

Abstract
Dhh1 is a highly conserved DEAD-box protein that has been implicated in many processes involved in mRNA regulation. At least some functions of Dhh1 may be carried out in cytoplasmic foci called processing bodies (P-bodies). Dhh1 was identified initially as a putative RNA helicase based solely on the presence of conserved helicase motifs found in the superfamily 2 (Sf2) of DEXD/H-box proteins. Although initial mutagenesis studies revealed that the signature DEAD-box motif is required for Dhh1 function in vivo, enzymatic (ATPase or helicase) or ATP binding activities of Dhh1 or those of any its many higher eukaryotic orthologues have not been described. Here we provide the first characterization of the biochemical activities of Dhh1. Dhh1 has weaker RNA-dependent ATPase activity than other well characterized DEAD-box helicases. We provide evidence that intermolecular interactions between the N- and C-terminal RecA-like helicase domains restrict its ATPase activity; mutation of residues mediating these interactions enhanced ATP hydrolysis. Interestingly, the interdomain interaction mutant displayed enhanced mRNA turnover, RNA binding, and recruitment into cytoplasmic foci in vivo compared with wild type Dhh1. Also, we demonstrate that the ATPase activity of Dhh1 is not required for it to be recruited into cytoplasmic foci, but it regulates its association with RNA in vivo. We hypothesize that the activity of Dhh1 is restricted by interdomain interactions, which can be regulated by cellular factors to impart stringent control over this very abundant RNA helicase.
Keywords: Gene Expression, RNA-binding Protein, RNA Helicase, RNA Metabolism, Translation
Introduction
The regulation of gene expression in eukaryotes is a complex series of events that is well coordinated by different cellular machineries. Central to this process is the regulation of mRNA metabolism. It begins with transcription followed by pre-mRNA processing in the nucleus, nucleo-cytoplasmic transport, translation, and finally, the controlled degradation of mRNA.
mRNA degradation is one of the important means of mRNA quality control. In eukaryotic cells two major pathways have evolved for mRNA degradation; they are non-sense-mediated decay, which is deadenylation-independent and the more ubiquitously used deadenylation-dependent pathway (1). Non-sense-mediated decay is the process of choice for quick and efficient removal of transcripts with non-sense codons, unspliced introns, or extended 3′-UTR (2). The deadenylation-dependent pathway begins with the removal of the 3′-poly(A) tail by 3′-5′ exonucleases Ccr4 and Pop2 or poly(A) nuclease (3–7). This is followed by removal of the 5′-m7-guanosine triphosphate cap by the Dcp2/Dcp1 decapping complex (8, 9). The binding of Dcp1/Dcp2 to the 5′ cap and decapping are enhanced by a complex of seven Lsm (like-Sm) proteins (Lsm1 through Lsm7), Pat1, Edc3, and Dhh1 (10–17). The final step in the pathway involves degradation of the decapped and deadenylated RNA by the 5′-3′ exonuclease Xrn1 and by the exosome that acts in the 3′-5′ direction (16, 17). Proteins involved in mRNA turnover are localized to distinct foci in the cytoplasm called processing bodies (P-bodies) or GW bodies (18–20). Although the formation of P-bodies have been initially associated with mRNA decay and storing translationally repressed RNA, recent studies have shown that they not terminal centers of RNA degradation, as a subset of translationally repressed RNA can reenter the active pool in response to cellular signals (11, 21–23).
One of the factors important for mRNA degradation and translational repression in yeast is the abundant DEAD-box-containing protein, Dhh1. Dhh1 localizes to P-bodies and stimulates mRNA decapping, possibly by interacting with the decapping enzyme Dcp1p (11, 24, 25). Dhh1, along with its interacting partners Pat1 and Edc3, is also involved in translational repression of mRNAs and has been shown to sequester RNAs into a non-translating pool that does not undergo degradation (22, 25, 26). Studies have linked Dhh1 to cell cycle regulation, where it is important for G1/S DNA-damage checkpoint recovery in Saccharomyces cerevisiae (27, 28). Recent studies have also shown that Dhh1 controls hyphal development (29). Thus, although not essential for normal cell growth, this protein plays a number of important functions.
Dhh1 also interacts with the evolutionary conserved Ccr4-Not complex (30, 31). Multiple roles have been assigned to the Ccr4-Not complex in regulating the life of mRNAs (32–34). First identified as a complex regulating transcription initiation, it was later shown that the Ccr4 subunit is the major poly(A) deadenylase (5, 6). Although the later observations raised questions about the function of Ccr4-Not in transcription, we recently confirmed that the Ccr4-Not complex interacts with elongating RNAPII and directly regulates transcription elongation (35). It appears that the Ccr4-Not complex, and not Ccr4 specifically, regulates mRNA decay because even the Not group of proteins has been found localized to P bodies and regulates mRNA half-lives (36). Thus, this complex provides a link to transcription in the nucleus and mRNA decay in the cytoplasm.
Dhh1 shows exceptional sequence conservation. For example, the human orthologue of Dhh1, Ddx6 (rck/p54), bears a 69% identity and 83% similarity to Dhh1. Ddx6 can complement the phenotypes of a dhh1Δ mutant (27), suggesting they play similar roles in regulating cell function in their respective organisms. Dhh1 belongs to the superfamily 2 (Sf2) of DEXD/H-box proteins (37). Sf2 helicases contain nine conserved helicase motifs. DEXD/H-box proteins have been reported to be components of various cellular machineries that regulate mRNA metabolism and are involved in mRNA splicing, transport, translation, decay, ribosome biogenesis, and transcription in both eukaryotes and prokaryotes (38–41). Many of these functions have been attributed to the ability of these proteins to act as helicases and disrupt RNA secondary structure. In addition, several DEXD/H-box proteins are shown to displace protein factors from single-stranded RNA without duplex unwinding (42, 43). The crystal structure of the core domain of Dhh1 was recently solved, and its domain structure compares well with that of other DEAD-box helicases like S. cerevisiae eIF4A, MjDEAD, Drosophila melanogaster Vasa, and hepatitis C virus NS3 (44). However, there are distinct differences in the relative orientation of the two globular (RecA-like) domains in Dhh1 compared with other DEXD/H proteins. It was proposed that interdomain interactions within Dhh1 might regulate its activity (44). The structure of Dhh1 has been insightful in predicting domain orientations and key residues that may be involved in enzymatic function; however, enzymatic activities of Dhh1 have not been observed. Many questions remain as to how the various biochemical activities of Dhh1 regulate the multiple functions of this important helicase. Specifically, does the unusual conformation of the Dhh1 predicted from the crystal structure regulate its biochemical activities and its cellular functions in vivo?
Here we provide the first analysis of Dhh1 ATPase and ATP binding activities and have identified key residues involved in ATP and RNA binding and ATP hydrolysis. Interestingly, Dhh1 has weaker ATPase activity compared with other DEAD-box helicases, and we present data arguing that the intermolecular interactions between the N- and C-terminal domains restrict its ATPase activity. Disruption of these interactions through mutagenesis increases ATP hydrolysis and enhances its mRNA metabolism and mRNA binding functions in vivo. Based on our results, we hypothesize that Dhh1 activity is restricted by interdomain interactions, which can be regulated by cellular factors to impart stringent control over this very abundant RNA helicase.
EXPERIMENTAL PROCEDURES
Nucleotides
The 3′ fluorescein-labeled oligonucleotides for RNA binding assays and RNA oligonucleotides for helicase assays were purchased from Dharmacon Research Dharmacon Research, Inc. (Lafayette, CO). DNA oligonucleotides used for mutagenesis were from Integrated DNA Technologies Inc. (Coralville, IA) or Invitrogen. [γ-32P]ATP (6000 Ci/mmol) was from PerkinElmer Life Sciences. [α-32P]ATP, [α-32P]UTP, [α-32P]CTP, [α-32P]GTP, and [α-32P]dATP (3000 Ci/mmol) were from PerkinElmer Life Sciences. Nucleotides were obtained from GE Healthcare. AMP-PNP,2 ADP, poly(U), poly(G), poly(C), and poly(A) were obtained from Sigma. RNA oligonucleotides were purified on denaturing 8 m urea polyacrylamide gels. Concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop spectrophotometer and using the appropriate calculated extinction coefficient.
Expression and Purification of Recombinant Dhh1
The coding sequence of DHH1 was amplified by PCR and cloned into pRSET-A (Invitrogen) to express the protein with a hexahistidine tag (His6) at the N terminus of the protein. This introduces an additional 37 amino acids, including the His6 into the protein. Mutant proteins were generated by PCR-generated site-directed mutagenesis. Dhh1 was expressed in Escherichia coli BL21-DE3 pLysS cells. Expression was induced with 1.0 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at room temperature. The cells were harvested, and the cell pellet was resuspended in 20 ml of buffer A (20 mm Tris-HCl (pH 7.4), 500 mm NaCl, 0.1% Triton X-100, and 10% glycerol) containing 100 μm PMSF, pepstatin, leupeptin, and chymostatin (5 μg/ml each), 1 mm benzamidine hydrochloride, and 10 mm imidazole. The cell suspension was sonicated on ice using a Branson Sonifer. The resulting cell lysate was clarified by centrifugation at 16,000 rpm for 30 min. The supernatant was incubated with NTA-cobalt-agarose (Clontech, Mountain View, CA) for 1 h at 4 °C. The resin was packed into a column and washed with 50 ml of buffer A containing 10 mm imidazole followed by a wash with Buffer A containing 20 mm imidazole. The protein was eluted using 250 mm imidazole in buffer B (20 mm Tris-HCl (pH 7.4), 250 mm NaCl, 0.1% Triton X-100, and 10% glycerol) containing 100 μm PMSF and 1 mm benzamidine hydrochloride. The peak fractions were pooled and dialyzed against 2 liters of buffer C (20 mm HEPES-NaOH (pH 7.4), 75 mm NaCl, 10% glycerol). The dialyzed sample was passed over a SP-Sepharose column. The column was washed with 20 ml of buffer C followed by 10 ml of buffer D (20 mm HEPES-NaOH (pH 7.4), 150 mm NaCl, 10% glycerol). The protein was eluted using buffer E (20 mm HEPES-NaOH (pH 7.4), 500 mm NaCl, 10% glycerol). The pure fractions were pooled and dialyzed against 2 liters of buffer B containing 1 mm dithiothreitol, 0.5 mm EDTA. The protein was stored at −80 °C.
