ∼2 kcal/mol. The evolution of the RFD value from 1.3 to 1.5 during the folding reaction is consistent with the blue-shifted fluorescence maximum (25), and both observations suggest a relatively nonpolar local environment for trp-102. This environment is likely created by the protein, and not the lipid, and suggests that trp-102 interacts closely with hydrophobic residues despite the presence of charged residues within its 7 Å radius. The structural markers for cation-π interactions, hydrogen-bonding, and local dielectric also indicate that the charged residues do not directly interact with trp-102.
Implications for folding mechanisms and protein stability
These results provide a structural perspective on the molecular interactions that may underlie the differences in free energies for the various single-tryptophan mutants. At the outset we acknowledge that Trp-to-Phe mutations have the effect of both removal of native tryptophan residues and addition of nonnative phenylalanine residues from key locations in the protein. In the case of Trp-7 and Trp-15, a prior study that probed the W7A and W15A mutants revealed that these tryptophan residues provide ∼4 and ∼2 kcal of stability, respectively (24). These values are consistent with our current and prior data on the quadruple mutants that preserve single tryptophan residues at positions 7 and 15 (26). In contrast, less is known about Trp-102 and Trp-170. Trp-170 is a novel, to our knowledge, mutant in which a native phenylalanine residue has been replaced by a nonnative tryptophan residue. A prior F170A mutant revealed that removal of the phenylalanine residue destabilized OmpA by 2.4 kcal (24). In our study, a single tryptophan residue at position 170 gave rise to the most stable mutant, with a decrease in free energy of only 0.9 kcal relative to wild-type. This phe-to-trp mutation at position 170 enhances the stability of OmpA by introduction of a hydrogen bond between trp-170 and a lipid molecule in a nonpolar environment, and we estimate that this interaction contributes up to ∼7 kcal toward the stability of OmpA based on comparison with the trp-free mutant. These values are only estimates of the contributions of individual residues to OmpA stability, but nonetheless, serve as reasonable guidelines for ongoing experimental and computational studies.
It is interesting to note that there is no spectroscopic evidence for transversal of the protein through the bilayer. For example, the fluorescence intensity at 330 nm and RFD values increase throughout the folding period as opposed to increase then decrease; the latter would be expected if the protein travels through a hydrophobic bilayer core and then stabilizes in the interfacial region. This monotonic nature could also result if the tryptophan residues are buried within the protein as it crosses the bilayer. However, this scenario is unlikely for tryptophan residues that are exposed to the lipid in the folded structure. Furthermore, the UVRR spectra indicate that some tryptophan residues interact with the lipid bilayer in a specific manner throughout the folding process. It appears that the common picture in which a membrane protein adsorbs and then inserts into a static bilayer is oversimplified. In contrast, our data provide evidence for lipid-assisted membrane protein folding, where stabilizing protein-lipid interactions occur on the same timescale as folding and insertion. In this view, the lipid plays a dynamic role in membrane protein folding, where the bilayer is able to provide the appropriate dielectric environment for the nascent folding protein throughout the reaction, as well as provide specific stabilizing forces. Our results suggest that formation of these important intermolecular interactions between tryptophan and the lipid headgroup plays a critical role in the folding process, and sheds light on the dynamical behavior of the bilayer. It is imperative that forthcoming computational and experimental models for membrane protein folding include these important lipid-assisted effects.
Summary
UVRR spectroscopy is a versatile tool for study of complex biological systems that provides a useful library of molecular and environmental markers. We have applied this structure-sensitive tool to obtain detailed information on anchoring tryptophan residues in the β-barrel membrane protein, OmpA. Our results reveal protein-lipid interactions that may constitute the molecular basis for the localization of tryptophan residues at the lipid bilayer/water interface. The formation of tryptophan-lipid hydrogen bonds in a nonpolar environment is energetically favorable, and cation-π interactions may be stronger than intramolecular hydrogen bonds. The ability of tryptophan residues to form stabilizing interactions is dependent on local environment, and this observation underscores the importance of site-specific probes. These results emphasize the need for a membrane protein folding paradigm that includes the dynamic role of the lipid. Hence, lipid-assisted membrane protein folding appears to be a more appropriate model for this complex in vitro process.
Acknowledgments
We thank Tiffany J. Rynearson for contributions at the early stage of this project, Annette Medina-Morales for assistance with the gel-shift assays, and Vanessa Oklejas for helpful discussions.
G.K. was supported by Heme and Blood Proteins Training Grant T32-DK007233. This work was supported by the National Science Foundation (CHE-0645720).
