

Abstract
Mutations in distal S6 were shown to significantly alter the stability of the open state of CaV2.3 (Raybaud, A., Baspinar, E. E., Dionne, F., Dodier, Y., Sauvé, R., and Parent, L. (2007) J. Biol. Chem. 282, 27944–27952). By analogy with KV channels, we tested the hypothesis that channel activation involves electromechanical coupling between S6 and the S4S5 linker in CaV2.3. Among the 11 positions tested in the S4S5 linker of domain II, mutations of the leucine residue at position 596 were found to destabilize significantly the closed state with a −50 mV shift in the activation potential and a −20 mV shift in its charge-voltage relationship as compared with CaV2.3 wt. A double mutant cycle analysis was performed by introducing pairs of glycine residues between S4S5 and S6 of Domain II. Strong coupling energies (ΔΔGinteract > 2 kcal mol−1) were measured for the activation gating of 12 of 39 pairs of mutants. Leu-596 (IIS4S5) was strongly coupled with distal residues in IIS6 from Leu-699 to Asp-704. In particular, the double mutant L596G/I701G showed strong cooperativity with a ΔΔGinteract ≈6 kcal mol−1 suggesting that both positions contribute to the activation gating of the channel. Altogether, our results highlight the role of a leucine residue in S4S5 and provide the first series of evidence that the IIS4S5 and IIS6 regions are energetically coupled during the activation of a voltage-gated CaV channel.
Keywords: Biophysics, Calcium Channels, Computer Modeling, Energetics, Neurobiology, Activation Gating, Electrophysiology, Structure-Function
Introduction
Voltage-dependent Ca2+ channels are membrane proteins that play a critical role in promoting Ca2+ influx in response to membrane depolarization in excitable cells (1–6). The CaVα1 subunits of voltage-dependent Ca2+ channels are evolutionarily related to the α subunit of KV channels with a single polypeptidic chain carrying four domains of six transmembrane segments (S1-S6). Although the overall identity at the primary sequence level is quite low between CaV and KV channels, it improves significantly for transmembrane segments. By homology with the three-dimensional structures of KcsA, MthK, KVAP, KirBac, and KV1.2 channels (7–11), the four S4 transmembrane segments are thus believed to act as the voltage sensor, whereas the S6 helices line the channel pore of CaVα1.
Both in KV and CaV channels, the question as to how the S4 motion is transmitted to the S6 helix to open the gate upon depolarization remains heavily studied. In the electromechanical coupling model based upon the KV1.2 crystal structure (12), the S4S5 linker, which is located within atomic proximity (4–5 Å) of the S6 helix, interacts with the latter in the closed state of the channel. The movement of the S4 voltage sensor is likely to induce a conformational change that pulls the S4S5 linker away from the S6 inner helix ultimately resulting in ions flowing through the pore. The closed conformation of the channel is postulated to be stabilized by hydrophobic interactions at the level of the “closed bundle” in S6 (13–15).
Experimental evidence supporting a role for the S4S5 linker in the voltage dependence of activation of KV channels is steadily mounting. Introduction of the S4S5 linker from KcsA disrupts the voltage-dependent activation of KV2.1 (16, 17). In hERG, cross-linking studies have shown that residues located in the S4S5 linker and in the S6 helix are in atomic proximity and that gating currents were greatly affected as a result of disulfide bridge formation (18). Furthermore, mutations of a unique glycine residue (Gly-546) within the N-terminal end of the S4S5 linker were shown to stabilize the closed state by ∼1.6–4.3 kcal/mol and restricted the voltage sensor movement (19).
The potential coupling mechanism between the S4S5 linker and the S6 helix has yet to be explored in CaV channels. In contrast to KV channels, CaV channels are structurally asymmetrical with four distinct albeit homologous S4S5 linkers and four distinct S6 helices. We have already shown that glycine substitutions in the distal S6 regions of domains I, II, III, and IV altered the relative stability between the open and closed states (20). Mutations within IIS6 were found to impact most significantly on the activation gating of CaV2.3. In particular, I701G shifted by −35 mV the voltage dependence of activation while slowing down inactivation and deactivation kinetics (20). These data indicated that IIS6 plays a unique role in the channel equilibrium between the closed and the open state(s) in CaV2.3.
To determine whether the S4S5 linker of Domain II (IIS4S5) is coupled with the S6 helix of the same domain (IIS6) during activation of CaV2.3, we performed a double mutant cycle analysis (21) within domain II of CaV2.3. Double mutant cycle analysis has often been proven to be a powerful tool for studying intra-protein interactions in CaV1.2 (22), KV (23, 24), and CNG (25) channels among others. Our assay, conducted by introducing pairs of glycine residues, one in the IIS4S5 linker and one in the IIS6 helix, has identified crucial points of energy interaction between these two regions. Notably Leu-596 in IIS4S5 appeared to be strongly coupled with IIS6 distal residues from Leu-699 to Asp-704 with ΔΔGinteract more negative than −2 kcal mol−1. Altogether, our results provide the first series of evidence that the IIS4S5 and IIS6 regions are coupled during the activation of CaV2.3.
EXPERIMENTAL PROCEDURES
Recombinant DNA Techniques
The human CaV2.3 (GenBank L27745) (26), the rat CaVβ3 (GenBank M88751) (27), and the rat brain CaVα2bδ (GenBank NM_000722) (28) were used. Point mutations were produced with the QuikChange XL-mutagenesis kit (Strategene, La Jolla, CA) using 39-mer primers on the full-length CaV2.3 clone as described elsewhere (20). Constructs were verified by automated double-stranded sequence analysis (Genomics Platform, IRIC, Université de Montréal, QC, Canada). Run-off transcripts were prepared using the T7 RNA Polymerase mMessage mMachine® transcription kit (Ambion, Austin, TX) and stored at −20 °C before use.
Functional Expression of CaV2.3
Oocytes were obtained from female Xenopus laevis clawed frogs as described previously (20, 29). Oocytes were injected with 46 or 4.6 nl of a solution containing cRNA coding for the CaVα1, CaVα2bδ, and CaVβ3 subunits in a 6:2:3 weight ratio. Coexpression with the ancillary CaVβ3, which is predominant in the brain (30), promotes strong functional expression (31, 32) and emphasizes closed-state inactivation (33) of CaV2.3. Functional expression of mutants was deemed significant with whole cell Ba2+ currents larger than −300 nA.