ATPase Assay
ATPase assays were typically performed at 30 °C in 25 mm MOPS (pH 7.0), 50 mm NaCl, 10 mm MgCl2, 4 μm Dhh1, 50 μm ATP, 4 μCi of [γ-32P]ATP, and 10 μg of polynucleotide substrates (Sigma). Reactions were initiated by the addition of ATP and quenched with EDTA after 1 h. Specific concentrations of substrate or enzyme along with any deviations from the above are indicated in the legends to Figs. 1 and 2. 3 μl of the quenched reaction was spotted onto PEI-cellulose TLC plates (EM Science, Gibbstown, NJ). TLC plates were developed in a buffer containing 40 mm LiCl and 100 mm formic acid, dried, and exposed to a PhosphorImager screen. TLC plates were visualized by using a PhosphorImager and quantified using the ImageQuant software (GE Healthcare) to determine the amount of ATP hydrolyzed to ADP and Pi. The pmol of ATP hydrolyzed by Dhh1 in 1 h were determined by calculating the ratio of released Pi to unhydrolyzed ATP.
FIGURE 1.

ATPase activity of Dhh1 is stimulated by RNA. A, purification of recombinant Dhh1 is shown. Recombinant His6-Dhh1 was purified on NTA-cobalt beads (Talon) followed by SP-Sepharose chromatography. An SDS-PAGE gel shows the different fractions during purification. E1-E4, elutions from NTA-cobalt beads or SP-Sepharose column. B, ATPase activity of Dhh1 is shown. An ATPase assay was performed at 30 °C for 60 min using 4 μm Dhh1 and increasing amounts of poly(U) (0–50 μg). Results are plotted as pmol of ATP hydrolyzed as a function of increasing poly(U) concentration. The data is shown as the average and S.E. of three experiments.
FIGURE 2.

ATPase activity of Dhh1 is stimulated by the mutation of residues involved in interdomain interactions. A, the crystal structure of Dhh1 with the locations Lys-91 and Thr-344 indicated in red and blue, respectively. The structure was analyzed using PyMOL software (PDB code 1S2M). B, ATPase activity of wild type and mutant Dhh1 is shown. ATPase assays were carried at 30 °C for 60 min using 4 μm protein and increasing amounts of poly(U) (0–50 μg). Results are plotted as pmol of ATP hydrolyzed as a function of increasing poly(U) concentration. C, RNA sequence requirement for ATPase activity is shown. ATPase activity was carried out using 4 μm protein and 10 μg of RNA indicated in the figure. D, nucleotide specificity of Dhh1 is shown. NTPase activity of wild type and mutant Dhh1 was carried at 30 °C for 60 min using 4 μm protein and 10 μg of poly(U) RNA. The data in panels C and D represent an average and S.E. of three experiments.
Nucleotide Cross-linking
Dhh1 (4 μm) was incubated with 10 μCi of α-32P-labeled nucleotides in 25 mm MOPS (pH 7.0), 50 mm NaCl, 10 mm MgCl2 at room temperature for 10 min. For experiments to study competition with cold nucleotides, 100 μm ATP, GTP, CTP, UTP, dATP, or ADP were added before cross-linking. The samples were then UV-cross-linked on ice (254 nm) for 6 min using a Stratalinker (Stratagene, La Jolla, CA). The samples were boiled in SDS-PAGE buffer and resolved in a 10% SDS-PAGE gel. The gels were stained with Coomassie Blue, dried and exposed to a PhosphorImager screen for detection. The gels were quantified using the ImageQuant software (GE Healthcare). The signals were corrected for the amount of protein in each sample.
Nucleic Acid Binding Assay
Experiments were performed using a Beacon fluorescence polarization system (GE Healthcare). Assays were performed by mixing 0.1 nm 3′-fluorescein-labeled poly(U) with increasing amounts of Dhh1 in binding buffer (25 mm MOPS (pH 7), 2 mm β-mercaptoethanol, 10 mm MgCl2, and 50 mm NaCl) followed by monitoring the change in fluorescence polarization (mP). The volume of Dhh1 or buffer added to the binding reaction was equal to the total reaction volume. All steps were performed in reduced light. To determine the dissociation constant (Kd), mP was plotted as a function of Dhh1 concentration, and the data were fit to a hyperbola using Kaleidagraph software (Synergy Software, Reading, PA).
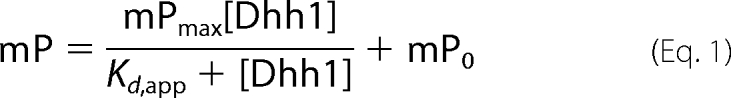 |
The above equation represents the equation of a rectangular hyperbola, where mP is the observed millipolarization, mPmax is maximum polarization, mP0 is polarization of RNA alone in the absence of Dhh1, and Kd,app is the apparent dissociation constant.
Helicase Assay
Helicase assays were carried out as described in a previous publication (45). 5 μm RNA oligonucleotides were annealed in 25 mm MOPS (pH 7.0), 50 mm NaCl by heating to 90 °C for 1 min and slowly cooling to room temperature using a Progene Thermocycler (Techne, Minneapolis, MN). The ratio of labeled to unlabeled strand was 1:1.2. All helicase assays were performed at 30 °C. 1 μm Dhh1 was mixed with 2 nm 32P-labeled unwinding substrate in 25 mm MOPS (pH 7.0), 50 mm NaCl, and 1 mm EDTA. Reactions were initiated by the addition of 10 mm ATP, 10 mm MgCl2, and 100 nm trapping strand (a 9-mer RNA that is complementary to the displaced strand) in 25 mm MOPS (pH 7.0), 50 mm NaCl. Reactions were quenched by adding EDTA and SDS to a final concentration of 100 mm and 0.33%, respectively. Products were resolved on native polyacrylamide gels.
Strains and Media
Strains used in this study are described in detail in supplemental Table 1). DHH1 containing 800 base pairs (bp) upstream and 400 bp downstream of the open reading frame was cloned into the yeast centromeric vector pRS414 from the plasmid pRS426-DHH1(46). Mutants of DHH1 were generated by site-directed mutagenesis using the QuikChange method. Plasmids with wild type and mutant DHH1 were transformed into strain YJR148 (27) and used in Figs. 4 and 5B. For the detailed analysis of some mutants, they were integrated into the genome. These mutants were cloned into the integrating vector pRS404, and the resulting plasmid was linearized by restriction endonuclease digestion and transformed into YJR148. Integration was confirmed by PCR and protein expression by Western blotting. For spot growth assays, cultures were grown overnight in synthetic drop out (SD) medium lacking tryptophan and diluted to an A600 = 1.0. 3-Fold serial dilutions of the culture were spotted on to SD-tryptophan plates. Gene deletions were performed as previously described using a PCR-mediated gene disruption strategy (47). Integrated mutants were grown in 1% yeast extract, 2% peptone, 2% dextrose (unless indicated otherwise), and 20 μg/ml adenine sulfate.
FIGURE 4.

Phenotypic analysis of Dhh1 mutants. A, a schematic shows the conserved domains in Dhh1 with the position of residues that were mutated. B, analysis of the expression of Dhh1 mutants is shown. Western blotting using anti-Dhh1 antibody is shown. The amount of TATA-binding protein (TBP) was used as a loading control. C, yeast cells expressing wild type or mutant DHH1 were grown in SD-tryptophan, and 3-fold serial dilutions of cultures were spotted onto SD-tryptophan plates (control) or the same medium containing 75 mm hydroxyurea (HU) or 0.01% methyl methane sulfonate (MMS). UV sensitivity was measured by exposing a plate to 60 J/m2 UV radiation. For testing heat sensitivity, plates were incubated at 37 °C. Control plates were incubated at 30 °C. The mutants were tested in two groups; a wild type strain and deletion mutant was analyzed in each group.
FIGURE 5.

All activities of Dhh1 are required for mRNA decay. A, shown is a half-life estimation of GAL1. Cells were grown overnight to an A600 of 0.8 in 1% yeast extract, 2% peptone, and 20 μg/ml adenine sulfate supplemented with 2% galactose and then shifted to 4% dextrose-containing medium to repress GAL1 transcription. Samples were collected at different time points. Blots were probed with GAL1 and loading control, ScR1. GAL1 signal was normalized to the amount of ScR1 signal to correct for loading. To calculate half-life, a 0-min time point for each set was set to 100%, and the log of the amount of transcript was plotted as a function of time. B, shown is accumulation of ECD1 mRNA, a known target of Dhh1. Northern blot analysis to detect EDC1 and ScR1 (loading control) transcripts was carried out. EDC1 signal was normalized to the amount of ScR1. Values were normalized to the wild type signal, which was set to 1. Bars represent the average and S.D. of at least three experiments.
RNA Isolation and Analysis of RNA by Northern Blotting
RNA isolation and northern blotting were carried out as previously described (48). Total RNA (15 μg) was separated on 1% formaldehyde gel and transferred to nitrocellulose membrane (Hybond-N+; Amersham Biosciences) via capillary action. After UV-cross-linking and a ≥4-h prehybridization at 65 °C, radioactively labeled gene-specific probes were added. Signal was detected using PhosphorImager screen, scanned with the Typhoon system, and quantified using ImageQuant software (GE Healthcare). scR1, an RNA polymerase III-transcribed gene, was used as a loading control.