Supporting Material
References
- 1.White S.H., Wimley W.C. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 2.Popot J., Engelman D.M. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- 3.Tamm L.K., Hong H., Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim. Biophys. Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Booth P.J., Templer R.H., Lorch M. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 2001;36:501–603. doi: 10.1080/20014091074246. [DOI] [PubMed] [Google Scholar]
- 5.Lee A.G. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 6.MacKenzie K.R. Folding and stability of alpha-helical integral membrane proteins. Chem. Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 7.Wimley W.C., Creamer T.P., White S.H. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 8.Hessa T., Kim H., von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 9.Roseman M.A. Hydrophobicity of the peptide C=O …H-N hydrogen-bonded group. J. Mol. Biol. 1988;201:621–623. doi: 10.1016/0022-2836(88)90642-0. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Tal N., Sitkoff D., Honig B. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J. Phys. Chem. B. 1997;101:450–457. [Google Scholar]
- 11.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Babakhani A., Gorfe A.A., McCammon J.A. Peptide insertion, positioning, and stabilization in a membrane: insight from an all-atom molecular dynamics simulation. Biopolymers. 2007;85:490–497. doi: 10.1002/bip.20698. [DOI] [PubMed] [Google Scholar]
- 13.Yau W.-M., Wimley W.C., White S.H. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 14.Killian J.A., von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 15.Reithmeier R.A.F. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 1995;5:491–500. doi: 10.1016/0959-440x(95)80034-4. [DOI] [PubMed] [Google Scholar]
- 16.Gallivan J.P., Dougherty D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millefiori S., Alparone A., Vanella A. Electronic and vibrational polarizabilities of the twenty naturally occurring amino acids. Biophys. Chem. 2008;132:139–147. doi: 10.1016/j.bpc.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Dornmair K., Kiefer H., Jahnig F. Refolding of an integral membrane protein. J. Biol. Chem. 1990;265:18907–18911. [PubMed] [Google Scholar]
- 19.Kleinschmidt J.H., Tamm L.K. Time-resolved distance determination by tryptophan fluorescence quenching: probing intermediates in membrane protein folding. Biochemistry. 1999;38:4996–5005. doi: 10.1021/bi9824644. [DOI] [PubMed] [Google Scholar]
- 20.Hong H., Tamm L.K. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc. Natl. Acad. Sci. USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramakrishnan M., Qu J., Marsh D. Orientation of beta-barrel proteins OmpA and FhuA in lipid membranes. Chain length dependence from infrared dichroism. Biochemistry. 2005;44:3515–3523. doi: 10.1021/bi047603y. [DOI] [PubMed] [Google Scholar]
- 22.Kleinschmidt J.H. Folding kinetics of the outer membrane proteins OmpA and FomA into phospholipid bilayers. Chem. Phys. Lipids. 2006;141:30–47. doi: 10.1016/j.chemphyslip.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Kim J.E., Arjara G., Winkler J.R. Probing folded and unfolded states of outer membrane protein A using steady-state and time-resolved tryptophan fluorescence. J. Phys. Chem. B. 2006;110:17656–17662. doi: 10.1021/jp061991r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong H., Park S., Tamm L.K. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J. Am. Chem. Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 25.Kleinschmidt J.H., den Blaauwen T., Tamm L.K. Outer membrane protein A of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry. 1999;38:5006–5016. doi: 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez K.M., Gable J.E., Kim J.E. Effects of tryptophan microenvironment, soluble domain, and vesicle size on the thermodynamics of membrane protein folding: Lessons from the transmembrane protein OmpA. Biochemistry. 2008;47:12844–12852. doi: 10.1021/bi800860k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez K.M., Neary T.J., Kim J.E. UV resonance Raman spectroscopy of folded and unfolded states of an integral membrane protein. J. Phys. Chem. B. 2008;112:9507–9511. doi: 10.1021/jp800772j. [DOI] [PubMed] [Google Scholar]
