

Abstract
The plasticity of macrophages is evident from their dual role in inflammation and resolution of inflammation that are accompanied by changes in the transcriptome and metabolome. Along these lines, we have previously demonstrated that the micronutrient selenium increases macrophage production of arachidonic acid (AA)-derived anti-inflammatory 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and decreases the proinflammatory PGE2. Here, we hypothesized that selenium modulated the metabolism of AA by a differential regulation of various prostaglandin (PG) synthases favoring the production of PGD2 metabolites, Δ12-PGJ2 and 15d-PGJ2. A dose-dependent increase in the expression of hematopoietic-PGD2 synthase (H-PGDS) by selenium and a corresponding increase in Δ12-PGJ2 and 15d-PGJ2 in RAW264.7 macrophages and primary bone marrow-derived macrophages was observed. Studies with organic non-bioavailable forms of selenium and the genetic manipulation of cellular selenium incorporation machinery indicated that selenoproteins were necessary for H-PGDS expression and 15d-PGJ2 production. Treatment of selenium-deficient macrophages with rosiglitazone, a peroxisome proliferator-activated receptor γ ligand, up-regulated H-PGDS. Furthermore, electrophoretic mobility shift assays indicated the presence of an active peroxisome proliferator-activated receptor-response element in murine Hpgds promoter suggesting a positive feedback mechanism of H-PGDS expression. Alternatively, the expression of nuclear factor-κB-dependent thromboxane synthase and microsomal PGE2 synthase was down-regulated by selenium. Using a Friend virus infection model of murine leukemia, the onset of leukemia was observed only in selenium-deficient and indomethacin-treated selenium-supplemented mice but not in the selenium-supplemented group or those treated with 15d-PGJ2. These results suggest the importance of selenium in the shunting of AA metabolism toward the production of PGD2 metabolites, which may have clinical implications.
Keywords: Gene Expression, Inflammation, Macrophages, Metabolism, Prostaglandins, Selenium, Class Switching, Resolution
Introduction
The essential trace nutrient selenium plays a major role as a key component of the antioxidant machinery in all cell types. This ability of selenium is attributed, in part, to the co-translational incorporation of the amino acid selenocysteine (Sec)3 into the active site of selenoproteins, many of which have important enzymatic functions (1). Proteins of the glutathione peroxidase (GPX) and thioredoxin reductase families are well characterized selenoenzymes that are involved in the regeneration of antioxidant systems and the maintenance of intracellular redox state and membrane integrity, as well as gene regulation by redox control of binding of transcription factors to DNA (2). Epidemiological studies suggest that inadequate selenium levels and associated reactive oxygen and nitrogen species production are linked to increased incidence of cardiovascular diseases, viral infections, including progression of HIV-AIDS and Alzheimer disease, infertility, and most notably cancer (2–5). Supra-nutritional doses of selenium have demonstrable anti-carcinogenic effects in a variety of cancers through mechanisms such as perturbation of tumor cell metabolism, induction of apoptosis, and inhibition of angiogenesis (6). Also, supplementation of selenium has been shown to improve the health status of patients suffering from inflammatory conditions such as septic shock, autoimmune thyroiditis, pancreatitis, allergic asthma, and rheumatoid arthritis (2, 7–9). These anti-inflammatory actions can be partly explained by the antioxidant role of various selenoproteins, but the precise mechanisms in the context of specific cell types involved in inflammation need to be explored.
Macrophages are cells of the immune system that are central to the progression of a physiological, protective inflammatory response into a pathological, tissue-destructive inflammatory state. An imbalance between the production of reactive oxygen and nitrogen species and the scavenging anti-oxidant mechanisms forms the basis for such progression (10). In addition to the oxidative (“respiratory”) burst reaction, classical macrophage activation is also characterized by the production of several mediators such as IL-1, IL-6, TNFα, prostaglandin E2 (PGE2), and thromboxane A2 (TXA2) (11). PGE2, TXA2, PGD2, and PGD2 downstream products, Δ12-PGJ2 and 15d-PGJ2, are the major eicosanoids derived from the fatty acid arachidonic acid (AA) in macrophages (12), whereas PGI2 is predominantly produced by vascular endothelial and smooth muscle cells that express high levels of PGI2 synthase (PGIS) (13). The initial step of prostaglandin (PG) synthesis involves formation of PGH2 from AA by the action of cyclooxygenase (COX)-1 and COX-2, which is subsequently acted upon by specific PG synthases, microsomal PGE synthase-1 (mPGES-1), thromboxane synthase (TXAS), and PGD synthase (PGDS), to form PGE2, TXA2, and PGD2, respectively (12). Studies have shown that during the resolution phase of inflammation, the AA metabolism shifts from the production of PGE2 toward that of PGD2 and its downstream double-dehydration product, 15d-PGJ2 (13, 14). This warrants further research into the regulation of the AA-COX pathway, which includes the various PG synthase enzymes. Oxidative stress in cells activates the transcription factor nuclear factor-κB (NF-κB). NF-κB serves as a key transcription factor for mPGES-1 and TXAS, leading to the up-regulation of PGE2 and TXA2, respectively (14, 15). Our laboratory has previously shown that selenium supplementation of macrophages down-regulated NF-κB with a corresponding increase in the activation of PPARγ, thus affecting the production of PGE2 (16–18).