RNA Immunoprecipitation (RIP)
RIP assays were based on a previous publication (49), with the changes described below. Yeast cultures (100 ml) were grown at 30 °C in synthetic dropout medium lacking tryptophan and containing 4% dextrose to an absorbance (600 nm) of 0.8 and then shifted to rich media containing 1% yeast extract, 2% peptone, 4% dextrose, and 20 μg/ml adenine sulfate and grown for 1 h at 30 °C. For the induction of stress, cells were treated with 0.03% methyl methane sulfonate for 1 h before cross-linking. Cells were cross-linked with 1% (v/v) formaldehyde at room temperature for 15 min. Formaldehyde was quenched with glycine (125 mm). All buffers were prepared in diethyl pyrocarbonate-treated water. Cells were resuspended in 500 μl of cold FA-lysis RIP buffer (50 mm HEPES-KOH (pH 7.5), 150 mm NaCl, 1% Triton X-100, and 0.1% sodium deoxycholate supplemented with 2 μg/ml leupeptin, 3 μg/ml aprotinin, 2 μg/ml pepstatin A, 1 μg/ml chymostatin, 1 mm benzamidine-HCl, 0.5 mm PMSF). The cells were distributed into two microcentrifuge tubes, and 300 μl of glass beads were added. Tubes were mixed at the highest speed on a vortex mixer for 45 min at 4 °C. The lysate was transferred to a 15-ml tube and brought up to a volume of 1.8 ml using FA-lysis RIP buffer. The lysate was sonicated in a Bioruptor sonicator (Diagenode, Philadelphia) for 2–30-s pulses and then transferred back into microcentrifuge tubes. The lysates were clarified by two 30-min spins at 14,000 rpm at 4 °C. The protein concentration of the extracts ranged between 7 and 10 mg/ml. 500 μl of the whole cell extract was diluted with an equal volume of FA-lysis RIP buffer, and MgCl2 and CaCl2 were added to 25 and 5 mm, respectively. RNase-free DNase I was added to 4 units per ml (Worthington, Lakewood, NJ), and the extract was incubated for 60 min at 30 °C. EDTA was added to 50 mm, and the whole cell extract was cooled on ice and centrifuged at 14,000 rpm. An aliquot (100 μl) was removed for the input sample. The remainder was added to tubes containing 20 μl of protein A-Sepharose beads (GE Healthcare) containing the equivalent of 2 μl of anti-Dhh1 polyclonal antiserum. The antibody was raised to full-length Dhh1 in rabbits. The beads were incubated overnight at 4 °C with agitation. The beads were washed 3 times with FA-lysis RIP buffer, twice using FA-wash buffer 2 (50 mm HEPES-KOH (pH 7.5), 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.5 m NaCl, 0.5 m PMSF, 1 mm benzamidine-HCl), twice using FA-wash buffer 3 (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, 10 mm Tris-HCl (pH 8.0), 0.5 m PMSF, 1 mm benzamidine-HCl), and twice in TE buffer (10 mm Tris-HCl, 1 mm EDTA). All washes were performed with ice-cold cold buffers. The immune complexes were eluted off the beads for 20 min at 65 °C in 450 μl of elution buffer (25 mm Tris-HCl (pH 7.5), 1 mm EDTA, 0.2 m NaCl, and 0.5% SDS). Proteinase K was added to 100 μg/ml, and the eluted material was incubated at 55 °C for 30 min and then 4–5 h at 65 °C to reverse the cross-links. The RNA was purified by acid-phenol (pH 4.8)/chloroform (1:1) extraction and ethanol-precipitated in the presence of 20 μg glycogen. The pellet was resuspended in diethyl pyrocarbonate-treated water and converted to cDNA using random 9-mer primers and avian myeloblastosis virus reverse transcriptase as recommended by the manufacturer (Promega, Madison, WI). Transcripts were detected using gene-specific primers by semiquantitative PCR. The primers to the PYK1 ORF were described in previous publications (50, 51). PCR products were analyzed by electrophoresis in agarose gels and ethidium bromide staining, scanned with the Typhoon system (GE Healthcare), and quantified by using ImageQuant software. Percentage immunoprecipitate (IP) values were calculated using the following product: (IP signal/input signal) × 100. Background was determined by conducting the RIP on Δdhh1 cells, and these values were subtracted from the specific signals. Error bars represent the S.E. of at least three repetitions.
Live Cell Imagining of Dhh1
Cells containing wild type or mutant DHH1-GFP integrated at its original genomic locus were transformed with a centromeric vector expressing DCP2-RFP (pRP1186) (52). Yeast cultures were grown to A600 of 0.4–0.6 in the appropriate synthetic dropout (SD) media. Cells were collected by brief centrifugation, resuspended in fresh synthetic dropout medium ± 2% glucose, incubated at room temperature for 15 min, and spun down. Cells were resuspended in of the initial volume and spotted on slides. The slides were immediately examined under a microscope at room temperature. All images were acquired using an Olympus Fluoview 1000 microscope system running FV1000 Fluoview software, with an Olympus 100×, oil-immersion 1.4 NA objective. Images were stored as 512 × 512 pixel files. All experimental images were captured as Z-stacks of 5–8 images and compiled. ImageJ was used to measure the number and size of P bodies as described previously (52).
RESULTS
Dhh1 Has RNA-stimulated ATPase Activity
Recombinant His6-Dhh1 was expressed in E. coli, isolated on NTA-cobalt beads, and further purified by SP-Sepharose chromatography. The use of NTA-cobalt beads and ion-exchange chromatography was necessary to purify Dhh1 away from a contaminating E. coli protein with nucleic acid-stimulated ATPase activity.3 The contaminating ATPase was also stimulated by DNA, indicating that it is not RNA-specific.3 Fig. 1A, shows the purification of the protein through the SP column. ATPase assays were carried out at 30 °C with increasing amounts of poly(U) RNA. Dhh1 has very low ATPase activity in the absence of RNA, but a ∼10-fold stimulation in activity was observed in the presence of RNA (Fig. 1B).
Although Dhh1 displayed RNA-dependent ATPase activity, the level of activity was significantly less than that reported for other DEXD/H-box proteins, such as eIF4A and Ded1 (39, 53, 54). Furthermore, a side by-side comparison of the ATPase activities of Dhh1 and the hepatitis C virus NS3 RNA helicase suggests that Dhh1 has weaker ATPase than better-studied DEAD-box proteins (supplemental Fig. 1). The difference in activity was unexpected because Dhh1 has all of the signature motifs and conserved catalytic residues found in other well studied DEXD/H-box proteins. We speculated that there may be biological significance for the weaker enzymatic activity and explored the possible causes. The crystal structure of the core domain of Dhh1 revealed extensive interdomain interactions between the C-terminal and N-terminal lobes of the protein, which positions Dhh1 into a closed conformation (44). Such interactions are lacking in eIF4A and mjDEAD, DEAD-box proteins with more robust ATPase activity than Dhh1 (see Ref. 44 and supplemental Fig. 2). More specifically, Leu-342 and Thr-344 of motif V in the C-terminal domain of Dhh1 hydrogen bond with Lys-91 of motif I in the N-terminal domain (Fig. 2A). Furthermore, the side chain of Arg-345 in motif V stacks against that of Arg-89 in motif I and Leu-343 in motif V and make van der Waals interactions with Gly-93 and Thr-94 in motif I. Current models propose that binding of ATP and RNA by DEXD/H-box proteins results in a conformational change, bringing the C- and N-terminal domains closer, and rotation of these domains occurs during the ATP hydrolysis cycle (39, 55). Given the interactions between and N-terminal and C-terminal RecA-like domains observed in the crystal structure of the core domain Dhh1, it is possible that these interactions may hinder the movement of the two domains with respect to each other and limit ATPase activity. To test this hypothesis, we have mutated Thr-344 of motif V and Lys-91 of motif I to alanines to disrupt the hydrogen bonding between these residues. This is predicted to weaken the interdomain interactions and may increase the ATPase activity of the mutant. Furthermore, we analyzed a mutant in the first conserved aspartic acid in the “DEAD box” of Dhh1, D195A, as a control and to confirm that Dhh1 is a bona fide DEAD-box ATPase. As expected, the level of ATPase activity of the D195A mutant was negligible, and the little that was observed may originate from a trace amount of contaminating E. coli ATPase in our preparations (Fig. 2B). A double D195A/E196A mutant had a similar amount of activity as the single D195A mutant (not shown). Interestingly, mutation of both Lys-91 and Thr-344 to alanine residues resulted in a ∼2.5–3-fold increase in the ATPase activity over that of wild type Dhh1 (Fig. 2B). These results support the hypothesis that disrupting the contacts made between the N- and C-terminal RecA-like domains of Dhh1 would increase ATPase activity and suggest that the interdomain interactions may limit its ability to hydrolyze ATP.
Next, we evaluated the RNA sequence and nucleotide specificity requirements for the ATPase activity of Dhh1. The ATPase activities of wild type Dhh1 and the D195A and K91A/T344A mutants were examined in the presence of poly(U), poly(A), poly(C), and poly(G) ribonucleic acids. As shown in Fig. 2C, poly(U), poly(A), and poly(C) stimulated the ATPase activity of Dhh1 to roughly the same degree, but poly(G) was significantly less effective. However, the ineffectiveness of poly(G) is most likely due to its propensity to form G-quartets in solution rather than reflect selectivity against G residues (56).
Dhh1 Displays Specificity for Adenine-containing Nucleotides
Typical DEAD-box proteins display specificity for adenine-containing nucleotides and hydrolyze ribose and deoxyribose versions of adenine nucleotides equally well (39). To provide further evidence that Dhh1 is a typical DEAD-box ATPase, we examined the nucleotide specificity of the protein by measuring its ability to hydrolyze and bind to different nucleotides. The nucleotide specificity for hydrolysis was examined by conducting NTPase assays in the presence of poly(U) RNA and 50 μm cold nucleotide and a trace amount of corresponding α-32P-labeled versions. We analyzed the DEAD-box and interdomain interaction mutants (see above) in parallel as controls. As shown in Fig. 2D, wild type Dhh1 hydrolyzed ATP and dATP, but not GTP, CTP, or UTP. Similar to what was observed when ATP was used as a substrate, the K91A/T344A mutant displayed enhanced hydrolysis of dATP compared with wild type protein but no hydrolysis of the other nucleotides. Thus, even though disrupting the interdomain interactions within Dhh1 enhanced its ATPase activity, these mutations did not alter the nucleotide specificity of the protein.
The nucleotide binding specificity of Dhh1 was examined by UV-cross-linking of radiolabeled nucleotides to the protein. The amount of NTP cross-linked was quantified, and the signals for each sample were corrected for the amount of protein in the gel and normalized to the signal from wild type Dhh1. To distinguish signals attributed to nucleotide binding versus nonspecific cross-linking, we analyzed a mutant with substitutions in highly conserved residues within the Q-motif of Dhh1 (F66R/Q73A). The Q-motif, especially the conserved phenylalanine and glutamine residues, has been shown to participate in nucleotide binding in other DEAD-box proteins, and co-crystal structures of DEAD-box helicases with nucleotide analogs revealed that these residues make contact with the adenine base (57–60). We used the D195A mutant as a control, as this mutant should bind ATP but not hydrolyze it. It has been reported that the analogous DEAD-box mutation in eIF4A enhanced nucleotide cross-linking, presumably by preventing hydrolysis of the nucleotide (54, 61). If the cross-linking to Dhh1 is specific, we expect that cross-linking would be reduced and enhanced in the Q-motif and DEAD-box mutants, respectively. As shown in Fig. 3, A and B, radiolabeled ATP cross-linked to wild type Dhh1, and mutation of the Q-motif residues Phe-66 and Gln-73 resulted in a reduction in ATP cross-linking. We interpret the level of cross-linking remaining in this mutant to be background levels. In addition, as observed with eIF4A (54), the DEAD-box mutation (D195A) increased cross-linking of ATP to Dhh1 ∼2-fold.