Whole Cell Recording and Data Acquisition in Oocytes
Wild-type and mutant channels were screened at room temperature for macroscopic Ba2+ currents, 2 to 4 days after RNA injection, using a two-electrode voltage-clamp amplifier (OC-725C, Warner Instruments) as described earlier (20, 29, 34). Oocytes were routinely injected with 23 nl of a 50 mm EGTA (Sigma) solution 0.5 to 2 h before the experiments. The pCLAMP 10 software (Molecular Devices, Sunnyvale, CA) was used for on-line data acquisition and analysis. A series of 500-ms voltage pulses (5 mV steps) was applied at a frequency of 0.2 Hz from a holding potential of −120 mV in most cases. The holding potential was −140 mV for mutants that show a strong negative shift in the voltage dependence of inactivation.
Cut-open Oocyte Recordings
Gating currents were recorded using the cut-open oocyte voltage-clamp technique (35, 36) with a CA-1B amplifier (Dagan Corp., Minneapolis, MN). The membrane of the oocyte exposed to the bottom chamber was permeabilized by a brief treatment with 0.1% saponin. The external solution contained 2 mm CoCl2, 110 mm NaOH, and 10 mm Hepes, titrated to pH 7.0 with methanesulfonic acid (37). The internal solution in contact with the oocyte cytoplasm was either 120 mm tetraethyl ammonium, 10 mm Hepes, 0.5 mm EGTA, and 5 mm MgSO4, titrated to pH 7.0 with methanesulfonic acid; or 110 mm potassium glutamate and 10 mm Hepes, titrated to pH 7.0 with KOH. Data acquisition and analysis were performed using the Digidata 1322A 16-bit system (Molecular Devices). Gating currents were filtered at 1 kHz and digitized at 10 kHz. Holding potential was −120 mV and 30-ms voltage steps were evoked from −100 to 60 mV by 5-mV increments. Linear components arising from charging the membrane and leak currents were eliminated by a P/−-4 protocol. Gating charge versus voltage (Q-V) relations were obtained by integrating the OFF gating currents and then fitting single Boltzmann functions (Equation 1) according to
where parameter e is the elementary electric charge; k is the Boltzmann constant; z is the effective valence.
Data Analysis
Activation parameters were estimated from the peak I–V curves obtained for each channel combination and are reported as the mean of individual measurements ± S.E. Briefly, the I–V relationships were normalized to the maximum amplitude and fitted to a Boltzmann equation with E0,5,act being the midpotential of activation as described elsewhere (20, 29, 38). The estimation of E0,5,act using nonstationary measurements rests upon the assumption that the transition to the open state is much faster than the transition to the inactivated state.
The free energy of activation was calculated using the midactivation potential by Equation 2.
Deactivation time constants and inactivation parameters were calculated as described in detail under supplemental “Experimental Procedures”. Statistical analyses were performed using the one-way analysis of variance fitting routine for two independent populations included in Origin 7.0. Data were considered statistically significant at p < 0.01.
Double Mutant Cycle Analysis
The coupling or interaction energy ΔΔGinteract was calculated as,
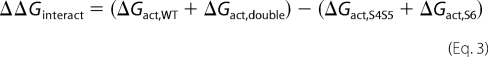 |
where ΔGact,wt is the free energy of activation of the wild-type channel, ΔGact,double is the free energy of activation of the double glycine mutant, ΔGact,S4S5 is the free energy of activation of the single glycine mutant in the S4S5 linker, and ΔGact,S6 is the free energy of activation of the single glycine mutant in S6.
ΔΔGinteract = 0 suggests that the activation energies are purely additive hence that the residues are not functionally coupled. A value of ΔΔGinteract more positive than +1 kcal mol−1 or more negative than −1 kcal mol−1 (|ΔΔGinteract| > 1) was considered significantly different in similar studies (24, 25). However, given the experimental variations associated with the measurements, we used a threshold of |ΔΔGinteract| > 2 kcal mol−1.
Homology Modeling of CaV2.3
An analysis based on the SAMT02 algorithm (39) confirmed KV 1.2 as a suitable template to model the S4S5 linker and S6 segments of domain II of CaV2.3. The computer-based molecular model of CaV2.3, going from the IIS4 helix to the end of IIS6 was obtained with Modeler 9 version 4 using the molecular coordinates of KV1.2 (Protein Data Bank code 2A79). Details are provided under supplemental “Experimental Procedures”.
RESULTS
Single Point Glycine Mutations in IIS4S5
A glycine scan was performed in the S4S5 linker of domain II to identify residues most likely to play a role during the activation of CaV2.3. Because of the absence of a side chain, glycine mutations most likely minimize steric constraints, whereas generally decreasing the degree of hydrophobicity of the S4S5 linker. We used the predictive SAM-T02 algorithm (39) to evaluate the structural impact of introducing glycine residues in the S4S5 linker of domain II in CaV2.3 channels. As seen under supplemental Fig. S1, single point mutations are not predicted to significantly alter the predicted secondary structure of this region.
Glycine residues were thus introduced as single point mutations between residues 592 and 602 in the S4S5 linker of domain II (IIS4S5) (Fig. 1B). The ΔGact and ΔGinact (ΔG for the activation and inactivation, respectively) of these 11 consecutive single point mutations are reported in Table 1. As shown in Fig. 2, all mutant channels activated in response to membrane depolarization. From the histogram in Fig. 3A, it appears that the ΔGact and ΔGinact for most mutants (L592G, V593G, V594G, S595G, S598G, S599G, and K601G) were not significantly different from CaV2.3 wt. In contrast, the ΔGact were significantly altered by glycine residues introduced at positions 596, 600, and 602 with a clear leftward shift in the voltage dependence of activation. It is noteworthy that no glycine mutants shifted ΔGact toward positive voltages. L596G experienced the most significant change in the ΔGact value from −1.0 ± 0.1 kcal mol−1 (n = 108) for CaV2.3 wt to −6.5 ± 0.7 kcal mol−1 (n = 6) for L596G yielding a ΔΔGact = −5.5 ± 0.8 kcal mol−1. This singular effect was specific for the glycine mutation. Other nonconservative mutations at this position, L596D and L596P, were activated at significantly lower membrane potentials with ΔGact = −2.5 ± 0.2 and −2.7 ± 0.2 kcal mol−1, respectively.
FIGURE 1.