Lipocalin-type PGDS and hematopoietic PGDS (H-PGDS) are two isoforms that catalyze the conversion of PGH2 to PGD2. While the former is expressed mainly in the CNS, the latter is found in immune cells like macrophages and mast cells, suggesting a role of H-PGDS in the regulation of inflammation (19, 20). PGD2 and 15d-PGJ2 have been shown to possess anti-inflammatory properties, which are conferred via different mechanisms, including the activation of DP2 receptor, PPARγ activation, and covalent modification of cysteine thiols in target proteins such as IKKβ and p65 of the NF-κB signaling axis (20, 21). However, not much is known about the molecular regulation of the glutathione (GSH)-dependent sigma-class glutathione S-transferase, H-PGDS. Studies from multiple laboratories, ours included, suggest that AA pathway is redox-sensitive (17, 22, 23). Selenium has been shown to modulate the expression of TXAS and prostacyclin synthase (PGIS) in endothelial cells (24). Furthermore, our laboratory has previously reported that selenium exerts its anti-inflammatory effects in activated macrophages via the enhanced production of 15d-PGJ2 in a COX-1-dependent manner (25). This led us to the hypothesis that H-PGDS is regulated by the redox state of macrophages, which depends largely on their selenium status as well as the differential modulation of PPARγ and NF-κB. Here, we demonstrate that selenium supplementation of macrophages differentially regulates the expression of mPGES-1, TXAS, and H-PGDS to favor the shunting of AA through the H-PGDS pathway where selenoproteins are indispensable. Furthermore, we present data that validate the shunting of PG metabolism using a mouse model of leukemia upon infection by Friend virus (FV), a retrovirus that infects hematopoietic progenitors in the bone marrow and spleen to cause development of leukemia (26). In this model, resistance of selenium-supplemented mice to FV infection was overcome with indomethacin treatment, although treatment with 15d-PGJ2, but not PGE2, completely rescued the selenium-supplemented mice despite inhibition of the AA pathway. Taken together, our data highlight the clinical significance of selenium-dependent shunting of AA toward PGD2 metabolites.
MATERIALS AND METHODS
Reagents
Sodium selenite, methylseleninic acid (MSA), l-selenomethionine (Se-Met), bacterial lipopolysaccharide (Escherichia coli 0111:B4), ebselen (2-phenyl-1,2-benzoselenazol-3-one), indomethacin, and DMSO were obtained from Sigma. 1,4-Phenylenebis(methylene) selenocyanate (p-XSC) was provided by Dr. S. Amin, Pennsylvania State University College of Medicine (Hershey, PA). CAY10526, HQL-79, 15d-PGJ2, 11-deoxy-16,16-dimethyl-PGE2, and rosiglitazone were purchased from Cayman Chemicals (Ann Arbor, MI). Recombinant l-methioninase (rMETase) and parthenolide (PTN) were from Wako Chemicals (Richmond, VA) and Calbiochem, respectively.
Cell Culture and Animals
Murine macrophage-like cell line RAW264.7 (ATCC, Manassas, VA) was cultured in Dulbecco's modified Eagle's medium (Invitrogen) containing 5% fetal bovine serum (ATCC), 2 mm l-glutamine (Cellgro, Manassas, VA), penicillin-G (100 units/ml), and streptomycin (100 μg/ml; Invitrogen). For routine culture, the cells were passaged every 3 days at a ratio of 1:10 as per the recommendations by ATCC. For experiments, the cells were seeded at a density of ∼500,000/well of a 6-well plate and cultured for 2–4 days depending on the treatments. Selenite and Se-Met were dissolved in sterile water. p-XSC, ebselen, parthenolide, CAY10526, rosiglitazone, and 15d-PGJ2 were dissolved in DMSO. The final concentration of DMSO in the cell culture medium was 0.1% v/v. Human embryonic kidney (HEK293T) cells were used for extracting nuclear proteins for the EMSA. They were cultured in DMEM containing 10% FBS, 2 mm l-glutamine, and penicillin/streptomycin. Primary murine macrophages (BMDMs) were prepared by extracting femoral bone marrow plugs from ∼3-month-old C57BL/6 mice maintained on selenium-deficient (0.01 ppm selenite), selenium-adequate (0.1 ppm selenite), and selenium-supplemented (0.4 ppm selenite) diets for 12 weeks. These cells were then cultured in the same medium as described earlier with the addition of 20% L929 fibroblast-conditioned medium (as a source of granulocyte-macrophage colony-stimulating factor) for 1 week. Macrophages from selenium-deficient, selenium-adequate, and selenium-supplemented mice were cultured in defined media containing 7, 100, or 250 nm selenium (as selenite). For the FV-leukemia model, BALB/c mice (∼3 months old), maintained on selenium-deficient or selenium-supplemented diet (0.4 ppm selenite) for 12 weeks, were retro-orbitally injected with FV as described previously (27). These mice were dissected on day 15 post-infection, and signs of splenomegaly as well as WBC counts in the peripheral blood, characteristic of leukemia in this model (26, 28), were examined. Hematological analysis was performed on an Advia 120 hematology autoanalyzer equipped with a veterinary software program. The COX inhibitor indomethacin was administered to the mice at a concentration of 0.00325% (w/v) in drinking water, as described previously (29), from 2 weeks prior to FV injection until 2 weeks post-infection, when the animals were euthanized. 15d-PGJ2 or 11-deoxy-16,16-dimethyl-PGE2 was exogenously administered at a concentration of 0.025 mg/kg/day (dissolved in PBS) intraperitoneally from day 7 to 14 post-infection to the respective groups. The special diets were formulated based on the recommended rodent diets from American Institute of Nutrition and purchased from Harlan-Teklad (Madison, WI).
Glutathione Peroxidase Activity Assay
Cells cultured under different concentrations of sodium selenite (0–250 nm) were harvested and lysed with the mammalian protein extraction reagent (M-PER; Pierce) containing protease inhibitor mixture and 1 mm PMSF. Whole cell lysates were used for the GPX assay. The GPX activity was measured using H2O2 and NADPH, and the activity was expressed as nanomoles of NADPH oxidized per min per mg of protein in a spectrophotometric assay as described earlier (30).
Arginase Activity Assay
Arginase activity, assessed by a colorimetric assay that detects urea production generated by arginase hydrolysis of l-Arg, has been described previously (31). The selenium-deficient and selenium-adequate RAW264.7 macrophages assayed were cultured as described earlier. Absorbance at 560 nm was recorded on a Packard plate reader. A urea standard calibration curve (0–1 μmol; y = 9E-05x + 0.0007, R2 = 0.99) was used to calculate the arginase activity. Enzyme activity is expressed as micromoles of urea produced per mg of protein (31).