FIGURE 3.

Analysis of UV-cross-linking of nucleotides to Dhh1. A, assays contained 4 μm protein and 10 μCi of α-32P-radiolabled nucleotide. Samples were cross-linked on ice (254-nm UV radiation) for 6 min and resolved in a 10% SDS-PAGE gel. The gel was stained with Coomassie Blue, dried, and exposed to a PhosphorImager screen. The 32P signal is shown on the left, and the Coomassie Blue-stained gel is shown on the right. B, quantification shown is of ATP cross-linking from three experiments. Data are expressed as relative ATP cross-linking compared with that of wild type Dhh1, which was set to 1.0. Cross-linking was corrected for the amount of total protein detected in the Coomassie Blue-stained gels. C, relative cross-linking of wild type and mutant proteins to NTPs is shown. In each case cross-linking to wild type Dhh1 is set to 1. D, nucleotide competition experiments are shown. Wild type Dhh1 was incubated with excess cold nucleotide indicated in the panel on the x axis. Radiolabeled ATP was added, and cross-linking was carried out as described in panel A.
Next, we analyzed the cross-linking of different nucleotides to Dhh1 and its mutant derivatives. Because the level of background cross-linking to the different nucleotides can vary depending on purity and the chemical nature of the nucleotide bases, we compared the cross-linking of the nucleotides to the wild type protein and the Q-domain mutant. Background cross-linking would be equal in both versions of Dhh1. Consistent with the data showing that Dhh1 can hydrolyze dATP, the cross-linking of this nucleotide was similar to that of ATP. In addition, cross-linking of dATP to the Q-motif mutant was reduced (Fig. 3C). On the other hand, although some incorporation of radiolabeled CTP, GTP, and UTP into Dhh1 was observed, the levels were equal for the wild type protein and the two mutant derivatives (Fig. 3C). This suggests that the cross-linking of CTP, UTP, and GTP arises from the nonspecific interaction of these nucleotides with the protein and represents background. The failure to detect specific cross-linking of radiolabeled CTP, GTP, and UTP to Dhh1 is consistent with the NTPase assays showing that Dhh1 cannot hydrolyze these nucleotides (Fig. 2D).
Because the cross-linking efficiency of nucleotides differs, we addressed nucleotide specificity by competition analysis. Cross-linking was carried out with radiolabeled ATP and 200-fold cold competitor nucleotides. Excess cold ATP, dATP, and ADP competed equally well in the cross-linking assay, reducing the level to 40–50% of that observed without competitor. The amount of cross-linking detected in the presence of excess adenine-containing nucleotides is approximately the same as that observed to the Q-motif mutant in the absence of competitor (Fig. 3D); thus, this likely represents background levels. GTP competed reasonably well but CTP and UTP less so. Even though some competition for binding was observed, Dhh1 was incapable of hydrolyzing these nucleotides (Fig. 2D). Overall, our analysis indicates that Dhh1 has a strong preference for adenine-containing nucleotides.
Dhh1 Binds RNA with High Affinity
Next, we sought to characterize the RNA binding properties of Dhh1. Dhh1 displays RNA-dependent ATPase activity, and a previous report showed that it binds to poly(U) RNA using a semiquantitative, filter binding assay (44). The published account used only a fixed amount of RNA in the filter binding assays and did not measure the affinity or length requirement for binding. We used the highly sensitive and quantitative fluorescence polarization assay to measure the RNA binding affinity and length dependence. Dhh1 was titrated into a buffer containing a 3′-fluorescein-labeled poly(U) oligomer, and mP was recorded. The dissociation constant (Kd) for the binding of Dhh1 to RNA was calculated by plotting mP as a function of Dhh1 concentration and fitted to a rectangular hyperbola.
Dhh1 bound a 20-base polyuridine RNA (rU20) with high affinity, displaying a Kd of ∼2 nm (Table 1). Thus, Dhh1 has a very high affinity for RNA. Cheng et al. (44) reported that the trypsin sensitivity of Dhh1 changed in the presence of ATP, suggesting that it undergoes a conformational change upon binding the nucleotide. The consequences of the conformational change are not known, but it could affect RNA binding affinity by altering the positioning of the two lobes that form the RNA binding cleft. Thus, we examined if nucleotides affected RNA binding by conducting binding assays in the presence and absence of ATP, ADP, and AMP-PNP. No changes in affinity of Dhh1 for RNA were observed in the presence of ATP, and only a slight change was detected when ADP or AMP-PNP was included (Table 1). Therefore, even though there is evidence that ATP binding changes the conformation of Dhh1, this does not affect RNA binding affinity significantly.
TABLE 1.
Dhh1 binds RNA in vitro with high affinity
Wild type Dhh1 or mutant proteins were titrated into a binding assay containing 0.1 nm 3′ fluorescein-labeled RNA or single-stranded DNA. Binding was measured by monitoring the change in mP. The data were fit to a hyperbola, and a dissociation constant was calculated from the curve. Dissociation constants for binding of Dhh1 to U20 (20-mer poly(U) containing a 3′ fluorescein). Binding was carried out under different conditions; that is, the presence/absence of Mg2+ and ATP. Values are expressed as the average and S.E. of three experiments.
| Conditions | Dissociation constant |
|---|---|
| nm | |
| Protein + MgCl2 | 1.8 ± 0.4 |
| Protein + MgCl2 + ATP | 2.2 ± 0.8 |
| Protein + MgCl2 + ADP | 3.9 ± 0.5 |
| Protein + MgCl2 + AMP-PNP | 3.6 ± 0.4 |
The crystal structure of the core domain of Dhh1 was solved without RNA, but co-crystal structures of other DEXD/H-box proteins with RNA have been solved. We, therefore, used the crystal structure of D. melanogaster Vasa, a homologous DEAD-box protein, to conduct structure-guided mutagenesis to provide evidence that Dhh1binds RNA similarly. Vasa binds ∼10 bases of RNA by bending it into a groove formed between the two globular domains of the protein (58). Importantly, the locations of the charged residues in Vasa that contact RNA are conserved in Dhh1 and are located in a cleft formed between the N- and C-terminal domains of the core of Dhh1 (44). To provide evidence that Dhh1 recognizes RNA through a similar mechanism, we analyzed the RNA length requirement and conducted mutagenesis of a subset of these conserved residues (see below). Fluorescence polarization experiments were conducted using poly(U) probes of differing lengths (rU5, rU7, rU8, rU10, rU12, and rU20). The data presented in Table 2 show that the affinity of Dhh1 for RNA is length-dependent and generally increased with increasing lengths of nucleic acid. Interestingly, there was a significant decrease in affinity when the length of RNA was reduced from 12 to 10 nucleotides (Table 2). The affinity dropped further when the RNA was shortened to eight nucleotides. Thus, the length requirement for RNA binding detected in the binding assays generally agrees with the minimal number of nucleotides contacting the protein in the co-crystal structures of analogous DEXD/H-box proteins and RNA (58, 62). However, Dhh1 must make contact with more than 10 nucleotides of RNA because increasing the number of nucleotides from 10 to 12 or 20 led to a further increase in affinity. Finally, we examined the binding of Dhh1 to a single-stranded deoxyribose nucleic acid probe. Dhh1 bound to a poly(dT20) probe with high affinity (∼77 nm), but this is ∼30-fold lower than the rU20 probe (Table 2). However, it is unclear if Dhh1 binds single-stranded DNA in vivo.
TABLE 2.
Length dependence of RNA binding
Binding assays were conducted as described in Table 1 and under “Experimental Procedures.” Dissociation constants for the binding of Dhh1 to sequences containing different lengths of poly ribose uridine (rU) and polydeoxyribose thymidine (dT) nucleic acids are reported.
| RNA length | Dissociation constant |
|---|---|
| nm | |
| rU5 | 968 ± 317 |
| rU7 | 577 ± 112 |
| rU8 | 405 ± 54 |
| rU10 | 81 ± 8 |
| rU12 | 5.4 ± 0.5 |
| rU20 | 2.7 ± 0.6 |
| dT20 | 77 ± 9 |
It was reported that mutation of basic residues lining the potential RNA binding cleft in Dhh1 reduced RNA binding (44). Specifically, mutation of Arg-89 or Lys-91 in motif I, Arg-345 or Gly-346 in motif V, and Arg-370 in motif VI reduced RNA binding when assayed by a filter binding assay. We verified and extended these studies by conducting mutagenesis of residues in Dhh1 implicated in RNA binding. Fluorescence polarization binding assays largely confirmed that mutation of the residues comprising the proposed RNA binding cleft reduced RNA binding. However, a quantitative measure of Kd values revealed that the effects of these mutations were not as severe as what was implied from the filter binding assay (44). Arginine 322 and serine 340 were also targeted for mutagenesis. These amino acids correspond to Arg-528 and Thr-546 of Vasa, which were shown to make direct contact with RNA in its co-crystal structure with RNA (58). Mutation of these residues individually did not affect RNA binding significantly (Table 3). However, when both residues were mutated to alanines, RNA binding affinity was reduced 4-fold compared with wild type protein (Kd ∼ 82 nm for wild type protein versus ∼320 nm for the R322A/S340A mutant). Collectively, the length dependence for RNA binding and mutagenesis studies suggest that the RNA binding pocket of Dhh1 lies between the N- and C-terminal domains and that it binds RNA similar to the related DEAD-box protein Vasa.
TABLE 3.