A, predicted alignment of the four S6 segments of CaV2.3 with the S6 segment from KV1.2. The top row shows the primary sequence of the S6 segment from Protein Data Bank 2A79 file for the atomic coordinates of KV1.2. The primary sequences of IS6, IIS6, IIIS6, and IVS6 of CaV2.3 were aligned with S6 of KV1.2 using the T-coffee algorithm (see supplemental “Experimental Procedures” and Ref. 18). The IIS6 residues used in the double mutants are underlined. The IS6, IIIS6, and IVS6 residues used in additional experiments are shaded (modified from Ref. 18). B, predicted alignment of the S4S5 linkers of CaV2.3 with the S4S5 linker from KV1.2. All linkers are exactly the same length. The top row shows the primary sequence of the S4S5 linker from Protein Data Bank 2A79 file for the atomic coordinates of KV1.2. The primary sequences of the IS4S5, IIS4S5, IIIS4S5, and IVS4S5 linkers of the CaV2.3 channel were aligned with S4S5 of KV1.2 (see “Experimental Procedures”).
TABLE 1.
Biophysical properties of IIS4S5 mutants in Cav2.3
Biophysical parameters of CaV2.3 wild-type and mutant channels were estimated with CaVα2bδ and CaVβ3 in the presence of a 10 mm Ba2+ solution as described elsewhere (29, 38, 41). The voltage dependence of inactivation was determined from the peak currents after 5-s depolarizing pulses from a holding potential of −120 mV (−140 mV for L596G). Activation properties (E0.5,act and z) were estimated from the mean I–V relationships and fitted to a Boltzmann equation where z is the slope factor. The data are shown with the mean ± S.E. of the individual experiments and the number of experiments appears in parentheses.
| CaV2.3/α2δ/β3 10 mm Ba2+ | Activation |
Inactivation |
Deactivation, max τ deact | |||
|---|---|---|---|---|---|---|
| E0.5,act | ΔGact | E0.5,inact | ΔGinact | R50 at 10 mV | ||
| mV | kcal mol−1 | mV | kcal mol−1 | Ms | ||
| CaV2.3 wt | −6.7 ± 0.4 (108) | −1.0 ± 0.1 | −63.5 ± 0.6 (74) | −4.3 ± 0.1 | 0.41 ± 0.02 (41) | 1.3 ± 0.2 (10) |
| L592G | −11 ± 1 (5) | −1.2 ± 0.1 | −63 ± 1 (5) | −3.6 ± 0.1 | 0.54 ± 0.01 (5) | 1.0 ± 0.1 (5) |
| V593G | −5 ± 1 (5) | −0.5 ± 0.2 | −68 ± 1 (5) | −3.8 ± 0.6 | 0.47 ± 0.03 (5) | 0.60 ± 0.02 (5) |
| V594G | −11 ± 1 (5) | −1.3 ± 0.1 | −55 ± 2 (4)a | −2.5 ± 0.1b | 0.73 ± 0.02 (5)b | 1.0 ± 0.1 (5) |
| S595G | −17 ± 2 (9)b | −1.7 ± 0.3 | −39 ± 1 (5)b | −2.5 ± 0.1b | 0.73 ± 0.03 (5)b | 0.7 ± 0.1(5) |
| L596G | −53 ± 1 (6)b | −6.5 ± 0.7b | −108 ± 3 (6)b | −4.7 ± 0.2 | 0.87 ± 0.03 (6)b | 21 ± 2 (6)b |
| L596D | −20 ± 1 (5)b | −2.5 ± 0.2b | −43 ± 1 (5)b | −3.0 ± 0.1b | 0.78 ± 0.03 (5)b | 2.5 ± 0.5 (5) |
| L596P | −20 ± 1 (5)b | −2.7 ± 0.2b | −58 ± 1 (5) | −3.4 ± 0.3a | 0.70 ± 0.02 (5)b | 11 ± 2 (5)b |
| M597G | −14 ± 1 (4)a | −1.5 ± 0.2 | −82 ± 1 (4)b | −3.6 ± 0.2 | 0.26 ± 0.07 (4) | 0.8 ± 0.1 (5) |
| S598G | −4 ± 2 (5) | −0.5 ± 0.2 | −59 ± 1 (5) | −3.4 ± 0.1a | 0.41 ± 0.02 (5) | 0.7 ± 0.2 (5) |
| S599G | −2 ± 1 (9)a | −0.3 ± 0.2 | −55 ± 1 (7)b | −4.1 ± 0.1 | 0.49 ± 0.02 (9) | 0.8 ± 0.1 (8) |
| M600G | −16 ± 1 (5)b | −2.1 ± 0.2a | −65 ± 1 (5) | −4.3 ± 0.1 | 0.33 ± 0.03 (5) | 0.8 ± 0.1 (4) |
| K601G | −5 ± 1 (5) | −0.6 ± 0.2 | −51 ± 1 (5)b | −3.1 ± 0.1b | 0.53 ± 0.03 (5) | 1.1 ± 0.1 (5) |
| S602G | −20 ± 1 (6) | −3.1 ± 0.4a | −70 ± 1 (5)a | −4.4 ± 0.2 | 0.58 ± 0.06 (6)a | 1.7 ± 0.4 (6) |
| I214G (IS4S5) | −9 ± 1 (5) | −0.7 ± 0.1 | −83.0 ± 0.2 (5)b | −3.3 ± 0.3a | 0.19 ± 0.05 (5)b | 1.1 ± 0.3 (5) |
| V1281G (IIIS4S5) | −4.6 ± 0.8 (5) | −0.5 ± 0.1 | −67 ± 1 (5) | −4.0 ± 0.1 | 0.29 ± 0.01 (5) | 1.1 ± 0.2 (5) |
| F1599G (IVS4S5) | −1 ± 1 (5)a | −0.1 ± 0.1 | −59 ± 2 (5) | −2.8 ± 0.3b | 0.49 ± 0.03 (5) | 4 ± 2 (5)a |
a p < 0.01 as compared with CaV2.3 wt.
b p < 0.001 as compared with CaV2.3 wt.
FIGURE 2.

Representative traces of whole cell currents obtained for the single IIS4S5 mutants in a 10 mm Ba2+ solution. A series of 500-ms voltage pulses were applied between −70 and +50 mV (5-mV steps) from a holding potential of −120 mV (except for L596G where the holding potential was −140 mV). Traces are shown from left to right, starting at the top of the figure, for Cav2.3 wild-type, L592G, V593G, V594G, S595G, L596G, M597G, S598G, S599G, M600G, K601G, and S602G. Unless stated otherwise, all channels were co-expressed with the CaVα2bδ and CaVβ3 auxiliary subunits. Scales are 0.2 μA and 50 ms.
FIGURE 3.