SDS-PAGE and Immunoblotting
Whole cell lysates were prepared from RAW264.7 cells or BMDMs using M-PER containing protease inhibitors, and protein concentration was estimated by BCA reagent. An equal amount of protein (ranging from 5 to 25 μg) was electrophoresed on a 12.5% discontinuous SDS-polyacrylamide gel. Proteins were then transferred to a nitrocellulose membrane. The following primary antibodies were used to probe the membrane: anti-H-PGDS, anti-TXAS, anti-mPGES-1 (Cayman), anti-GPX1, anti-TR1 (Abcam, Cambridge, MA), and anti-GAPDH (Fitzgerald, Concord, MA). Appropriate secondary antibodies were used. Immunoreactive bands were visualized using the West Pico and/or West Femto chemiluminescence reagents obtained from Thermo Scientific (Rockford, IL). GAPDH was used as an internal control for normalization. Bands were evaluated by densitometry using the ImageJ software program (National Institutes of Health).
Real Time (Quantitative) and Semi-quantitative RT-PCR
RNA was isolated from RAW264.7 cells or BMDMs using the Isol-RNA lysis reagent (5 Prime, Gaithersburg, MD) as per its protocol. RNA purity and concentration were determined using UV spectroscopy. 2 μg of DNase (New England Biolabs)-treated RNA was used for reverse transcription reaction using High Capacity cDNA reverse transcriptase kit, per the manufacturer's instructions (Applied Biosystems, Foster City, CA). Prevalidated TaqMan probes for Hpgds, Mpges-1, Txas, and Pgis were purchased from Applied Biosystems (Foster City, CA). Gapdh probe was used as an internal control to normalize the data. Amplifications were performed using PerfeCTa quantitative PCR SuperMix master mix (Quanta Biosciences) in a 7300 real time PCR system (Applied Biosystems). ΔCt (CtGene − CtGapdh) was calculated for each sample and used for analysis of transcript abundance with respect to the untreated negative control as described previously (32). For the semi-quantitative RT-PCR, the sequences of the primers were as follows: mouse Hpgds sense 5′-ATGCCTAACTACAAACTGCTT-3′ and antisense 5′-CTAGAGTTTTGTCTGTGGCCT-3′; mouse Gpx1 sense 5′-ACAGTCCACCGTGTATGCCTTC-3′ and antisense 5′-CTCTTCATTCTTGCCATTCTCCTG-3′; and mouse β-actin sense 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and antisense 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. PCR was carried out using 0.2 μm of primers, 2.5 mm MgCl2, 0.2 mm of each deoxyribonucleotide triphosphate, 1.25 units of GoTaq DNA polymerase (Promega, Madison, WI), and 2.5 ng of template cDNA. After initial denaturation at 95 °C for 2 min, PCR was continued with amplification cycles of 30 s at 95 °C, 30 s at 56 °C, and 1 min at 72 °C, followed by elongation for 5 min at 72 °C. The yield of PCR products was tested to be in the linear range, and optimal cycle number was chosen. The PCR products were separated by electrophoresis on a 1% agarose gel and visualized by UV transillumination. β-Actin was used as an internal control for normalization. Bands were evaluated by densitometry using the ImageJ program.
Transfections and Knockdowns
siRNA for SectRNA synthetase was obtained as ON-TARGET plus SMART pool (Thermo Scientific Dharmacon). 2 μg of siRNA was transfected into 2 × 106 RAW264.7 cells using Nucleofector Solution V according to the manufacturer's specifications (Amaxa Biosystems). AllStars Negative Control siRNA (Qiagen) was used in equal amounts as a control. Selenite was added 12 h after transfection at a final concentration of 100 nm, and cells were harvested at 36 h after transfection for analysis by immunoblotting. The pU6-TetO4m4 vector containing shRNA target sequence, 5′-GACGTAGAGTTGGCATACC-3′ (nucleotides 694–712) for selenophosphate synthetase 2 (SPS2), was obtained from Dr. Dolph L. Hatfield, NCI, National Institutes of Health, Bethesda. The control vector was designated pU6Tet control, which consisted of the same construct minus the target sequence. For transfection, RAW264.7 macrophages were seeded into 6-well plates (1 × 106 cells/well) without antibiotics. After 24 h, cells were transfected with either shSPS2 or empty vector using TransIT®-LT1 (Mirus Bio) according to the manufacturer's specifications. The constructs were used at a final concentration of 2.5 μg/well. Forty eight hours after transfection, cells were left in the media with or without 250 nm selenite for 48 h followed by 12 h of 1 μg/ml LPS stimulation. Cells and media supernatants were collected for 15d-PGJ2 quantitation, and the corresponding cell lysates were used for Western immunoblotting analyses. SPS2 knockdown stable RAW264.7 cells were also prepared by transfecting the shSPS2 or the empty vector as described above. Resistant clones were selected in 100 μg/ml hygromycin and then expanded, followed by culturing them in a selenite-containing media to examine the knockdown of selenoproteins. Western blot analysis of SPS-2 confirmed ∼80% knockdown. Approximately 60% confluent HEK293T cells in a 60-mm culture dish were transfected with 6 μg of pCDNA3.1 containing human PPARγ full-length cDNA (provided by Dr. Jack Vanden Heuvel, Pennsylvania State University) using TransIT®-293 (Mirus Bio) transfection reagent. Rosiglitazone was added 24 h later to a final concentration of 2 μm for the expressed PPARγ to translocate into the nucleus. After 24 h, cells were harvested to prepare nuclear extracts.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay
Cytoplasmic fraction was separated from the nuclei by lysing the HEK293T cells in a buffer containing 10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.1% Nonidet P-40, 0.1 mm DTT, 1 mm PMSF, 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 μg/ml pepstatin, followed by centrifugation at 10,000 × g for 10 min at 4 °C. The nuclear pellet was then lysed with a buffer containing 20 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 420 mm NaCl, 0.2 mm EDTA, and protease inhibitors (see above). The lysates were sonicated (three pulses) at 60% power on ice and centrifuged at 14,000 × g for 10 min at 4 °C. Concentrations of proteins in this nuclear extract were determined using BCA reagent (Pierce). The DNA sequences for the sense strand of the oligonucleotides included the following: consensus PPRE (cPPRE) 5′-GGTGAGGAGGGGAAGGGTCAGTGTG-3′; PPRE1 5′-CTTATTAGGGTTAAAGGTCTAAA-3′; PPRE2 5′-TGAGGTGAGGTTAATGGACACAG-3′; PPRE3 5′-TGTAGAAGGGAGAAAGCTCAAAA-3′; and PPRE4 5′-ATGCACGAGTCCAAAGTTCAGCC-3′. Complementary strands were annealed, and 4 pmol/μl of the resultant double-stranded oligonucleotide was labeled with [γ-32P]ATP (3000 Ci/mol at 10 mCi/ml) using T4 polynucleotide kinase (New England Biolabs). 32P-Labeled oligonucleotides were separated using gel filtration G-25 macro spin columns (The Nest Group Inc., Southborough, MA). For the binding reaction, 10 μg of nuclear extracts were incubated for 20 min at room temperature with 5× gel-shift binding buffer (50 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 2.5 mm DTT, 2.5 mm EDTA, 250 mm NaCl, 20% glycerol, 500 ng/ml BSA), 50 μg/ml poly(dI-dC), and 32P-labeled PPRE oligonucleotides (20,000 cpm). In addition, a competitive binding assay between labeled PPRE1 and unlabeled consensus PPRE (at different ratios) was also performed. 2 μg of anti-PPARγ antibody (Cayman) was used for the supershift reaction. The samples were analyzed by PAGE (% T = 4) at 120 V for 45–50 min in TBE buffer. After drying, the gel was exposed to x-ray film overnight at −80 °C. The PPARγ bands were confirmed by competition using a >100-fold excess of corresponding unlabeled oligonucleotide.