Comparison of biochemical activities of Dhh1 and its mutant derivatives
The mutants of Dhh1 are listed in the table. The mutants are shown in the table and are grouped according the contributions they make to the function of the protein, predicted by its comparison to analogous DEAD-box RNA helcases. ATPase activities were carried out as described before. Moles of ATP hydrolyzed by the proteins are shown. ATP binding was determined by UV-cross-linking assays, and the cross-linking of ATP to wild type Dhh1 was set to 1.0. Cross-linking of ATP by mutant proteins is expressed relative to that of wild type Dhh1. RNA binding was assayed using 0.1 nm 3′ fluorescein-labeled single-stranded 10-mer poly(U). Dissociation constants (nm) calculated from binding curves are tabulated. Errors represent the average and S.D. of at least three independent experiments. ND, not determined.
| Activity | Dhh1 | ATP hydrolyzed | Relative cross-linking to ATP | Dissociation constant for rU10 binding |
|---|---|---|---|---|
| pmol | nm | |||
| WT Dhh1 | 90 ± 10 | 1 | 82 ± 9 | |
| ATP binding | Q73A | 75 ± 11 | 0.66 ± 0.2 | 85 ± 4 |
| Q73A/F66R | 42 ± 14 | 0.51 ± 0.1 | 91 ± 12 | |
| ATP hydrolysis | K96A | 31 ± 5 | 0.85 ± 0.03 | ND |
| D195A | 35 ± 4 | 2.15 ± 0.3 | 120 ± 20 | |
| RNA binding | R322A | 76 ± 20 | 0.92 ± 0.2 | 128 ± 10 |
| S340A | 76 ± 23 | 0.84 ± 0.3 | 69 ± 3 | |
| R322A/S340A | 26 ± 10 | 1.13 ± 0.2 | 316 ± 20 | |
| R370A | 130 ± 45 | 0.91 ± 0.1 | 402 ± 45 | |
| Interdomain interactions | K91A | 124 ± 27 | 1.19 ± 0.1 | 219 ± 13 |
| T344A | 109 ± 21 | 1.08 ± 0.1 | 48 ± 5 | |
| K91A/T344A | 270 ± 75 | 1.07 ± 0.2 | 136 ± 21 | |
| G346A | 33 ± 8 | 1.02 ± 0.1 | 68 ± 7 |
We examined the ATPase activity of the RNA binding mutants. The assays were carried out with saturating amounts of RNA (20 μg) to diminish the contributions of reduced RNA binding on activity. All but one RNA binding mutant displayed ATPase activities similar to wild type Dhh1 (Table 3). The small changes we observed in most mutants were within experimental error. However, the double R322A/S340A mutant showed no specific ATPase activity, hydrolyzing ATP to the same level as a DEAD-box (D195A) and ATP binding mutant (Q73A/F66R). The reduction in ATPase activity cannot be explained by reduced ATP binding; the mutant cross-linked to ATP as well as wild type Dhh1 or by the reduced affinity for RNA. The latter point is supported by comparison of the two mutants with the greatest reduction in RNA binding. The R322A/S340A mutant displayed a slightly higher affinity for RNA than the R370A mutant, yet the R370A mutant showed robust ATPase activity when assayed in the presence of saturating amounts of RNA. This suggests that Arg-322 and Ser-340 have additional roles in ATP hydrolysis. Evidence for this possibility comes from studies of Drosophila DEAD-box protein Vasa. The orthologous residues in Vasa are important for RNA binding, ATPase, and helicase activities. The crystal structure of Vasa with RNA indicates that these residues are important for bending the RNA in the binding cleft. The bending of the RNA may be important for the ability of Vasa and Dhh1 to hydrolyze ATP; a similar model has been proposed by others when describing the mechanism of other DEXD/H helicases (38). Interestingly, the double mutant K91A/T344A mutant bound RNA somewhat better than the single K91A mutant. Although Lys-91 contributes to RNA binding via an interaction with the negatively charged RNA, the increased flexibility caused by disruption of the interdomain interactions in the K91A/T344A mutant may allow the RNA to make contacts with other residues in the presumed RNA binding cleft, leading to higher affinity for RNA.
Dhh1 Does Not Display Helicase Activity in Vitro
Our studies thus far show that Dhh1, like many DEXD/H-box proteins, binds to RNA and hydrolyzes ATP in an RNA-dependent manner. We next sought to examine if Dhh1 has helicase activity. It cannot be taken for granted that Dhh1 displays helicase activity because many DEXD/H-box proteins do not display helicase activity. We used an assay to detect the unwinding of double-stranded RNA containing either a 5′ or 3′ overhang. Our previous experiments have shown that Dhh1 can bind a 12-mer RNA with high affinity, so we designed duplex (8 base) substrates to incorporate a 12-nucleotide single-stranded RNA overhang. We titrated various concentrations of wild type Dhh1 (0.3, 0.6, and 1.0 μm) into the assay and carried out experiments with 1 μm K91A/T344A and D195A/E196A mutants. The K91A/T344A mutant was tested to determine whether enhancing ATPase activity could activate helicase activity. We failed to observe unwinding of short double-stranded RNAs with templates containing either a 5′ or 3′ overhang (supplemental Fig. 1). As a control we used the hepatitis C virus NS3 protein that unwinds double-stranded RNA with a 3′ overhang. Under our reaction conditions 0.5 μm NS3 specifically unwound double-stranded RNA with a 3′ overhang but not a substrate with a 5′ overhang, the same specificity that was described earlier (45) (supplemental Fig. 1). Because Dhh1 bound more strongly to a 20-mer RNA, we repeated the assay using substrate with a 20 nucleotide ssRNA overhang either at the 5′ or 3′ ends but failed to observe any Dhh1-dependent unwinding of substrates (not shown). Once again, hepatitis C virus NS3 unwound the substrate with a 20-mer 3′ overhang (not shown). Thus, Dhh1 did not display helicase activity under the conditions used here.
Phenotypic Screening of Dhh1 Mutants
Biochemical analysis of Dhh1 has identified residues that are required for ATPase activity, RNA binding, and ATP binding. Earlier studies have shown that a DHH1 null mutant is viable but displays sensitivities to stress conditions including heat and DNA-damaging agents such as methyl methane sulfonate, hydroxyurea, and UV irradiation (27, 28, 30). Mutations to the DEAD-box or residues implicated in RNA binding could not complement the temperature sensitivity of a dhh1Δ strain (44). We applied the same analysis to the mutants we characterized biochemically and examined their ability to complement the DNA damage sensitivity of the Δdhh1 strain. It is a formal possibility that the DNA damage resistance functions of Dhh1 may require different functional domains than those required for heat stress. Wild type Dhh1 or mutant proteins were expressed under the control of its own promoter from low copy plasmids in a dhh1Δ null strain. A schematic of the mutants in relation to the nine conserved functional motifs of Dhh1 is shown in Fig. 4A. Furthermore, we verified by Western blotting that each of the Dhh1 mutants accumulated to levels equal to that of wild type Dhh1 (Fig. 4B). As a whole, the data show that mutating any of the conserved domains of Dhh1 resulted in DNA damage and heat stress sensitivity. Those showing the strongest phenotypes typically displayed severely impaired ATPase activity. The R322A mutation, which reduced RNA binding and ATPase weakly, still showed sensitivity to stress conditions. However, reducing RNA binding by as much as 5-fold did not affect the stress resistance of the R370A mutant. This suggests that either the amount of RNA within the cell is saturating or, more likely, other factors in the cell compensate for its reduced affinity for RNA. This mutant did display a weak RNA turnover defect (see below), however, suggesting that mutating this residue does have a subtle affect on Dhh1 function. The K91A/T344A and the K91A mutants, which displayed increased ATPase activity, were not sensitive to stress. Interestingly, the K91A/T344A mutant showed slightly better growth at 37 °C than cells expressing wild type Dhh1 (Fig. 4C). The growth advantage of this mutant at 37 °C is modest but reproducible.
The biochemical assays used Dhh1 protein retaining the His6-tag and additional amino acids incorporated into the N terminus. In contrast, the genetic analysis was carried out in cells expressing the natural from of Dhh1. It is a formal possibility the His6 tag changed the activity of the protein in vitro. However, proteins generally tolerate tags in the N terminus, and the biochemical activities and the expected phenotypes in vivo of the Dhh1 mutants correlated very well (also see below).
Mutation of the Functional Domains of Dhh1 Affect RNA Turnover
We isolated mutants with defects in the biochemical activities of Dhh1, and next we examined the requirement for these functions in mRNA decay, recruitment into cytoplasmic foci, and binding to mRNA in vivo. Representative DHH1 mutants that displayed specific biochemical defects were chosen for further analyses. The effect of these mutations on RNA turnover was determined by measuring the levels of GAL1 mRNA over time after repressing transcription with dextrose. GAL1 displayed an mRNA half-life of ∼11 min in wild type cells, and deleting DHH1 extended the half-life to 22 min (Fig. 5A); these values agree with previous reports (11). We observed that mutations in Dhh1 affecting ATP hydrolysis (D195A/E196A), RNA binding (R322A/S340A), and ATP binding (F66R/Q73A) resulted in a doubling of half-life of the GAL1 mRNA, similar to that observed in the dhh1Δ mutant (Fig. 5A). On the other hand, the half-life of GAL1 mRNA in the K91A/T344A mutant was reproducibly shorter than that measured in cells expressing wild type Dhh1, but the difference from the wild type value is within experimental error (not shown). This suggests that disrupting the interdomain interactions within Dhh1 and enhancing its ability to hydrolyze ATP increased its function in vivo. To further substantiate our results showing that mutations in Dhh1 reduced mRNA turnover in vivo, we studied the steady state levels of EDC1 mRNA, which is sensitive to the loss of Dhh1 (36, 44). Because monitoring steady state mRNA levels is more straightforward, more mutants were examined. Deletion of DHH1 caused a 2–2.5-fold increase in EDC1 mRNA (Fig. 5B and Ref 44). Here we observed that mutants that affect ATPase activity (D195A, D195A/E196A, G346A, and K96A), ATP binding (F66R/Q73A), or RNA binding (R322A/S340A and R370A) led to an increased accumulation of EDC1 mRNA, indicating impaired decay functions in these mutants (Fig. 5B). Interestingly, the steady state levels of EDC1 mRNA were slightly reduced in mutants containing substitutions in residues involved in forming the interdomain interactions (K91A and K91A/T344A) between the N- and C-terminal lobes. The reduction in mRNA may be the result of enhanced turnover of EDC1 mRNA. These results indicate that disrupting any of the biochemical activities of Dhh1 leads to defects in mRNA turnover and that the interdomain interactions within Dhh1 may limit its ability to contribute to the turnover mRNAs in vivo (see below also).