A, histogram of the ΔG for activation and ΔG for inactivation gating (respectively, ΔGact and ΔGinact) of the glycine mutants between positions 592 and 602 in the S4S5 linker in domain II (IIS4S5) of CaV2. 3. ΔGact and ΔGinact were obtained as described under “Experimental Procedures.” The ΔGact of L596G, M600G, and S602G were significantly more negative than CaV2.3 wt at p < 0.001 (one-way analysis of variance). In contrast, the ΔGinact of V594G, S595G, S598G, K601G, and S602G were significantly more positive than CaV2.3 wt at p < 0.01 (one-way analysis of variance). B, gating currents were measured with the cut-open oocyte technique. Normalized gating charge was plotted against test voltages for CaV2.3 wt and L596G. The voltage dependence of the gating charge or V0.5,Q values were obtained from fitting the traces with a single Boltzmann equation as described under “Experimental Procedures.” V0.5,Q values were 6 ± 1 mV (n = 19) for CaV2.3 wt and −24 ± 1 mV (n = 10) for L596G. C, the mean time constants of deactivation (obtained from fitting current traces from monoexponential functions) for CaV2.3 wt and the IIS4S5 glycine mutants (592–602) were plotted against the test potential. Numerical values are found in Table 1. L596G yielded a significantly longer deactivation time constant (p < 0.001) than the CaV2.3 wt at all voltages more positive than −80 mV. D, representative tail currents for CaV2.3 wt and L596G. Currents were activated during 8-ms conditioning depolarizing voltage pulses at 0 mV for the wt and −40 mV for L596G. Deactivation was recorded during subsequent repolarization to test potentials from −120 to 0 mV (10-mV increments) as illustrated on the pulse protocol.
The voltage dependence of the Q/Qmax curve for L596G was also significantly shifted toward negative voltages with V0.5,Q = −17 ± 1 mV (n = 4) as compared with V0.5,Q = 6 ± 1 mV (n = 19) for CaV2.3 wt (Fig. 3B). L596G displayed slower inactivation kinetics with a R50 = 0.87 ± 0.03 as compared with R50 = 0.41 ± 0.02 for CaV2.3 wt (Table 1), a significantly slower deactivation time constant (Fig. 3, C and D) but little change in the ΔGinact. These data are compatible with L596G either destabilizing the closed state or stabilizing the open state of CaV2.3. Similar observations were reported after mutations of IIS6 (supplemental Table S1 and Ref. 20) raising the possibility that the two regions contribute synergically to channel activation.
Leu-596 Is Coupled to Distal IIS6
To investigate the hypothesis that L596G in the S4S5 linker is specifically coupled to the distal S6 region of domain II, we performed a double mutant cycle analysis of these two regions. The double mutant cycle analysis provides a way for isolating the energetics of specific pairwise interactions (21, 23). We opted to introduce pairs of glycine residues in IIS4S5 and IIS6, which had been shown to yield functional channels in our previous mutational analysis (20). The biophysical properties of a total of 39 double mutations produced between the distal IIS6 (699 to 704) (supplemental Table S1) and the 11 positions (592 to 602) in IIS4S5 were characterized from whole cell current recordings (supplemental Fig. S2).
Using Equation 3, ΔΔGinteract were calculated for the double mutants with the ΔGact and ΔGinact values reported in Table 2 and supplemental Table S2. Fig. 4 shows the histogram of ΔΔGinteract values for the activation of the 39 double mutants. The double mutant cycle analysis revealed cooperativity between L596G and 6 consecutive positions in the distal IIS6 (supplemental Fig. S3). Interaction was non-additive for the L596G double mutants with ΔΔGinteract values ranging from 3 to 6 kcal mol−1 (see Table 2 for exact values). ΔΔGinteract values larger than 1 kcal mol−1 are usually suggestive of a significant interaction (23, 24) and exceed the coupling energies calculated between IS6 and IIS6 in CaV1.2 (22). Fig. 5 shows the whole cell current traces, the average I/V curves, and the voltage dependence of the gating charge (Q/Qmax) of the L596G/I701G mutant. As seen, the gating charge curve for L596G/I701G was indistinguishable from either point mutations but significantly shifted to the left when compared with CaV2.3 wt. The absence of additivity in some the L596G/IIS6 mutants could thus arise in part from the dominant effect of L596G on the gating of CaV2.3. A similar observation can be made for I701G, the gating of which was not significantly modulated by any mutation in IIS4S5, save for M597G. The negative shift in the Q/V curve of I701G is reminiscent of the negative shift reported after mutation of the equivalent residue in S6 of Shaker KV channels (36). Altogether, these data suggest that Leu-596 and Ile-701 participate in a concerted fashion to the activation gating of CaV2.3.
TABLE 2.
Activation gating properties of the double mutants IIS4-S5/IIS6 in Cav2.3
Activation gating properties of CaV2.3 wild-type and double mutant channels were estimated with CaVα2bδ and CaVβ3 in the presence of a 10 mm Ba2+ solution as described elsewhere (29, 38, 41). Activation properties (E0.5,act and z) were estimated from the mean I–V relationships and fitted to a Boltzmann equation where z is the slope factor. The data are shown with the mean ± S.E. of the individual experiments and the number of experiments appears in parentheses. The biophysical properties of the L699G to A703G mutants were previously described (20) and are shown under “supplemental Table 1 along D704G. Ln Ω factor and ΔΔGinteract were calculated for double mutants as indicated under “Experimental Procedures” with ΔGact values (in kcal mol−1) for L699G = −2.3 ± 0.2 (n = 13), A700G = −1.8 ± 0.2 (n = 14), I701G = −4.7 ± 0.3 (n = 24), A702G = −1.2 ± 0.1 (n = 20), V703G = −3.6 ± 0.2 (n = 15). A ln Ω value of 0 indicates the absence of coupling.