PG Quantitation
The concentrations of PGE2 and TXB2 in culture media supernatants of BMDMs and RAW264.7 cells treated with selenium as indicated and treated with 1 μg/ml LPS were determined by enzyme immunoassay according to the manufacturer's instructions (Cayman) and normalized to total cellular protein. 15d-PGJ2 was quantitated from supernatants from RAW264.7 cells transiently transfected with shSPS2 by using enzyme immunoassay kit from Assay Designs (Ann Arbor, MI). In most cases, the values were normalized to total cell protein.
LC-MS Analysis of 15d-PGJ2 and Δ12-PGJ2 Production
The RAW264.7 or BMDM cell culture supernatants were acidified with HCl (1 n) to pH 3.0 and extracted with ethyl acetate. The compounds 15d-PGJ2 and Δ12-PGJ2 were analyzed using an LC-MS/MS system comprising of Shimadzu LC20AD UFLC pumps and API2000 triple quadruple mass spectrometer set to negative mode with a temperature of 200 °C for the confirmation of molecular ions. The solvent system used was methanol/H2O (70:30) with 0.1% acetic acid at a flow rate of 0.15 ml/min. The MS analysis was performed on API2000 set to scan mode (m/z 100–350) for authentic standards, and molecular ions m/z 315.6 (M − H+) for 15d-PGJ2 and 333.5 (M − H+) for Δ12-PGJ2, respectively, were used for quantitation. As an internal control to calculate extraction efficiency, 200 ng of deuterated 15d-PGJ2(d4) (Cayman Chemicals) was used, which showed a 70% extraction efficiency. Standard calibration curves for 15d-PGJ2 and Δ12-PGJ2 were set up for the quantitation, and calculations were performed based on the following equations: y = 76.831x − 608.42; r2 = 0.9927 (for 15d-PGJ2) and y = 7.2516x + 172.34; r2 = 0.9871 (for Δ12-PGJ2). As described earlier, the values were normalized to total cell protein.
Statistical Analysis
The results are expressed as means ± S.E. The differences between groups were analyzed by Student's t test using GraphPad Prism. The criterion for statistical significance was p < 0.05.
RESULTS
Selenium Up-regulates H-PGDS Expression and Its Downstream Metabolites in RAW264.7 Macrophages
To examine the regulation of H-PGDS by selenium, RAW264.7 macrophages cultured for 3 days in various concentrations of exogenously added selenium (0–500 nm selenium as selenite) were used. Analysis of the cell lysates displayed a dose-dependent increase in the cytosolic GPX enzyme activity (Fig. 1A). Although the level of GPX was very low in selenium-deficient conditions, 100 nm selenium induced an ∼10-fold increase in the GPX activity and protein expression (Fig. 1, A and B). Using this model, we examined the ability of selenium to up-regulate expression of H-PGDS. As seen with GPX activity, the expression of H-PGDS also increased as a function of selenium concentration, with saturation at 150–250 nm selenium. We observed an ∼3-fold increase in protein levels of H-PGDS at 250 nm (Fig. 1B). In concert with the increased levels of H-PGDS, downstream metabolites of PGD2, Δ12-PGJ2, and 15d-PGJ2 also showed a similar dose-dependent increase with selenite in the cell culture supernatants using quantitative LC/MS-based assays (Fig. 1C). At all the doses of selenium tested, the concentration of Δ12-PGJ2 was much higher than its dehydrated metabolite 15d-PGJ2.
FIGURE 1.

Sodium selenite up-regulates H-PGDS expression and its downstream metabolites in RAW264.7 macrophages. RAW264.7 cells were cultured for 3 days in different concentrations of sodium selenite and the cell lysates were used for GPX1 activity assay (A) and representative immunoblot (IB) of n = 3 (B). Densitometric ratios normalized to GAPDH are shown below for GPX1 and H-PGDS in the form of mean fold changes with respect to “Se 0” ± S.E. are shown. C represents a LC-MS quantitation of Δ12-PGJ2 and 15d-PGJ2 from cell culture supernatants, normalized to total cell protein (n = 3).
Selenite Differentially Regulates the Expression of PG Synthases in RAW264.7 Cells and BMDMs
To investigate the role of selenium in the shunting of the AA-COX pathway, the expression of two other additional PG synthases, mPGES-1 and TXAS, which are highly expressed in macrophages, were examined by immunoblotting and real time RT-PCR. With an increase in selenium, the expression of mPGES-1 and TXAS showed a dose-dependent decrease at both protein and transcript levels. However, transcript levels of Hpgds increased as a function of selenium, in agreement with the protein level as described earlier (Fig. 2, A and B). Analysis of the expression levels of Pgis showed an interesting biphasic regulation in that an initial increase in the transcript was seen with 50 and 100 nm selenium (Fig. 2B). However, further addition of selenium (>100 nm) decreased the levels of Pgis. Analysis of protein expression of PG synthases in primary BMDMs isolated from the bone marrow of mice on diets containing different selenium concentrations showed a dose-dependent increase in H-PGDS, whereas mPGES-1 and TXAS expression was decreased at 100 and 500 nm of selenium relative to selenium-deficient cells (Fig. 2C).