The Localization of Dhh1 into Cytoplasmic Foci Is Regulated by ATP and RNA Binding and Intermolecular Interactions
The role of Dhh1 in mRNA turnover and translational repression has been attributed to its localization to cytoplasmic foci called P-bodies, where it interacts with components of the mRNA decapping machinery (Dcp1/Dcp2), Pat1, and Xrn1 (11). Dhh1 is localized throughout the cytoplasm in a punctate pattern and can be redistributed into larger discrete foci upon cell stress (25). It is not known which activities of Dhh1 are required for its localization to P-bodies. To address this question, we analyzed Dhh1 and its mutants' ability to form P-bodies in resting and stressed cells. We constructed strains containing C-terminal GFP-tagged Dhh1 at its genomic locus and transformed the wild type and mutant strains with a plasmid carrying RFP-tagged Dcp2, a reliable and easily visualized component of yeast P-bodies (52). Stress was induced by starving cells of dextrose, a condition that strongly and rapidly induces P-body formation (25, 52). The stressed or unstressed cells were immediately imaged using a confocal fluorescence microscope to visualize Dhh1- and Dcp2-containing foci (25). In addition to monitoring Dhh1 localization in cytoplasmic foci, we conducted RIP assays to monitor the association of Dhh1 with mRNAs in vivo. The procedure uses formaldehyde to rapidly cross-link cells in culture to preserve RNA-protein interactions and provides an effective method to measure the association of Dhh1 with mRNAs. In conjunction with monitoring Dhh1 localization, RIP can correlate mRNA binding with P-body formation in cells. Furthermore, as we will demonstrate by the correlation between Dhh1 cross-linking and the extent of foci formation in wild type and mutant cells, this method provides a more quantitative surrogate to measure the recruitment of Dhh1 into RNA-containing foci.
In wild type cells very few cells displayed Dhh1-containing foci under the resting condition (+dextrose), but the foci were increased in number and size when cells were deprived of dextrose and the fraction of cells with foci increased (Fig. 6). The Dhh1 colocalized almost completely with the Dcp2-RFP, consistent with previous results showing that Dhh1 localizes to P-bodies in stressed cells (25) (Fig. 6). Correlating with the increase in Dhh1-containing foci in response to stress, there was an increase in the cross-linking of Dhh1 to a model mRNA, PYK1, in stressed cells (Fig. 7).
FIGURE 6.

The localization of Dhh1 mutants to cytoplasmic foci requires residues involved in ATP and RNA binding. Dhh1-GFP and Dcp2-RFP were analyzed by confocal fluorescent microscopy. Log phase wild type and Dhh1 mutants expressing endogenous GFP- tagged Dhh1 protein and Dcp2 (pRP1186) were dextrose-deprived for 15 min, and live cell images were obtained. Dhh1-GFP, Dcp2-RFP, and merged images are shown in −dextrose conditions. Dhh1-GFP images are shown in +dextrose condition. Exposures displayed in the panels were chosen so that the outline of the cells can be seen.
FIGURE 7.

RNA binding in vivo is affected by mutations in Dhh1. RIP analysis to study in vivo mRNA binding by Dhh1 is shown. Dhh1-cross-linked RNA was extracted and converted to cDNA. Binding of Dhh1 to PYK1 mRNA was analyzed by PCR using primers to the ORF of PYK1. Percent IP was calculated by correcting the IP RNA value for the input RNA value. The experiments shown in the left and right panels were conducted at different times; thus, the mutants in each panel should be compared with the wild type data conducted in parallel.
An analysis of the localization of Dhh1 mutants yielded interesting results. First, in the ATPase-defective mutant (D195A/E196A), an increase in the size and number of Dhh1-containing foci was observed in resting cells (Fig. 6). Under stress, an increase in foci intensity was observed. The differences in foci between this mutant and wild type cells were not obvious under stress conditions. However, the size and number of P-bodies may have been close to saturation under the stressed condition; therefore, the differences between the wild type and mutant are not easily observed. Dcp2-RFP fluorescence overlapped that of Dhh1, indicating that the ATPase activity of Dhh1 is not required for Dcp2 to shuttle into P-bodies. Examination of the association of the DEAD-box mutant with mRNA using RIP revealed an increase over wild type in resting cells, but even a more dramatic increase was observed in stressed cells. The cross-linking of the DEAD-box mutant was 2-fold higher than that observed in wild type cells. The increase in size and number of foci and the elevated association of the mutant protein with mRNA suggest that the ATPase activity and mRNA decay functions of Dhh1 are not required for it to shuttle mRNAs into P-bodies. However, the failure to hydrolyze ATP may prevent the decay or release of the mRNAs and an accumulation of Dhh1 and mRNAs into P-bodies. This hypothesis is supported by the observation that impairing mRNA decay by deleting decapping enzyme Dcp1 or the exonuclease Xrn1 likewise led to an accumulation of Dhh1 into P-bodies in unstressed cells (25, 64).
The GFP signal of the Q-motif mutant is similar and may be slightly weaker than the wild type cells in the unstressed condition. However, a decrease in the number and size of Dhh1-containing foci was observed when the cells were stressed by dextrose deprivation compared with wild type cells (Fig. 6). Calculation of the average size of mutant Dhh1-containing foci revealed that they are on average half the size of those observed in wild type cells (0.12 versus 0.25 μm for the wild type). The RIP experiments suggest that the association of the F66R/Q73A mutant was similar to that of wild type Dhh1 in resting cells but that the stress-induced increase in its association with mRNAs was nearly abolished in this mutant (Fig. 7). These results are a little surprising, as the Q-motif mutant displays no ATPase activity and impaired mRNA decay functions similar to the DEAD-box mutant, and yet its localization and RNA association patterns are different from the DEAD-box mutant. Thus, the localization and RIP studies suggest that ATP binding is required for Dhh1 to bind mRNA in vivo and transport into P-bodies. This suggests that ATP binding per se, and not hydrolysis specifically, plays a role in regulating Dhh1 (see “Discussion”).
Similar to the ATP binding mutant, the RNA binding mutant (R322A/S340A) also showed weaker background staining in unstressed cells and formed smaller foci when cells were stressed (Fig. 6). The foci formed by this mutant were also about half the size of those formed by wild type Dhh1 (0.14 versus 0.24 um). RNA-IP experiments revealed that this mutant displayed a 2-fold reduction in its cross-linking to PYK1 mRNA both in the stressed and unstressed conditions (Fig. 7). These two pieces of data argue that the interaction of Dhh1 with RNA is required for its recruitment into P-bodies. Furthermore, because this mutant is defective for mRNA decay (Fig. 5), simply altering mRNA turnover and accumulating RNAs in the cell cannot cause the recruitment of Dhh1 into cytoplasmic foci. Furthermore, the pattern of Dcp2 fluorescence in this mutant was similar to that of the wild type cells, indicating that reducing Dhh1 RNA binding does not significantly affect the recruitment of Dcp2 into P-bodies.
Mutating residues involved in the interdomain interactions enhanced ATPase activity, imparted a slight growth advantage to cells undergoing heat stress, and increased mRNA turnover in vivo. These phenotypes suggest that interdomain interactions limit Dhh1 activity in vivo. Interestingly, we observed an increase in foci formation in a greater fraction of unstressed cells expressing the K91A/T344A mutant (Fig. 6). A slight increase in the number and intensity of P-bodies under stressed conditions was observed, but the increase was not robust. Similar to the DEAD-box mutant, foci formation may be saturated in cells under these conditions, and enhancement is not detected. The most striking result was observed in the RIP experiments. Disrupting interdomain interactions led an ∼3-fold increase in the cross-linking of the mutant to PYK1 mRNA in unstressed cells, and the level increased further upon stress (Fig. 7). The increased association of the mutant with mRNA is not caused by differences in the expression of the mutant or an increase in its affinity for RNA. This mutant accumulates in the cells to the same level as wild type Dhh1 (Fig. 4B), and it has a slightly lower affinity for RNA (Table 3). These results argue that interactions between the N- and C-terminal RecA-like domains in the core of Dhh1 may play an important regulatory function in vivo, perhaps preventing Dhh1 from binding mRNAs and shuttling into P-bodies, thereby restricting mRNA decay in unstressed cells.
DISCUSSION
Dhh1 Is a Bona Fide DEAD-box Protein with ATPase Activity
The classification of Dhh1 as a DEAD-box RNA helicase was based on the presence of well annotated motifs characteristic of this family of enzymes (37). It was surprising that a characterization of the ATPase activity of Dhh1 or any of its orthologues has not been described. This is especially true given that the protein has been expressed in quantities sufficient for structural studies, and a preliminary analysis of its RNA binding abilities was conducted (44). Comparison of the crystal structure of Dhh1 to those of other RNA helicase revealed a distinct difference in the organization and interactions between the two N- and C-terminal lobes of the core domain. Significant interactions between residues in the N- and C-terminal domains were observed in the crystal structure Dhh1, which were not seen in other DEAD-box proteins whose structures have been solved (44). These interactions have the potential to “lock” Dhh1 into an inactive conformation. The prevailing hypothesis of how DEAD-box proteins hydrolyze ATP is that the binding of ATP and RNA to the protein results in a conformational change, bringing the N- and C-terminal domains together. ATP hydrolysis is coupled to the concomitant movement of these domains with respect to each other (55). The previous failures to demonstrate ATPase activity and the interdomain interactions detected in the crystal structure of Dhh1 raised the possibility that it is not a typical DEAD-box protein or that the gene evolved over time to lose its ATPase and helicase activities. Thus, it was important to show that Dhh1 is a bona fide DEAD-box protein with ATPase activity.
A comparison of the ATPase activity of Dhh1 to that of other well characterized RNA helicases (53, 54, 65, 66) indicates that it is a weak ATPase. We surmised that the interdomain interactions could restrict the movement of the N- and C-terminal domains of Dhh1 and, hence, reduce its ATPase activity. Analysis of a double K91A/T344A mutant and to a lesser extent the single mutants showed that disrupting interdomain interactions increased ATPase activity. In addition to providing an explanation for the weak ATPase activity of the wild type protein, this result also corroborates the existing models that predict that the enzymatic activity of DEAD-box proteins requires the coordinated movement of the two RecA-like helicase domains. The ATPase activity of the interdomain interaction mutant was still less than that observed for more robust helicases. This is not surprising because there are multiple residues that form interdomain interactions between the two lobes of Dhh1 (44). Mutagenesis of more residues to further disrupt the interaction between the N and C lobes to increase activity to a higher level is not practical as some of the residues may play other roles in the function of the protein, such as RNA binding. Any gains in ATPase activity caused by disrupted interdomain interactions might be offset by reducing other activities required for ATPase activity.