| CaV2.3/α2δ/β3, 10 mm Ba2+ | Activation |
Deactivation, max τ deact | |||
|---|---|---|---|---|---|
| E0.5,act | ΔGact | ln(Ω) | ΔΔGinteract | ||
| mV | kcal mol−1 | kcal mol−1 | ms | ||
| L592G/I701G | −49 ± 1 (4)a | −4.6 ± 0.5a | 2 ± 1 | 1 ± 1 | 11 ± 2 (4)a |
| V593G/L699G | −30 ± 2 (5)a | −3.7 ± 0.5a | −3 ± 1 | −2 ± 1 | 6 ± 1 (5)a |
| V593G/A700G | −25 ± 1 (4)a | −3.5 ± 0.3a | −4 ± 1 | −2.2 ± 0.8 | 13 ± 2 (4)a |
| V593G/I701G | −53 ± 2 (5)a | −6.6 ± 0.4a | −2 ± 1 | −1 ± 1 | 9 ± 1 (5)a |
| V593G/A702G | −30 ± 1 (5)a | −3.6 ± 0.4a | −5 ± 1 | −2.9 ± 0.8 | 6 ± 1 (5)a |
| V593G/V703G | −20 ± 3 (5)a | −2.3 ± 0.4b | −1 ± 1 | 0.8 ± 0.9 | 4 ± 2 (6) |
| V594G/L699G | −29 ± 1 (5)a | −3.3 ± 0.3a | −1 ± 1 | −0.7 ± 0.7 | 4.0 ± 0.2 (5)a |
| V594G/A700G | −19 ± 1 (4)a | −2.3 ± 0.3b | 0 ± 1 | −0.2 ± 0.7 | 1.3 ± 0.3 (4) |
| V594G/I701G | −53 ± 1 (5)a | −6.6 ± 0.6a | −1 ± 1 | −1 ± 1 | 11 ± 2 (5)a |
| V594G/A702G | −14 ± 2 (6)a | −1.9 ± 0.4 | −1 ± 1 | −0.4 ± 0.7 | 1.2 ± 0.2 (6) |
| V594G/V703G | −24 ± 1 (5)a | −3.0 ± 0.2a | −1 ± 1 | 0.9 ± 0.6 | 1.5 ± 0.2 (5) |
| V594G/D704G | −18 ± 1(5)a | −2.5 ± 0.3a | 2 ± 1 | −0.3 ± 0.3 | 1.6 ± 0.3 (5) |
| S595G/L699G | −25 ± 1 (5)a | −3.8 ± 0.5a | −1 ± 1 | −1 ± 1 | 5 ± 1 (5)a |
| S595G/A700G | −26 ± 1 (6)a | −3.8 ± 0.4a | −2 ± 1 | −1 ± 1 | 2.6 ± 0.6 (6) |
| S595G/I701G | −61 ± 1 (5)a | −7.2 ± 0.5a | −1 ± 1 | −1 ± 1 | 1.0 ± 0.5 (5) |
| S595G/A702G | −17 ± 2 (5)a | −2.7 ± 0.6a | −1 ± 1 | −1 ± 1 | 8 ± 1 (5)a |
| S595G/V703G | −25 ± 1 (3)a | −2.3 ± 0.1b | 4 ± 1 | 2 ± 1 | 1.3 ± 0.1 (5) |
| L596G/L699G | −24 ± 2 (5)a | −2.4 ± 0.5b | 9 ± 2 | 5 ± 2 | 2.1 ± 0.3 (5) |
| L596G/A700G | −22 ± 1 (5)a | −3.0 ± 0.3a | 7 ± 2 | 4 ± 1 | 1.4 ± 0.2 (5) |
| L596G/I701G | −53 ± 1 (5)a | −5.6 ± 0.3a | 10 ± 2 | 6 ± 1 | 8 ± 2 (5)a |
| L596G/A702G | −26 ± 1 (5)a | −3.6 ± 0.3a | 5 ± 2 | 3 ± 1 | 4.7 ± 0.6 (5)a |
| L596G/V703G | −25 ± 1 (5)a | −3.7 ± 0.2a | 9 ± 2 | 5 ± 1 | 2.5 ± 0.4 (5)b |
| L596G/D704G | −21 ± 1 (5)a | −2.8 ± 0.3a | 8 ± 2 | 5 ± 1 | 2.8 ± 0.2 (5)b |
| M597G/L699G | −30 ± 1 (5)a | −3.3 ± 0.3a | −1 ± 1 | −0.5 ± 0.8 | 2.2 ± 0.4 (5) |
| M597G/A700G | −26 ± 1 (5)a | −3.0 ± 0.1a | −1 ± 1 | −0.7 ± 0.6 | 0.9 ± 0.1 (5) |
| M597G/I701G | −13 ± 1 (6)a | −1.5 ± 0.2 | 8 ± 1 | 4.8 ± 0.8 | 1.1 ± 0.1 (4) |
| M597G/A702G | −20 ± 1 (5)a | −2.2 ± 0.1b | −1 ± 1 | −0.5 ± 0.5 | 2 ± 1 (5) |
| M597G/V703G | −27 ± 1 (5)a | −3.7 ± 0.3a | −1 ± 1 | −0.3 ± 0.9 | 2.0 ± 0.3 (5)a |
| M597G/D704G | −22 ± 1 (5)a | −2.4 ± 0.1a | 0 ± 1 | 0.0 ± 0.5 | 1.0 ± 0.2 (5) |
| S598G/L699G | −20 ± 1 (5)a | −2.0 ± 0.1b | 0 ± 1 | −0.2 ± 0.6 | 2.5 ± 0.2 (5)b |
| S598G/A700G | −20 ± 1 (5)a | −2.4 ± 0.1a | −2 ± 1 | −1.1 ± 0.7 | 1.4 ± 0.2 (5) |
| S598G/I701G | −51 ± 2 (5)a | −6.5 ± 0.6a | −2 ± 2 | −1 ± 1 | 7 ± 2 (5)a |
| S598G/A702G | −17 ± 1 (5)a | −1.9 ± 0.2 | −2 ± 1 | −1.2 ± 0.6 | 3.6 ± 0.3 (5)a |
| S598G/V703G | −27 ± 1 (5)a | −3.7 ± 0.3a | −1 ± 1 | −0.6 ± 0.8 | 2.2 ± 0.2 (5) |
| S598G/D704G | −14 ± 1 (5)a | −1.6 ± 0.2 | 0 ± 1 | −0.2 ± 0.6 | 1.0 ± 0.1 (5) |
| S599G/I701G | −42 ± 1 (6)a | −4.7 ± 0.3a | 1 ± 1 | 0.4 ± 0.9 | 13 ± 1 (5)a |
| M600G/I701G | −53 ± 1 (5)a | −6.2 ± 0.5a | 1 ± 1 | 1 ± 1 | 25 ± 2 (5)a |
| K601G/I701G | −40 ± 1 (5)a | −4.1 ± 0.2a | 2 ± 1 | 1 ± 1 | 14 ± 3 (4)a |
| S602G/I701G | −52 ± 1 (4)a | −6.0 ± 0.1a | 3 ± 1 | 1.9 ± 0.9 | 20 ± 2 (4)a |
a p < 0.001 as compared with CaV2.3 wt.
b p < 0.01 as compared with CaV2.3 wt.
FIGURE 4.