- 28.Shafaat H.S., Sanchez K.M., Kim J.E. Ultraviolet resonance Raman spectroscopy of a membrane-bound beta-sheet peptide as a model for membrane protein folding. J. Raman Spectrosc. 2008;40:1060–1064. [Google Scholar]
- 29.Huang C.-Y., Balakrishnana G., Spiro T.G. Early events in apomyoglobin unfolding probed by laser T-jump/UV resonance Raman spectroscopy. Biochemistry. 2005;44:15734–15742. doi: 10.1021/bi051578u. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed A., Beta I.A., Asher S.A. UV-resonance Raman thermal unfolding study of trp-cage shows that it is not a simple two-state miniprotein. J. Am. Chem. Soc. 2005;127:10943–10950. doi: 10.1021/ja050664e. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Mendieta I.R., Spence G.R., Smith D.A. Ultraviolet resonance Raman studies reveal the environment of tryptophan and tyrosine residues in the native and partially unfolded states of the E colicin-binding immunity protein Im7. Biochemistry. 2005;44:3306–3315. doi: 10.1021/bi047746k. [DOI] [PubMed] [Google Scholar]
- 32.Harada I., Takeuchi H. Raman and ultraviolet resonance Raman spectra of proteins and related compounds. In: Clark R.J.H., Hester R.E., editors. Spectroscopy of Biological Systems. John Wiley & Sons; Chichester: 1986. pp. 113–175. [Google Scholar]
- 33.Schlamadinger D.E., Gable J.E., Kim J.E. Hydrogen-bonding and solvent polarity markers in the UV resonance Raman spectrum of tryptophan: application to membrane proteins. J. Phys. Chem. B. 2009;113:14769–14778. doi: 10.1021/jp905473y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chi Z., Asher S.A. UV Raman determination of the environment and solvent exposure of tyr and trp residues. J. Phys. Chem. B. 1998;102:9595–9602. [Google Scholar]
- 35.Austin J.C., Jordan T., Spiro T.G. Ultraviolet resonance Raman studies of proteins and related model compounds. In: Clark A.R.J.H., Hester R.E., editors. Biomolecular Spectroscopy, Part. John Wiley and Sons; New York: 1993. pp. 55–127. [Google Scholar]
- 36.Surrey T., Jahnig F. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc. Natl. Acad. Sci. USA. 1992;89:7457–7461. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miura T., Takeuchi H., Harada I. Characterization of individual tryptophan side chains in proteins using Raman spectroscopy and hydrogen-deuterium exchange kinetics. Biochemistry. 1988;27:88–94. doi: 10.1021/bi00401a015. [DOI] [PubMed] [Google Scholar]
- 38.Harada I., Miura T., Takeuchi H. Origin of the doublet at 1360 and 1340 cm-1 in the Raman spectra of tryptophan and related compounds. Spectrochim. Acta [A] 1986;42A:307–312. [Google Scholar]
- 39.Miura T., Takeuchi H., Harada I. Tryptophan Raman bands sensitive to hydrogen bonding and side-chain conformation. J. Raman Spectrosc. 1989;20:667–671. [Google Scholar]
- 40.Kleinschmidt J.H., Tamm L.K. Secondary and tertiary structure formation of the beta-barrel membrane protein OmpA is syncrhonized and depends on membrane thickness. J. Mol. Biol. 2002;324:319–330. doi: 10.1016/s0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 41.Surrey T., Jahnig F. Kinetics of folding and membrane insertion of a beta-barrel membrane protein. J. Biol. Chem. 1995;270:28199–28203. doi: 10.1074/jbc.270.47.28199. [DOI] [PubMed] [Google Scholar]
- 42.Miller D., Charalambous K., Booth P.J. In vitro unfolding and refolding of the small multidrug transporter EmrE. J. Mol. Biol. 2009;393:815–832. doi: 10.1016/j.jmb.2009.08.039. [DOI] [PubMed] [Google Scholar]
- 43.Allen S.J., Curran A.R., Booth P.J. Controlling the folding efficiency of an integral membrane protein. J. Mol. Biol. 2004;342:1293–1304. doi: 10.1016/j.jmb.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 44.Clark E.H., East J.M., Lee A.G. The role of tryptophan residues in an integral membrane protein: diacylglycerol kinase. Biochemistry. 2003;42:11065–11073. doi: 10.1021/bi034607e. [DOI] [PubMed] [Google Scholar]
- 45.White S.H., Ladokhin A.S., Hristova K. How membranes shape protein structure. J. Biol. Chem. 2001;276:32395–32398. doi: 10.1074/jbc.R100008200. [DOI] [PubMed] [Google Scholar]
- 46.Persson S., Killian J.A., Lindblom G. Molecular ordering of interfacially localized tryptophan analogs in ester- and ether-lipid bilayers studied by H-2-NMR. Biophys. J. 1998;75:1365–1371. doi: 10.1016/s0006-3495(98)74054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaede H.C., Yau W.-M., Gawrisch K. Electrostatic contributions to indole-lipid interactions. J. Phys. Chem. B. 2005;109:13014–13023. doi: 10.1021/jp0511000. [DOI] [PubMed] [Google Scholar]