Dhh1 Does Not Show Helicase Activity in Vitro
The classical definition of an RNA helicase is a protein that binds ATP and RNA, hydrolyzes ATP, and unwinds duplexed RNA. Our studies thus far have confirmed all of these activities for Dhh1, except for the helicase activity. In our hands Dhh1 could not unwind double-stranded RNA under multiple assay conditions and using different substrates (supplemental Fig. 1 and data not shown). Our failure to detect helicase activity is not unusual. Many DEXD/H-box proteins have been classified as RNA helicases based on sequence and structural similarities to DNA helicases and their ability to hydrolyze ATP and bind RNA (39). However, only a subset of these proteins has been shown to unwind dsRNA in vitro (42, 43, 67, 68). It is clear that the ATPase activity is required for Dhh1 to carry out its functions in vivo, because mutants with substitutions in the DEAD-box or other residues required for ATP hydrolysis cannot restore the function of Dhh1 in stress resistance or mRNA decay. Like many other examples where helicase activity of a DEAD-box protein was not observed, it is unclear if this is due to technical limitations of the assay or that the protein uses the conformational change associated with ATP hydrolysis to carry out other functions. For example, a growing number of helicases have been shown to disrupt or remodel RNA-protein interactions (69). Our mutational analysis and the work of others indicates that Dhh1 regulates the shuttling of mRNAs between the translatable pool and the non-translatable pool contained in cytoplasmic foci (23, 26). Logically, this would require the assembly and disassembly of mRNAs into messenger ribonucleoprotein complexes. Because the transport of Dhh1 in and out of cytoplasmic foci is regulated by its RNA binding and ATPase activities, respectively, ATP hydrolysis may play a role in assembling and disassembling mRNP particles rather than unwind duplex RNA. Dhh1 orthologues may play similar functions. The Xenopus orthologue Xp54 has been shown to shuttle between the nucleus and cytoplasm in a developmentally regulated manner whereby it interacts with nascent transcripts in the nuclei of transcriptionally active oocytes and localizes to the cytoplasm in transcriptionally quiescent oocytes (70). Also, studies in Drosophila, trypanosomes and clam have shown that Dhh1 orthologues repress translation of maternal mRNAs during early development (71–73). An attractive model for the control of mRNAs by Dhh1 is that it does not act as a canonical helicase to unwind RNA but as a protein that binds the RNAs with high affinity and controls the transport of these RNAs into cytoplasmic foci, thereby sequestering them from the translation machinery.
Activities Required for mRNA Decay and P-body Formation
One of the aims of this study was to analyze how disabling the known biochemical activities of Dhh1 affect its functions in vivo. A synopsis of the phenotypes of the mutants is presented in Table 4. Previous studies have partially addressed this issue (44). We have expanded this analysis to include a greater variety of mutants, including those in the ATP binding Q-motif and residues involved in interdomain interactions. For the most part we have confirmed that the biochemical activities and conserved motifs in Dhh1 are required for stress resistance and the regulation of mRNA decay in vivo. Some Dhh1 mutants with amino acids substitutions in conserved domains failed to display strong phenotypes in the growth assays. This is not surprising, as mutation of these residues may not impair the activity of the protein to a sufficient level to result in an observable phenotype. Herein lies the importance of monitoring the biochemical activities of the mutants and confirming that they are expressed to normal levels. There were cases where mutating conserved residues led to a smaller decrease in biochemical function/activity than others, and these partially active mutants displayed weaker growth phenotypes in the growth assays (for example the R370A mutant).
TABLE 4.
Qualitative summary of the biochemical activities and phenotypes of Dhh1 mutants
The biochemical activities were derived from Table 3, RNA turnover was from Fig. 5, mRNA binding was from Fig. 7, and foci formation was from Fig. 6. Additional plus signs (+) indicate a gain of function, the minus sign (−) indicates a loss of function, and +/− designation indicates a reduction in function.
| Mutant class | Dhh1 status | ATPase | ATP cross-linking | RNA Affinity (in vitro) | RNA turnover | mRNA binding (RIP) | Foci formation |
|---|---|---|---|---|---|---|---|
| Wild type | + | + | + | + | + | + | |
| Q-motif ATP binding | Q73A/F66R | - | - | + | - | +/− | +/− |
| DEAD-box ATP hydrolysis | D195A/E196A | - | ++ | + | - | ++ | ++ |
| RNA binding/ATP hydrolysis | R322A/S340A | - | + | +/− | - | +/− | +/− |
| Interdomain interactions | K91A/T344A | +++ | + | + | ++ | +++ | ++ |
Whereas the importance of residues required for RNA binding and ATPase activity (DEAD box) in mRNA decay and stress resistance functions was known (44), our characterization of the mutants has shed some light on the role of the biochemical activities in regulating mRNA decay, specifically the ability to bind RNAs in vivo and localize into cytoplasmic foci. Dhh1 and its metazoan orthologues are a major component of cytoplasmic processing bodies (23, 70). The function of these P-bodies is not clear, but they have been connected to both translational repression and mRNA decay (23). The size and abundance of P-bodies is regulated by stress but also requires RNA and translation (25). Blocking translation inhibits P-body formation, whereas impairing RNA decay causes increased P-body formation (26). This led to the hypothesis that mRNAs are directed to P-bodies for decay. However, data are mounting suggesting that decay can occur outside of P-bodies (74). Dhh1 is proposed to be a major regulator of RNA trafficking in and out of P-bodies (23, 26). It was not known which of the Dhh1 activities was required for it to be recruited to cytoplasmic foci. Here we show that the ATPase activity is not required for it to be recruited into cytoplasmic foci but may be required for its release from the foci. Inactivating Dhh1 ability to hydrolyze ATP actually leads to increased mRNA binding in vivo, an increase in the number of cells showing Dhh1-containing cytoplasmic foci in unstressed cells and a slight increase in the intensity of the foci that are produced when under stress (Figs. 6 and 7). The ability of Dhh1 to hydrolyze ATP is important for Dhh1 to release mRNAs and possibly cycle out of P-bodies. On the other hand, mutants in Dhh1 that weaken its binding to RNA in vitro correspondingly lead to reduced mRNA association and its ability to concentrate into cytoplasmic foci. A possible explanation for these observations is that Dhh1 binds mRNAs and transports them to cytoplasmic foci for degradation or translational repression, and activation of its ATPase activity by cellular factors localized in the foci results in its release from the RNAs and the P-body (Fig. 8).
FIGURE 8.

Model for the regulation of Dhh1 in vivo. Intermolecular interactions between the N- and C-terminal lobes (indicated by red line) limit ATP hydrolysis until Dhh1 is activated by cellular factors in response to cell stress. Breaking the intermolecular interactions allow rotation of the two domains, ATP hydrolysis, and release of the RNA (black line). Dhh1 is then released back into the free pool.
An interesting observation was made from the analysis of the Q-motif mutant (F66R/Q73A) that has impaired ATP binding and ATPase activity, especially when compared with that of the DEAD-box mutant. The in vivo mRNA binding and localization phenotypes are nearly opposite those of the DEAD-box mutant. The difference between these two mutants is that the DEAD-box mutant displays enhanced ATP binding (cross-linking), whereas the Q-motif mutant shows reduced binding. This raises the interesting possibility that the binding of ATP to Dhh1 regulates its function in vivo without the need to hydrolyze ATP. Interestingly, Cheng et al. (44) reported that the binding of ATP to Dhh1 changes its sensitivity to digestion by trypsin, suggesting the binding of ATP induces a conformational change in the protein. An ATP-induced conformational change in Dhh1 may regulate its localization in the cell directly or by altering its ability to associate with other cellular factors.
Interdomain Interactions Restrict Dhh1 Activity
We present evidence that interdomain interactions within Dhh1 at least in part attenuate its ATPase activity and restrict its function in vivo. Given that all the essential DEAD-box protein motifs are well conserved in Dhh1, this suggests that this may be an evolutionary adaptation with functional consequences. Dhh1 is an abundant protein, expressed at ∼40,000 copies per cell (75). Restricting its ATPase activity through intermolecular interactions may control its function in vivo. Interestingly, it has been shown that expression of Dhh1 from the highly active GAL1 promoter causes growth arrest (26). This observation suggests that unregulated or hyperactive Dhh1 activity is detrimental to cells.
The adaptations to Dhh1 that restricts its ATPase hydrolysis but allows it to bind both RNA and ATP could be a means to regulate this protein. For example, as mentioned above, Dhh1 may be involved in shuttling RNAs in and out of cellular compartments. These activities may require both RNA and ATP binding but not ATP hydrolysis. Inhibiting ATP hydrolysis may allow time for the Dhh1-RNA complex to be transported to its destination where it could be subsequently acted upon by cellular factors or post-translational modifications (Fig. 8). Interestingly, it was recently proposed that the ATPase activity of Xenopus p54 many regulate the balance between its association with decay versus translation factors (76). There are a growing number of examples where the activity of DEAD-box proteins is regulated by co-factors (38). The best characterized example of this is the regulation of eIF4A by its binding partner eIF4G. The ATPase activity of eIF4A is stimulated by eIF4G, and structural studies revealed that eIF4G stabilizes the helicase-competent conformation of eIF4A (63, 77). Although we do not have direct evidence that Dhh1 is regulated by another factor, it is known to associate with factors involved in mRNA decay and translation. Restricting its activity until it enters cytoplasmic foci (decay) or polysomes (translation) may allow tighter control of the protein, dictate its ability to regulate mRNA decay versus translation, and protect the cell from unregulated activity of this abundant DEAD-box protein.
Supplementary Material
Acknowledgments
We thank Jeff Coller and Roy Parker for the plasmids expressing P-body markers. We are grateful to Dr. Qixin Wang for supplying recombinant NS3 protein and advice on conducting the helicase assays. The members of the Center for Eukaryotic Gene Regulation and Center for RNA Biology are recognized for their comments during the completion of this work.
This work was supported, in whole or in part, by National Institutes of Health Grant GM58672 (to J. C. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3 and Table 1.