Histogram of the interaction energies (ΔΔGinteract) in kcal mol−1 for the activation of double mutants produced between IIS4S5 and IIS6 in CaV2.3. ΔΔGinteract were calculated as described under “Experimental Procedures” using the ΔGact values for the wt channel and the individual IIS4S5 mutants (see Table 1) as well as the ΔGact values of the individual IIS6 mutants (see supplemental Table S1). Numerical values for the gating properties of the double mutants are shown in Table 2. In the histogram, the double mutants are shown with the IIS4S5 mutant to the left of the IIS6 mutant (going from the N- to the C-terminal). As shown, the pairs of residues V593G/L699G, V593G/A700G, V593G/A702G, S595G/V703G L596G/L699G, L596G/A700G, L596G/I701G, L596G/A702G, L596G/V703G, L596G/D704G, M597G/I701G, and S602G/I701G displayed significant coupling during the activation gating of CaV2.3.
FIGURE 5.

A, whole cell current traces are shown from left to right for CaV2.3 L596G, I701G, and L596G/I701G in the presence of CaVα2bδ and CaVβ3. Current traces were obtained in 10 mm Ba2+ from a holding potential of −140 mV. B, normalized current-voltage relationships for CaV2.3 wt (dashed line), L596G (squares), I701G (circles), and L596G/I701G (diamonds) were obtained by fitting the experimental data to a modified Boltzmann equation (see “Experimental Procedures” and Refs. 20, 29, and 34)). C, normalized gating charge versus voltage plot for CaV2.3 wt (circles), L596G (squares), I701G (stars), and L596G/I701G (triangles). V0.5,Q values are 6 ± 1 mV (n = 19) for CaV2.3 wt, −20 ± 1 mV (n = 15) for I701G, −24 ± 1 mV (n = 10) for L596G, and −22 ± 1 mV (n = 9) for L596G/I701G.
Introducing a glycine residue at position 593 in IIS4S5 had little impact on the ΔGact of CaV2.3. Nonetheless, V593G was significantly coupled with few distal IIS6 residues (L699G, A700G, A702G) during channel activation with ΔΔGinteract values ranging from −2 to −3 kcal mol−1 (see Table 2 for exact values). V593G was not, however, coupled with I701G (supplemental Fig. S4). In addition to L596G, strong ΔΔGinteract were measured with M597G (supplemental Fig. S5) and S602G during the transition from the closed to the open state.
Stabilizing the Open State Decreases Inactivation Kinetics
Preferential closed-state inactivation of CaV2.3 predicts that fully activated channels will be slower to inactivate than those in intermediate closed states (33). This prediction is tested in Fig. 6 where the inactivation kinetics are plotted against the ΔGact of the L596G/IS6 and IIS4S5/I701G double mutants. As seen, double mutants with very negative ΔGact values tend to inactivate more slowly (higher R50 values). Coupling between S4S5 and S6 residues was also found to play a role in the closed state inactivation of KV4.2 channels (40). Hence, altering the stability of the closed state relative to the stability of the open state influences inactivation kinetics. This contrasts with the minimal changes in the activation properties of the CaV2.3 mutants reported after mutations in the I-II linker (29, 38, 41, 42). The ΔΔGinteract values for the inactivation of the L596G double mutants (but not the V593G double mutants) were smaller than the ΔΔGinteract values for activation (Fig. 7 and supplemental Table S2).
FIGURE 6.

A, correlation between the values of ΔGact and R50 for double mutations containing L596G. Values of R50 for L596G double mutants were obtained as described under “Experimental Procedures” and are reported as a function of their associated ΔGact. The linear regression was calculated with the routine included in Origin 7.0 and showed a strong negative correlation between these two values, with a R factor of −0.95. B, correlation between the values of ΔGact and R50 for double mutations containing I701G. Values of R50 for I701G double mutants were obtained as described under “Experimental Procedures” and are reported as a function of their associated ΔGact. The L596G/I701G mutant is indicated by a asterisk. The double mutant S598G/I701G is shown but not identified on the graph. The linear regression was calculated with the routine included in Origin 7.0 and showed a strong negative correlation between these two values, with a R factor of −0.85.
FIGURE 7.

A, histogram of the ΔΔGinteract for activation and inactivation of the V593G/IIS6 double mutants in CaV2.3. Residues Leu-699, Ala-700, and Ala-702 were significantly coupled with Val-593 during channel activation. As seen, ΔΔGinteract for inactivation were not significantly different from ΔΔGinteract for activation but for A702G. B, histogram of the ΔΔGinteract for activation and inactivation of the L596G/IIS6 double mutants in CaV2.3. Residues Leu-699, Ala-700, Ile-701, Ala-702, Val-703, and Asp-704 were significantly coupled with Leu-596 during channel activation. As seen, ΔΔGinteract for activation were stronger than ΔΔGinteract for inactivation.
Leu-589 Is Not Coupled with the Distal IIS6
The pivotal role played by a leucine residue in the gating of CaV2.3 is reminiscent of the situation in the Shaker channels where the heptad leucine motif in the S4S5 linker has been suggested to be important for its activation gating (43). The heptad motif being partially preserved between the S4S5 linker of Shaker channels and the IIS4S5 of CaV2.3, Leu-596 is predicted to be aligned with L3 in Shaker. In contrast, mutation of L2 to a valine (mutation V2) greatly destabilized the open state with a large positive shift in the activation potential (43). L2 of Shaker is predicted to be aligned with Leu-589 in CaV2.3. We investigated the gating properties of L589G and the double mutant L589G/I701G. In contrast to the observations in Shaker (43) and in hERG (19) channels, the activation gating of L589G was not significantly different from CaV2.3 wt (supplemental Table S3) and the double mutant L589G/I701G failed to display negative interaction energy.
DISCUSSION
In this study we have analyzed the role of the S4S5 linker of domain II in the activation gating of CaV2.3. Introducing glycine residues significantly affected ΔGact when inserted at positions 596, 600, and 602 with ΔΔGact ranging from −2 to −6 kcal mol−1. The negative shifts of ΔΔGact observed with the glycine mutants are compatible with either a destabilization of the closed state or a stabilization of the open state, a distinction that requires single-channel recordings and goes beyond the scope of our current investigation.
Mutations in IIS4S5 brought changes in the activation potential of CaV2.3 that were comparable with those reported in homotetrameric KV channels (24) including mutations within the heptad leucine motif of Shaker channels (43). In the latter, mutations of L3 (aligned with Leu-596) to a valine (V3) or alanine (A3) residue shifted the midpotential for activation by −6 and −20 mV, respectively (43). In contrast, the alanine mutation of Leu-251, also predicted to be aligned with Leu-596, introduced a +25 mV displacement in the activation potential of the KCNQ1 channel (24). Located in a more distal position, the E395A mutation (aligned with Ser-602), shifted by −20 mV the voltage dependence of the Q/Qmax curve of Shaker channels (36).