FIGURE 2.

Selenite differentially regulates the expression of PG synthases in RAW264.7 cells and BMDMs. RAW264.7 cells were treated with different concentrations of sodium selenite for 3 days. A and B represent immunoblot (IB) and real time RT-PCR results, respectively. Densitometric ratios from the immunoblots are shown as mean ± S.E. of n = 3. C shows the dose-response of protein expression of PG synthases on BMDMs from C57BL/6 mice maintained on selenium-deficient (0.01 ppm), selenium-adequate (0.1 ppm), and selenium-supplemented (0.4 ppm) diets. *, p < 0.05.
Different Forms of Selenium Have Variable Effects on Expression of PG Synthases in RAW264.7 Cells and BMDMs
To further understand the selenium-dependent regulation of the AA-COX pathway, the effects of four commonly used forms of selenium were compared. RAW264.7 cells were incubated with 0, 100, and 500 nm of selenium in the form of sodium selenite, MSA, p-XSC or Se-Met for 3 days and analyzed for protein and transcripts of H-PGDS and other PG synthases. GPX1 levels were also examined as a marker of selenoprotein status of these cells. Fig. 3, A and B, shows that MSA up-regulated Hpgds in a manner comparable with sodium selenite treatment, but p-XSC and Se-Met did not lead to a significant change in H-PGDS or GPX1 from the base-line levels. Conversely, expression of Mpges-1 and Txas was down-regulated with MSA but not Se-Met and p-XSC. Real time RT-PCR analyses on BMDMs (from selenium-deficient mice) treated with different forms of selenium ex vivo showed similar patterns of expression of PG synthases (Fig. 3C), an exception being the down-regulation of Txas by Se-Met. Quantification of the levels of PGE2 and TXB2 in RAW264.7 and BMDM cell culture supernatants by ELISA confirmed the results obtained from the expression analyses of PG synthases (Fig. 3, D and E). Analyses of the BMDM supernatants by LC/MS for Δ12-PGJ2 and 15d-PGJ2 revealed significantly high levels with the 500 nm selenite treatment and minor increases within the other groups as compared with the selenium-deficient samples (Fig. 3F). These results suggest that increased bioavailability of selenium, perhaps in the form of selenoproteins, is a critical determinant of selenium-dependent regulation of PG synthases in macrophages.
FIGURE 3.

Regulation of PG synthases by different forms of selenium. A represents the immunoblot (IB) of lysates from RAW264.7 cells treated with the indicated concentrations of sodium selenite, MSA, p-XSC, or Se-Met for 3 days. B and C represent real time RT-PCR analysis of transcripts of Hpgds, Txas, and Mpges in RAW264.7 and BMDMs treated with different forms of selenium, respectively. D and E show the levels of PGE2 and TXB2 in the cell culture supernatants from RAW264.7 and BMDMs, respectively, upon treatment with different forms of selenium. F represents mass spectrometric analysis of 15d-PGJ2 and Δ12-PGJ2 on supernatants from BMDMs treated with different forms of selenium as shown. All data shown are means ± S.E. of three independent observations. *, p < 0.05 when compared with the corresponding 0 Se sample.
Selenoprotein Synthesis Is Critical to Regulate the Expression of H-PGDS by Selenium
To examine if incorporation of selenium into selenoproteins was essential for the up-regulation of H-PGDS and subsequent formation of 15d-PGJ2, a genetic knockdown approach was used. Selenium-deficient RAW264.7 macrophages were transfected with either shSPS2 or control constructs and then maintained under selenium-deficient and selenium-supplemented (250 nm) conditions for 2 days followed by 1 μg/ml LPS stimulation for 12 h. The cell lysates were used for the analysis of the expression of SPS2, GPX1, and TR1 by immunoblotting. Although the expression of SPS2 (data not shown), GPX1, and TR1 was significantly knocked down in shSPS2-transfected cells despite the presence of selenium, there was no change in the expression of these proteins in cells transfected with the vector control construct (Fig. 4A). To confirm that SPS2 knockdown cells cannot produce 15d-PGJ2 due to the lack of selenoproteins, the cell supernatants were utilized for the analysis of 15d-PGJ2 production. The production of 15d-PGJ2 was almost completely inhibited in SPS2 knockdown cells even after supplementing with 250 nm selenium. In contrast, we observed a significant increase in the production of 15d-PGJ2 in those cells that were transfected with control constructs followed by selenite treatment (Fig. 4B). Evaluation of H-PGDS expression using the shSPS2 stably transfected macrophages that were cultured with 100 nm selenium showed a clear down-regulation of GPX1 as well as H-PGDS only in shSPS2 knockdown cells but not in the vector control (Fig. 4C). Furthermore, we genetically manipulated the expression of SectRNA synthetase, an enzyme that is critical in the pathway of Sec incorporation into selenoproteins (33). Western immunoblotting of cells that were subjected to siRNA-mediated knockdown of SectRNA synthetase clearly showed a substantial down-regulation of H-PGDS and GPX1 (Fig. 4D). Interestingly, treatment of Se-Met in presence of r-METase, an l-Met α,γ-lyase that releases selenium for incorporation into selenoproteins, led to the up-regulation of H-PGDS expression (Fig. 4E). To understand if the activity of GPX, one of the most abundantly expressed selenoproteins, was pivotal to up-regulate H-PGDS, ebselen, a small molecular mimetic of GPX1 (34), was used. RAW264.7 cells treated with different concentrations of ebselen showed a dose-dependent increase in levels of Hpgds transcripts in the absence of any increase in GPX1 (Fig. 4F). Taken together, these results provide further evidence in support of the important role of selenoproteins in the up-regulation of H-PGDS and subsequent shunting of the AA-COX pathway in macrophages.
FIGURE 4.