A. Dutta and J. C. Reese, unpublished information.
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- mP
- fluorescence polarization
- SD
- synthetic drop out
- RIP
- RNA immunoprecipitation
- IP
- immunoprecipitate
- NTA
- nickel-nitrilotriacetic acid
- RFP
- red fluorescent protein.
REFERENCES
- 1. Garneau N. L., Wilusz J., Wilusz C. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 113–126 [DOI] [PubMed] [Google Scholar]
- 2. Chang Y. F., Imam J. S., Wilkinson M. F. (2007) Annu. Rev. Biochem. 76, 51–74 [DOI] [PubMed] [Google Scholar]
- 3. Brown C. E., Tarun S. Z., Jr., Boeck R., Sachs A. B. (1996) Mol. Cell. Biol. 16, 5744–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dupressoir A., Morel A. P., Barbot W., Loireau M. P., Corbo L., Heidmann T. (2001) BMC Genomics 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tucker M., Staples R. R., Valencia-Sanchez M. A., Muhlrad D., Parker R. (2002) EMBO J. 21, 1427–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tucker M., Valencia-Sanchez M. A., Staples R. R., Chen J., Denis C. L., Parker R. (2001) Cell 104, 377–386 [DOI] [PubMed] [Google Scholar]
- 7. Yamashita A., Chang T. C., Yamashita Y., Zhu W., Zhong Z., Chen C. Y., Shyu A. B. (2005) Nat. Struct. Mol. Biol. 12, 1054–1063 [DOI] [PubMed] [Google Scholar]
- 8. Steiger M., Carr-Schmid A., Schwartz D. C., Kiledjian M., Parker R. (2003) RNA 9, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z., Jiao X., Carr-Schmid A., Kiledjian M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 12663–12668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonnerot C., Boeck R., Lapeyre B. (2000) Mol. Cell. Biol. 20, 5939–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coller J. M., Tucker M., Sheth U., Valencia-Sanchez M. A., Parker R. (2001) RNA 7, 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dunckley T., Tucker M., Parker R. (2001) Genetics 157, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mangus D. A., Amrani N., Jacobson A. (1998) Mol. Cell. Biol. 18, 7383–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tharun S., He W., Mayes A. E., Lennertz P., Beggs J. D., Parker R. (2000) Nature 404, 515–518 [DOI] [PubMed] [Google Scholar]
- 15. Tharun S., Parker R. (2001) Mol. Cell 8, 1075–1083 [DOI] [PubMed] [Google Scholar]
- 16. Houseley J., LaCava J., Tollervey D. (2006) Nat. Rev. Mol. Cell Biol. 7, 529–539 [DOI] [PubMed] [Google Scholar]
- 17. Long R. M., McNally M. T. (2003) Mol. Cell 11, 1126–1128 [DOI] [PubMed] [Google Scholar]
- 18. Eulalio A., Behm-Ansmant I., Izaurralde E. (2007) Nat. Rev. Mol. Cell Biol. 8, 9–22 [DOI] [PubMed] [Google Scholar]
- 19. Sheth U., Parker R. (2003) Science 300, 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheth U., Parker R. (2006) Cell 125, 1095–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balagopal V., Parker R. (2009) Curr. Opin. Cell Biol. 21, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brengues M., Teixeira D., Parker R. (2005) Science 310, 486–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parker R., Sheth U. (2007) Mol. Cell 25, 635–646 [DOI] [PubMed] [Google Scholar]
- 24. Fischer N., Weis K. (2002) EMBO J. 21, 2788–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teixeira D., Parker R. (2007) Mol. Biol. Cell. 18, 2274–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coller J., Parker R. (2005) Cell 122, 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bergkessel M., Reese J. C. (2004) Genetics 167, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Westmoreland T. J., Olson J. A., Saito W. Y., Huper G., Marks J. R., Bennett C. B. (2003) J. Surg. Res. 113, 62–73 [DOI] [PubMed] [Google Scholar]
- 29. Park Y. U., Hur H., Ka M., Kim J. (2006) Eukaryot. Cell. 5, 2120–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hata H., Mitsui H., Liu H., Bai Y., Denis C. L., Shimizu Y., Sakai A. (1998) Genetics 148, 571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maillet L., Collart M. A. (2002) J. Biol. Chem. 277, 2835–2842 [DOI] [PubMed] [Google Scholar]
- 32. Collart M. A., Timmers H. T. (2004) Prog. Nucleic Acid Res. Mol. Biol. 77, 289–322 [DOI] [PubMed] [Google Scholar]
- 33. Collart M. A. (2003) Gene 313, 1–16 [DOI] [PubMed] [Google Scholar]
- 34. Denis C. L., Chen J. (2003) Prog. Nucleic Acid Res. Mol. Biol. 73, 221–250 [DOI] [PubMed] [Google Scholar]
- 35. Kruk J. A., Dutta A., Fu J., Gilmour D. S., Reese J. C. (2011) Genes Dev. 25, 581–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muhlrad D., Parker R. (2005) EMBO J. 24, 1033–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strahl-Bolsinger S., Tanner W. (1993) Yeast 9, 429–432 [DOI] [PubMed] [Google Scholar]
- 38. Bleichert F., Baserga S. J. (2007) Mol. Cell 27, 339–352 [DOI] [PubMed] [Google Scholar]
- 39. Cordin O., Banroques J., Tanner N. K., Linder P. (2006) Gene. 367, 17–37 [DOI] [PubMed] [Google Scholar]
- 40. Rocak S., Linder P. (2004) Nat. Rev. Mol. Cell Biol. 5, 232–241 [DOI] [PubMed] [Google Scholar]
- 41. Jankowsky E., Fairman M. E. (2007) Curr. Opin. Struct. Biol. 17, 316–324 [DOI] [PubMed] [Google Scholar]
- 42. Bizebard T., Ferlenghi I., Iost I., Dreyfus M. (2004) Biochemistry 43, 7857–7866 [DOI] [PubMed] [Google Scholar]
- 43. Kikuma T., Ohtsu M., Utsugi T., Koga S., Okuhara K., Eki T., Fujimori F., Murakami Y. (2004) J. Biol. Chem. 279, 20692–20698 [DOI] [PubMed] [Google Scholar]
- 44. Cheng Z., Coller J., Parker R., Song H. (2005) RNA 11, 1258–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Q., Arnold J. J., Uchida A., Raney K. D., Cameron C. E. (2010) Nucleic Acid Res. 38, 1312–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reese J. C., Green M. R. (2001) Yeast 18, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 47. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 48. Reese J. C., Green M. R. (2003) Methods Enzymol. 370, 415–430 [DOI] [PubMed] [Google Scholar]
- 49. Selth L. A., Gilbert C., Svejstrup J. Q. (2009) Cold Spring Harb. Protoc. 2009, pdb prot5234 [DOI] [PubMed] [Google Scholar]
- 50. Psathas J. N., Zheng S., Tan S., Reese J. C. (2009) Mol. Cell. Biol. 29, 6413–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng S., Wyrick J. J., Reese J. C. (2010) Mol. Cell. Biol. 30, 3635–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buchan J. R., Nissan T., Parker R. (2010) Methods Enzymol. 470, 619–640 [DOI] [PubMed] [Google Scholar]
- 53. Iost I., Dreyfus M., Linder P. (1999) J. Biol. Chem. 274, 17677–17683 [DOI] [PubMed] [Google Scholar]
- 54. Pause A., Sonenberg N. (1992) EMBO J. 11, 2643–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singleton M. R., Dillingham M. S., Wigley D. B. (2007) Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 56. Williamson J. R. (1994) Annu. Rev. Biophys. Biomol. Struct. 23, 703–730 [DOI] [PubMed] [Google Scholar]
- 57. Shi H., Cordin O., Minder C. M., Linder P., Xu R. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17628–17633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sengoku T., Nureki O., Nakamura A., Kobayashi S., Yokoyama S. (2006) Cell 125, 287–300 [DOI] [PubMed] [Google Scholar]
- 59. Tanner N. K., Cordin O., Banroques J., Doère M., Linder P. (2003) Mol. Cell 11, 127–138 [DOI] [PubMed] [Google Scholar]
- 60. Zhao R., Shen J., Green M. R., MacMorris M., Blumenthal T. (2004) Structure 12, 1373–1381 [DOI] [PubMed] [Google Scholar]
- 61. Svitkin Y. V., Pause A., Haghighat A., Pyronnet S., Witherell G., Belsham G. J., Sonenberg N. (2001) RNA 7, 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Del Campo M., Lambowitz A. M. (2009) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65, 832–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Korneeva N. L., First E. A., Benoit C. A., Rhoads R. E. (2005) J. Biol. Chem. 280, 1872–1881 [DOI] [PubMed] [Google Scholar]
- 64. Teixeira D., Sheth U., Valencia-Sanchez M. A., Brengues M., Parker R. (2005) RNA 11, 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cordin O., Tanner N. K., Doère M., Linder P., Banroques J. (2004) EMBO J. 23, 2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schmid S. R., Linder P. (1991) Mol. Cell. Biol. 11, 3463–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang Y., Liu Z. R. (2002) J. Biol. Chem. 277, 12810–12815 [DOI] [PubMed] [Google Scholar]
- 68. Rogers G. W., Jr., Komar A. A., Merrick W. C. (2002) Prog. Nucleic Acid Res. Mol. Biol. 72, 307–331 [DOI] [PubMed] [Google Scholar]
- 69. Shibuya T., Tange T. Ø., Sonenberg N., Moore M. J. (2004) Nat. Struct. Mol. Biol. 11, 346–351 [DOI] [PubMed] [Google Scholar]
- 70. Weston A., Sommerville J. (2006) Nucleic Acids Res. 34, 3082–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Navarro R. E., Shim E. Y., Kohara Y., Singson A., Blackwell T. K. (2001) Development 128, 3221–3232 [DOI] [PubMed] [Google Scholar]
- 72. Nakamura A., Amikura R., Hanyu K., Kobayashi S. (2001) Development 128, 3233–3242 [DOI] [PubMed] [Google Scholar]
- 73. Minshall N., Thom G., Standart N. (2001) RNA 7, 1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu W., Sweet T. J., Chamnongpol S., Baker K. E., Coller J. (2009) Nature 461, 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 76. Minshall N., Kress M., Weil D., Standart N. (2009) Mol. Biol. Cell. 20, 2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oberer M., Marintchev A., Wagner G. (2005) Genes Dev. 19, 2212–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.