None of the CaV2.3 single mutations herein studied were seen to completely uncouple the voltage sensor movement from pore openings, unlike I384N and F484G in homotetrameric Shaker channels (44). This may not be surprising given that our current investigation was limited to domain II. Despite the inherently asymmetrical nature of the CaV2.3 pore, it can only be surmised at this time that each domain contributes equally to the coupling between voltage sensor and channel opening as it has been demonstrated in Shaker channels (45).
A double mutant cycle analysis was carried out to identify pairs of IIS4S5 and IIS6 residues that could participate to the channel activation as done previously in KV channels (12, 24, 46, 47). We had already shown that mutations carried out in the distal IIS6 impacted significantly on the relative stability of the closed state with negative shifts in the activation potential up to −40 mV (20). Our current analysis showed that Leu-596 in IIS4S5 and Ile-701 in IIS6 display significant energy coupling during channel activation. It is important to note that mutations of nearby residues in IIS4S5 displayed milder if any coupling with other residues in IIS6. The ΔΔGinteract values measured between L596G and I701G were found to be stronger than the ones reported between conservative hydrophobic mutations performed in KCNQ1 (24) and CNG (25) channels. This interaction could be specific to CaV2.3. Double mutant cycle experiments carried out in CaV1.2 and CaV2.1 with residues predicted to be aligned with Leu-596 and Ile-701 failed to show any significant energy coupling (supplemental Tables S4 and S5).
The molecular mechanism conveying CaV2.3 activation thus appears to rely in part on the electrochemical coupling between IIS4S5 and IIS6. Five of the six residues in IIS6 herein studied are hydrophobic suggesting that decreasing the hydrophobic content in either S4S5 or S6 might destabilize the closed state of the channel. Indeed, decreasing the hydrophobic content of IIS4S5 with L596D and L596P were also shown to alter the relative stability of the closed state in CaV2.3. The suggestion that the interaction between S4S5 and S6 is mostly hydrophobic (thus considered to be a weak interaction) is compatible with results obtained in Shaker-like (12, 46) and KCNQ1 (24) channels and is further supported by the recent observation that S4S5 peptides inhibit KCNQ1 in a state-dependent manner (47).
The predictive locations of Leu-596 and Ile-701 are shown in the homology model of CaV2.3 based upon the atomic coordinates of KV1.2 in the open state (Fig. 8). According to the model, it is plausible that Leu-596 and Ile-701 face each other in the protein. Nonetheless, the predicted distance of 10.6 Å between their C-α atoms (11.2 Å between their C-β atoms) makes a direct interaction unlikely. Leu-596 showed significant interaction with five other residues in distal IIS6 during the process that leads to CaV2.3 activation. This observation suggests that IIS6 may rotate outwardly, whereas the four S6 helices move away from one another. V593G, predicted to be facing away from Leu-596 in the computer-based model of the IIS4S5 linker, was also significantly coupled with a few positions in IIS6. Given the relative precision of the homology model, the possibility that Val-593 interacts with Ile-701 during channel activation cannot be completely ruled out. Cooperativity thus may reflect long range coupling of residues (21).
FIGURE 8.

Two different views (A, B) of the computer-based molecular model of domain II of CaV2.3 are shown in a schematic representation. Modeling of domain II was based upon the atomic coordinates of KV1.2 (Protein Data Bank code 2A79) and was achieved with Modeler 9v4 from the S4 to the end of S6. For sake of clarity, only the region going from the S4S5 linker to the end of S6 is shown. The regions in yellow correspond to the regions studied in our double mutant cycle analysis, namely residues Val-593 to Ser-598 in IIS4S5 and Leu-699 to Asp-704 in IIS6. Val-593 and Leu-596 (IIS4S5) are shown in stick representation in magenta and in blue, respectively, whereas Ile-701 (IIS6) is shown in red. The shortest distance between Leu-596 and Ile-701 was estimated to be 9.4 Å from the α-carbon of Leu-596 to the β-carbon of Ile-701. S.F. stands for selectivity filter and P.H. stands for pore helix. The figure was produced using PyMol (DeLano Scientific).
Nonetheless, many questions remain to be answered. Our study did not address whether the four voltage sensors act in a concerted or independent manner. It is thus impossible to argue that the S4S5/S6 coupling within domain II constitutes the obligatory rate-limiting voltage-dependent step for channel activation in CaV2.3. More importantly, it remains to be seen whether the cooperative mechanism in domain II requires atomic proximity between S4S5 and S6. These questions go beyond the scope of this current investigation and await further structural studies of CaV channels.
Supplementary Material
Acknowledgments
We thank Georges A. Haddad for help with the cut-open oocyte technique, Yolaine Dodier and Alexandra Raybaud for some constructs, Pierre-Olivier Demers-Giroux for help in characterizing mutants, Julie Verner for oocyte culture, Michel Brunette for computer maintenance, and Claude Gauthier for artwork.
Footnotes
This work was supported in part by Canadian Institutes of Health Research Grant MOP13390 and a grant from the Canadian Heart and Stroke Foundation (to L. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” “Results,” Figs. S1–S5, and Tables S1–S5.