Selenoprotein synthesis is critical to regulate the expression of H-PGDS by selenium. A shows the protein expression of selenoproteins GPX1 and TR1, and B shows the amounts of 15d-PGJ2 in cell culture supernatants upon transient knockdown of selenophosphate synthetase 2 (SPS2) in RAW264.7 cells using shSPS2, respectively. UT, untransfected cells. C shows the protein levels of GPX1 and H-PGDS in RAW264.7 cells stably transfected with shSPS2 or empty vector control. D represents the effects of transiently knocking down selenocysteine tRNA (SectRNA)-synthetase in selenite-supplemented RAW264.7 cells. E shows the effect on Hpgds transcript levels upon adding 0.1 unit/ml recombinant methioninase (r-Metase) along with selenomethionine to RAW264.7 cells. The effect of 24 h of treatment with the GPX-mimetic ebselen on Hpgds transcripts is shown in F. All data shown are representative of at least three independent experiments. IB, immunoblot.
PPARγ-dependent Transcription of H-PGDS
We speculated the presence of a positive-feedback regulation of H-PGDS by the endogenous PPARγ ligand, 15d-PGJ2. Examination of the murine Hpgds promoter for the presence of PPAR-response elements (PPREs) using MatInspector (Genomatix Software Gmbh, Munich, Germany) resulted in at least four putative binding sites at −1250, −3401, −4478, and −4896 bp for PPARγ found within 5 kb upstream of the transcription start site of Hpgds, which have a matrix similarity score of >0.75 when compared with a cPPRE (Fig. 5A). To examine the binding of these PPREs to PPARγ within the context of the promoter, we analyzed the binding of oligonucleotides containing each of these PPREs to HEK293T-expressed PPARγ. Gel shift analysis clearly showed that PPRE1 (−1250) interacted strongly with PPARγ, somewhat similar to the cPPRE, whereas PPRE3 (−4478) was relatively ineffective, and PPRE2 (−3401) and PPRE4 (−4896) showed minimal binding (Fig. 5B). We confirmed the specificity of PPARγ binding to PPRE1 by competing 32P-labeled PPRE1 oligonucleotide with varying amounts of unlabeled cPPRE oligonucleotide as well as by displaying a supershift using anti-PPARγ antibody (Fig. 5C). Having located functionally active PPREs (−1250 and −4896) in the murine Hpgds promoter, we studied the regulation of H-PGDS expression upon treatment of RAW264.7 macrophages with rosiglitazone, a bona fide agonist of PPARγ. Rosiglitazone significantly increased H-PGDS protein expression in selenium-deficient cells (Fig. 5D). These studies indicate that PPARγ plays a critical role in the regulation of H-PGDS expression, which is facilitated by selenium.
FIGURE 5.

Role of PPARγ in the expression of murine H-PGDS. A shows the comparison of the murine Hpgds promoter for putative PPREs using MatInspector and their matrix similarity scores with that of consensus PPRE. B shows electrophoretic gel shift assay demonstrating the binding of PPREs in the murine Hpgds promoter. Nuclear extract was prepared from HEK293T cells transfected with full-length PPARγ expression construct and stimulated with 2 μm rosiglitazone. Lanes 1, 3, 5–7, and 9 represent 32P-labeled PPREs with their corresponding “cold” competitor in an ∼100-fold excess, and lanes 2, 4, 6, 8, and 10 represent binding (*) with 32P-labeled PPREs alone, respectively. C shows EMSA with competitive binding of labeled PPRE1 with unlabeled consensus PPRE as well as anti-PPARγ-induced supershift. Lanes 1–8 represent PPRE1 with excess cold PPRE1, PPRE1, and PPRE1 and cold cPPRE (1:5); PPRE1 and cold cPPRE (1:25); PPRE1 and cold cPPRE (1:125),and binding of PPARγ to PPRE1 in the presence of cold PPRE1, the absence of cold competitor, and supershift (SS) of PPARγ-PPRE1 complex in the presence of anti-PPARγ antibody, respectively. D shows immunoblot (IB) on lysates from RAW264.7 cells treated with or without selenite and 1 μm rosiglitazone. Densitometric ratios from four representative Western blots are shown as mean of fold change ± S.E.
Inhibition of H-PGDS Blocks Selenium-mediated Up-regulation of PPARγ-dependent Arginase-1 Production
Having previously shown that there is increased nuclear translocation of PPARγ in the presence of selenium in macrophages (25), and knowing that macrophage alternative activation marker arginase-1 (Arg-I) is induced by PPARγ (35) and increases in CyPGs in selenium-treated cells, we studied the effect of selenium on the activation of arginase-1 and the role of H-PGDS in this process. Treatment of RAW264.7 macrophages with 100 nm selenium increased the arginase-1 activity up to ∼7-fold compared with the selenium-deficient cells in the presence of IL-4, which is a Th2 cytokine essential for alternative activation. A similar increase in the transcript levels of Arg-I was also observed (data not shown). Interestingly, pretreatment of cells with an H-PGDS inhibitor, HQL-79, completely blocked the selenium-mediated increase in arginase-1 (Fig. 6). Treatment of selenium-supplemented cells with HQL-79 also inhibited the production of Δ12-PGJ2 and 15d-PGJ2 (data not shown).
FIGURE 6.

Inhibition of H-PGDS blocks the selenium-mediated production of Arg-1 in macrophages. RAW264.7 cells in selenium-deficient conditions or 100 nm selenite were pretreated with 25 μm HQL-79 for 2 h followed by stimulation with IL-4 (5 ng/μl; 20 h) or LPS (1 μg/ml; 12 h). Arginase activity was determined. Values are means ± S.E. of n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001 indicate significant differences when compared with their selenium-deficient counterparts. a–c represent significant differences (p < 0.05) between vehicle and HQL-79 treated groups. UT, untransfected cells.
Control of NF-κB Activation Shunts the AA Pathway toward PGJ2 Production
Given that NF-κB is known to regulate the expression of mPGES-1 and TXAS (14, 15), we examined whether inhibition of NF-κB in selenium-deficient cells provides an alternative route of metabolism of PGH2, possibly through H-PGDS, which provides sufficient 15d-PGJ2 to up-regulate H-PGDS. To test this potential feedback activation, RAW264.7 macrophages were treated with 1 μm parthenolide (PTN) for 1 h before stimulation with 1 μg/ml LPS. Inhibition of NF-κB with PTN up-regulated H-PGDS expression (transcripts and protein) under selenium-deficient conditions. An increase in H-PGDS was accompanied by a decrease in TXAS and mPGES-1 expression (Fig. 7, A and B). Similarly, inhibition of mPGES-1 by CAY10526 in these cells, followed by LPS stimulation, resulted in an increase in H-PGDS expression (Fig. 7, A and B) as well as increased Δ12-PGJ2 and 15d-PGJ2 levels in the supernatants, when analyzed by LC-MS (Fig. 7C).