- 48.Sanderson J.M. Refined models for the preferential interactions of tryptophan with phosphocholines. Org. Biomol. Chem. 2007;5:3276–3286. doi: 10.1039/b707502b. [DOI] [PubMed] [Google Scholar]
- 49.Huysmans G.H.M., Baldwin S.A., Radford S.E. The transition state for folding of an outer membrane protein. Proc. Natl. Acad. Sci. USA. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J., Bosco D.A., Kelly J.W. Localized thermodynamic coupling between hydrogen bonding and microenvironment polarity substantially stabilizes proteins. Nat. Struct. Biol. 2009;16:684–691. doi: 10.1038/nsmb.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joh N.H., Min A., Bowie J.U. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shan S., Herschlag D. The change in hydrogen bond strength accompanying charge rearrangement: Implications for enzymatic catalysis. Proc. Natl. Acad. Sci. USA. 1996;93:14474–14479. doi: 10.1073/pnas.93.25.14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takano K., Scholtz J.M., Pace C.N. The contribution of polar group burial to protein stability is strongly context-dependent. J. Biol. Chem. 2003;278:31790–31795. doi: 10.1074/jbc.M304177200. [DOI] [PubMed] [Google Scholar]
- 54.Sun H., Greathouse D.V., Koeppe R.E. The preference of tryptophan for membrane interfaces: insights from N-methylation of tryptophans in gramicidin channels. J. Biol. Chem. 2008;283:22233–22243. doi: 10.1074/jbc.M802074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Wel P.C.A., Reed N.D., Koeppe R.E. Orientation and motion of tryptophan interfacial anchors in membrane-spanning peptides. Biochemistry. 2007;46:7514–7524. doi: 10.1021/bi700082v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen F.N.R., Jensen M.O., Nielsen C.H. Interfacial tryptophan residues: a role of the cation-pi effect? Biophys. J. 2005;89:3985–3996. doi: 10.1529/biophysj.105.061804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Norman K.E., Nymeyer H. Indole localization in lipid membranes revealed by molecular simulations. Biophys. J. 2006;91:2046–2054. doi: 10.1529/biophysj.105.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burley S.K., Petsko G.A. Aromatic-aromatic interaction: a mechanism of protein structural stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 59.Guvench O., Brooks C.L., III Tryptophan side chain electrostatic interactions determine edge-to-face vs parallel-displaced tryptophan side chain geometries in the designed beta-hairpin “trpzip2”. J. Am. Chem. Soc. 2005;127:4668–4674. doi: 10.1021/ja043492e. [DOI] [PubMed] [Google Scholar]
- 60.Bunte S.W., Jensen G.M., Jalkanen K.J. Theoretical determination of the vibrational absorption and Raman spectra of 3-methylindole and 3-methylindole radicals. Chem. Phys. 2001;265:13–25. [Google Scholar]
- 61.Jensen G.M., Goodin D.B., Bunte S.W. Density functional and MP2 calculations of spin densities of oxidized 3-methylindole: models for tryptophan radicals. J. Phys. Chem. 1996;100:954–959. [Google Scholar]
- 62.Okada A., Miura T., Takeuchi H. Protonation of histidine and histidine-tryptophan interactions in the activation of the M2 ion channel from influenza A virus. Biochemistry. 2001;40:6053–6060. doi: 10.1021/bi0028441. [DOI] [PubMed] [Google Scholar]
- 63.Wen Z.Q., Thomas G.J., Jr. Ultraviolet-resonance Raman spectroscopy of the filamentous virus Pf3: interactions of Trp-38 specific to the assembled virion subunit. Biochemistry. 2000;39:146–152. doi: 10.1021/bi992018w. [DOI] [PubMed] [Google Scholar]
- 64.Schlamadinger D.E., Daschbach M.M., Kim J.E. UV resonance Raman study of cation-pi interactions in an indole crown ether. J. Raman Spectrosc. 2010 doi: 10.1002/jrs.2781. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xue Y., Davis A.V., O'Halloran T.V. Cu(I) recognition via cation-pi and methionine interactions in CusF. Nat. Chem. Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanderson J.M., Whelan E.J. Characterization of the interactions of aromatic amino acids with diacetyl phosphatidylcholine. Phys. Chem. Chem. Phys. 2004;6:1012–1017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.