REFERENCES
- 1. Birnbaumer L., Campbell K. P., Catterall W. A., Harpold M. M., Hofmann F., Horne W. A., Mori Y., Schwartz A., Snutch T. P., Tanabe T. (1994) Neuron 13, 505–506 [DOI] [PubMed] [Google Scholar]
- 2. Catterall W. A. (2000) Annu. Rev. Cell Dev. Biol. 16, 521–555 [DOI] [PubMed] [Google Scholar]
- 3. Cribbs L. L., Lee J. H., Yang J., Satin J., Zhang Y., Daud A., Barclay J., Williamson M. P., Fox M., Rees M., Perez-Reyes E. (1998) Circ. Res. 83, 103–109 [DOI] [PubMed] [Google Scholar]
- 4. Perez-Reyes E., Cribbs L. L., Daud A., Lacerda A. E., Barclay J., Williamson M. P., Fox M., Rees M., Lee J. H. (1998) Nature 391, 896–900 [DOI] [PubMed] [Google Scholar]
- 5. Randall A., Benham C. D. (1999) Mol. Cell. Neurosci. 14, 255–272 [DOI] [PubMed] [Google Scholar]
- 6. Snutch T. P., Reiner P. B. (1992) Curr. Opin. Neurobiol. 2, 247–253 [DOI] [PubMed] [Google Scholar]
- 7. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., MacKinnon R. (1998) Science 280, 69–77 [DOI] [PubMed] [Google Scholar]
- 8. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) Nature 417, 515–522 [DOI] [PubMed] [Google Scholar]
- 9. Jiang Y., Lee A., Chen J., Ruta V., Cadene M., Chait B. T., MacKinnon R. (2003) Nature 423, 33–41 [DOI] [PubMed] [Google Scholar]
- 10. Kuo A., Gulbis J. M., Antcliff J. F., Rahman T., Lowe E. D., Zimmer J., Cuthbertson J., Ashcroft F. M., Ezaki T., Doyle D. A. (2003) Science 300, 1922–1926 [DOI] [PubMed] [Google Scholar]
- 11. Long S. B., Campbell E. B., Mackinnon R. (2005) Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 12. Long S. B., Campbell E. B., Mackinnon R. (2005) Science 309, 903–908 [DOI] [PubMed] [Google Scholar]
- 13. Armstrong C. M. (2003) Sci. STKE re10. [DOI] [PubMed] [Google Scholar]
- 14. Hackos D. H., Chang T. H., Swartz K. J. (2002) J. Gen. Physiol. 119, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yifrach O., MacKinnon R. (2002) Cell 111, 231–239 [DOI] [PubMed] [Google Scholar]
- 16. Lu Z., Klem A. M., Ramu Y. (2001) Nature 413, 809–813 [DOI] [PubMed] [Google Scholar]
- 17. Lu Z., Klem A. M., Ramu Y. (2002) J. Gen. Physiol. 120, 663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferrer T., Rupp J., Piper D. R., Tristani-Firouzi M. (2006) J. Biol. Chem. 281, 12858–12864 [DOI] [PubMed] [Google Scholar]
- 19. Van Slyke A. C., Rezazadeh S., Snopkowski M., Shi P., Allard C. R., Claydon T. (2010) Biophys. J. 99, 2841–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raybaud A., Baspinar E. E., Dionne F., Dodier Y., Sauvé R., Parent L. (2007) J. Biol. Chem. 282, 27944–27952 [DOI] [PubMed] [Google Scholar]
- 21. Horovitz A. (1996) Fold. Des. 1, R121–R126 [DOI] [PubMed] [Google Scholar]
- 22. Kudrnac M., Beyl S., Hohaus A., Stary A., Peterbauer T., Timin E., Hering S. (2009) J. Biol. Chem. 284, 12276–12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ranganathan R., Lewis J. H., MacKinnon R. (1996) Neuron 16, 131–139 [DOI] [PubMed] [Google Scholar]
- 24. Labro A. J., Boulet I. R., Choveau F. S., Mayeur E., Bruyns T., Loussouarn G., Raes A. L., Snyders D. J. (2011) J. Biol. Chem. 286, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kusch J., Zimmer T., Holschuh J., Biskup C., Schulz E., Nache V., Benndorf K. (2010) Biophys. J. 99, 2488–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneider T., Wei X., Olcese R., Costantin J. L., Neely A., Palade P., Perez-Reyes E., Qin N., Zhou J., Crawford G. D., Smith R. G., Appel S. H., Stefani E., Birnbaumer L. (1994) Receptors Channels 2, 255–270 [PubMed] [Google Scholar]
- 27. Castellano A., Wei X., Birnbaumer L., Perez-Reyes E. (1993) J. Biol. Chem. 268, 3450–3455 [PubMed] [Google Scholar]
- 28. Williams M. E., Feldman D. H., McCue A. F., Brenner R., Velicelebi G., Ellis S. B., Harpold M. M. (1992) Neuron 8, 71–84 [DOI] [PubMed] [Google Scholar]
- 29. Berrou L., Dodier Y., Raybaud A., Tousignant A., Dafi O., Pelletier J. N., Parent L. (2005) J. Biol. Chem. 280, 494–505 [DOI] [PubMed] [Google Scholar]
- 30. Birnbaumer L., Qin N., Olcese R., Tareilus E., Platano D., Costantin J., Stefani E. (1998) J. Bioenerg. Biomembr. 30, 357–375 [DOI] [PubMed] [Google Scholar]
- 31. Parent L., Schneider T., Moore C. P., Talwar D. (1997) J. Membr. Biol. 160, 127–140 [DOI] [PubMed] [Google Scholar]
- 32. Meza U., Thapliyal A., Bannister R. A., Adams B. A. (2007) Mol. Pharmacol. 71, 284–293 [DOI] [PubMed] [Google Scholar]
- 33. Patil P. G., Brody D. L., Yue D. T. (1998) Neuron 20, 1027–1038 [DOI] [PubMed] [Google Scholar]
- 34. Raybaud A., Dodier Y., Bissonnette P., Simoes M., Bichet D. G., Sauvé R., Parent L. (2006) J. Biol. Chem. 281, 39424–39436 [DOI] [PubMed] [Google Scholar]
- 35. Stefani E., Bezanilla F. (1998) Methods Enzymol. 293, 300–318 [DOI] [PubMed] [Google Scholar]
- 36. Batulan Z., Haddad G. A., Blunck R. (2010) J. Biol. Chem. 285, 14005–14019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olcese R., Neely A., Qin N., Wei X., Birnbaumer L., Stefani E. (1996) J. Physiol. 497, 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berrou L., Bernatchez G., Parent L. (2001) Biophys. J. 80, 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karplus K., Karchin R., Draper J., Casper J., Mandel-Gutfreund Y., Diekhans M., Hughey R. (2003) Proteins 53, Suppl, 6, 491–496 [DOI] [PubMed] [Google Scholar]
- 40. Barghaan J., Bähring R. (2009) J. Gen. Physiol 133, 205–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berrou L., Klein H., Bernatchez G., Parent L. (2002) Biophys. J. 83, 1429–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Findeisen F., Minor D. L., Jr. (2009) J. Gen. Physiol. 133, 327–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCormack K., Tanouye M. A., Iverson L. E., Lin J. W., Ramaswami M., McCormack T., Campanelli J. T., Mathew M. K., Rudy B. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 2931–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haddad G. A., Blunck R. (2011) J. Gen. Physiol. 137, 455–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gagnon D. G., Bezanilla F. (2010) J. Gen. Physiol. 136, 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pathak M. M., Yarov-Yarovoy V., Agarwal G., Roux B., Barth P., Kohout S., Tombola F., Isacoff E. Y. (2007) Neuron 56, 124–140 [DOI] [PubMed] [Google Scholar]
- 47. Choveau F. S., Rodriguez N., Ali F. A., Labro A. J., Rose T., Dahimène S., Boudin H., Le Hénaff C., Escande D., Snyders D. J., Charpentier F., Mérot J., Baró I., Loussouarn G. (2011) J. Biol. Chem. 286, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.