FIGURE 7.

Inhibition of NF-κB shunts the AA pathway toward PGJ2 production. A and B represent real time PCR and immunoblot (IB) analyses, respectively, for Hpgds, Txas, and Mpges-1 expression in selenium-deficient RAW264.7 cells and were treated with 1 μm PTN or 20 μm CAY10526 for 1 h followed by stimulation with 1 μg/ml LPS for 2 h. C shows the effect of CAY10526 on the production of 15d-PGJ2 and Δ12-PGJ2 in selenium-deficient RAW264.7 cells. All data are representative of n = 3, mean ± S.E. shown.
Selenium Supplementation Prevents Development of Disease in FV Murine Leukemia Model
To demonstrate the significance of selenium in an in vivo disease model with regard to the AA pathway, we used the FV murine leukemia model, where the infection of mice with the retrovirus leads to the expansion of a clone of infected hematopoietic precursors in the spleen and bone marrow that results in the development of acute leukemia (26). The disease is characterized by splenomegaly within 2 weeks of FV infection, accompanied by a significant rise in the WBC count (28). Selenium-deficient FV-infected BALB/c mice showed pathological changes demonstrated by the ∼12-fold increase in spleen size (Fig. 8, A and B) and ∼4-fold increase in the WBC counts (Fig. 8C), although FV-infected mice maintained on selenium-supplemented diets showed no signs of disease. Interestingly, administration of indomethacin to selenium-supplemented mice caused splenomegaly with an increase in WBC counts upon FV infection as in the selenium-deficient mice (Fig. 8, D–F). Daily intraperitoneal administration of 15d-PGJ2 at 0.025 mg/kg to a group of indomethacin-treated selenium-supplemented mice rescued them from the FV leukemia, although administration of a stable PGE2 analog failed to rescue these mice (Fig. 8, D–F). The dose of 15-PGJ2 used in the above experiment was based on a dose-response study to determine the minimal dose required to completely alleviate the signs of the disease (data not shown). Taken together, these studies indicate the critical role of selenium-mediated shunting of the AA pathway in an in vivo pathogenesis model.
FIGURE 8.

Role of selenium-dependent shunting of AA pathway in alleviating the development of FV leukemia. BALB/c mice maintained on selenium-deficient (Se-def) and selenium-supplemented (0.4 ppm selenite) diets for 12 weeks were infected with FV. The mice were euthanized 2 weeks post-infection. A and B show the spleens and spleen weights, respectively. C shows the WBC counts on blood samples from each group. D and E show spleens and spleen weights, respectively, from selenium-supplemented indomethacin (Indometh) (0.00325% w/v in drinking water)-treated BALB/c mice. 15d-PGJ2 or PGE2 analogs were administered intraperitoneally at a concentration of 0.025 mg/kg/day from day 7 to day 14 post-infection in respective groups. F shows WBC counts on blood samples from each group. n = 4 mice in each group. *, p < 0.05.
DISCUSSION
Epidemiological evidence suggests that the benefits of the essential micronutrient selenium in optimal health are, in part, contributed by the ability of selenium to alleviate inflammatory signaling pathways (2, 5). Selenium might impart these anti-inflammatory properties via different selenoproteins, but the mechanisms have not been completely understood. These selenoproteins form an important component of the cellular redox regulatory machinery, especially in oxidative stress-prone macrophages. Inhibition of the activation of the NF-κB signaling axis and its effect on the down-regulation of its pro-inflammatory gene products such as COX-2, mPGES-1, and TXAS are well known. We have earlier shown that selenium can inhibit NF-κB activation in macrophages (16–18). Given the fact that macrophages, among other immune cells, have the enzymatic machinery to metabolize AA to all PG classes, including TXA2, and that PGD2 metabolites produced by these cells have the ability to modulate NF-κB and PPARγ pathways (19–21), we explored the possibility of selenium regulating the production of different PGs at the transcription level. In this study, we report that bioavailability of selenium for incorporation into selenoproteins is essential to up-regulate the expression of H-PGDS and down-regulate the expression of mPGES-1 and TXAS. The resultant increase in H-PGDS shunts the metabolism of the AA-derived COX product, PGH2, from pro-inflammatory PGE2 and TXA2 toward the anti-inflammatory and pro-resolution product 15d-PGJ2. As a proof-of-concept of the translatability of these studies, we have utilized an FV-infection leukemia model. Our study demonstrates the protective effect of selenium to be inhibited by indomethacin treatment of mice, although subsequent treatment of such mice with 15d-PGJ2, but not PGE2, completely inhibited the disease. These studies corroborate the current ex vivo and previous studies of the in vivo production of 15d-PGJ2 in selenium-supplemented mice (25) as a critical mechanism underlying the anti-leukemic effect of selenium. However, the exact mechanism and targets of selenium or endogenous 15d-PGJ2 remain to be characterized.
The dose-dependent increase in the activity and expression of GPX1 with sodium selenite reinforces the fact that GPX1 is a reliable marker of cellular selenium status in macrophages. The saturation of GPX1 activity and expression at 100 nm selenium in the form of selenite added to selenium-deficient cells demonstrate the sensitivity of the selenoprotein synthesis machinery to bioavailable selenium. Therefore, we subjected selenium-deficient primary BMDMs and RAW264.7 cells to different doses (0–500 nm) of selenite to examine if there was an effect on the transcription of H-PGDS, which is the rate-limiting step for the formation of PGD2 and its stable metabolites, Δ12-PGJ2 and 15d-PGJ2. The dose-dependent up-regulation of H-PGDS along with the increased production of its downstream products by selenite, in both primary and immortalized macrophages, demonstrate that the cellular selenium status plays a pivotal role in regulating the expression of proteins outside the selenoproteome. Higher levels of Δ12-PGJ2 as compared with 15d-PGJ2 in the cell culture supernatant of primary cells than those in the immortalized cells are consistent with those reported for eicosanoids in general by Rouzer et al. (36). Also, the increased accumulation of Δ12-PGJ2 than its dehydrated product (15d-PGJ2) is also in agreement with the reports on the thermodynamically unfavorable conversion of Δ12-PGJ2 to 15d-PGJ2 in the presence of proteins (37, 38). Conversely, the dose-dependent down-regulation of mPGES-1 and TXAS and their pro-inflammatory products PGE2 and TXA2 at similar concentrations of selenite indicate a favorable skewing of AA-COX pathway metabolites. This regulation by selenium could be a direct consequence of the change in the redox state of the cell affecting the transcription of many diverse redox-sensitive proteins or an indirect consequence of the activation of signaling pathway(s), which eventually regulates H-PGDS expression. Along these lines, treatment of selenium-supplemented cells with buthionine-sulfoximine (an inhibitor of GSH synthesis) completely abrogated the selenium-dependent increase in 15d-PGJ2 (data not shown). Taken together, our data argue that selenium in the form of selenoproteins appears to be crucial for this effect. Evidence that selenium can modulate PGIS expression in endothelial cells (24) prompted us to look at the regulation of this enzyme in our macrophage model. We found that PGIS is expressed in insignificant quantities, which is consistent with the fact that PGIS is predominantly expressed in endothelial cells and smooth muscle cells (13).
Interestingly, our studies demonstrate that selenium in different forms variably affects the expression of PG synthases. In cell culture models, sodium selenite and MSA are known to make selenium easily bioavailable for the formation of Sec, which is incorporated into selenoproteins during translation (3). However, Se-Met and p-XSC do not release selenium for selenoprotein synthesis as reflected by their inability to increase GPX1 expression above the base-line levels (Fig. 3A). The expression of H-PGDS follows the same pattern as GPX1 for all of these forms of selenium, which suggests that selenoproteins appear to be involved in the up-regulation of H-PGDS expression. Similar observations obtained from primary BMDM at the level of transcripts and end products suggest that the regulation of PG synthases by selenium, in the form of selenoproteins, is not just a feature of immortalized cell lines such as the RAW264.7 macrophage-like cell line. Apart from using genetic methods to knock down the expression of selenoprotein, we have taken the absence of γ-lyase in our in vitro culture system to our advantage to provide support for the role of selenoproteins in AA metabolism. This may not be the case at the organismal level where γ-lyase is expressed mostly in the kidney to metabolize these compounds to contribute, in part, to the anti-carcinogenic properties of such compounds as p-XSC (39, 40). Alternatively, it is also possible that these compounds may target signaling pathways even without involving selenoproteins to impart their beneficial actions. Compounds like Se-Met require an enzymatic reaction involving a γ-lyase to release selenium. Thus, when the macrophages were treated with Se-Met and r-METase, there was a dose-dependent increase in H-PGDS, which continues to support the involvement of selenoproteins to play a key role in the expression of H-PGDS. Furthermore, the genetic knockdown of two critical enzymes in the selenoprotein incorporation pathway, SPS-2 and SectRNA synthetase, also led to the down-regulation of H-PGDS even in the presence of selenium. These results make a compelling case to identify the selenoprotein(s) that are crucial for the regulation of H-PGDS expression and subsequent production of the potent anti-inflammatory mediator 15d-PGJ2. Based on our studies with NF-κB inhibitors, we speculate that changes in the redox status of cells, as a result of the modulation of selenoprotein expression, could likely be the underlying mechanism. The ability of GPX-mimetic ebselen to up-regulate H-PGDS in selenium-deficient macrophages also indicates that redox modulatory pathways are likely to contribute to the shunting of AA. These are very interesting aspects that justify exploration in the future.
The regulation of H-PGDS at the transcriptional level and the already documented finding from our laboratory that selenium supplementation of macrophages leads to an increase in activation of PPARγ (25) mediated by the increased levels of an endogenous PPARγ ligand, 15d-PGJ2, led us to examine the PPARγ-dependent regulation of the murine H-PGDS gene. Our results from the PPARγ gel shift assays clearly indicate that PPARγ binds to the promoter of Hpgds at one or more of the four PPREs, with the PPRE at −1250 bp (PPRE1) being the strongest. Likewise, the up-regulation of H-PGDS by the prototypic PPARγ ligand rosiglitazone, in the absence of selenium, indicates that H-PGDS is indeed a PPARγ-dependent gene. These findings also open an intriguing possibility of feedback regulation of H-PGDS by 15d-PGJ2 in the selenium-supplemented macrophages. It is possible that in addition to PPARγ, PPARβ could also play a critical role in the up-regulation of H-PGDS because PPARβ is highly expressed in macrophages and is known to possess a variety of anti-inflammatory properties (41, 42). However, preliminary promoter analyses for PPARβ-binding sites and the inability of PPARβ-specific synthetic agonists to activate H-PGDS expression suggest that the binding activity is perhaps restricted to PPARγ (data not shown).
Besides being a PPARγ ligand, 15d-PGJ2 has been shown to inhibit the activation of pro-inflammatory genes by covalently binding specific cysteine residues in IKKβ, a pivotal enzyme in the canonical pathway of NF-κB activation (43). Thus, in selenium-supplemented macrophages, the up-regulation of H-PGDS leads to the enhanced production of 15d-PGJ2, which not only acts in a positive feedback loop to up-regulate H-PGDS transcription in a PPARγ-dependent manner, but also inhibits NF-κB target genes, including mPGES-1 and TXAS enzymes, in LPS-stimulated selenium-supplemented macrophages. To provide more evidence for the shunting of the PG synthesis pathway, we used parthenolide (PTN), a well known NF-κB inhibitor, to inhibit mPGES-1 and TXAS expression to create an increase in the availability of PGH2 for H-PGDS. Alternatively, inhibition of mPGES-1 by CAY10526 also mimicked the effect of PTN with respect to increased H-PGDS expression and subsequent Δ12-PGJ2 and 15d-PGJ2 production. Taken together, the subsequent up-regulation of H-PGDS under selenium-deficient conditions strongly supports the possibility of a feed-forward loop involving H-PGDS → 15d-PGJ2 → PPARγ → H-PGDS (Fig. 9).
FIGURE 9.
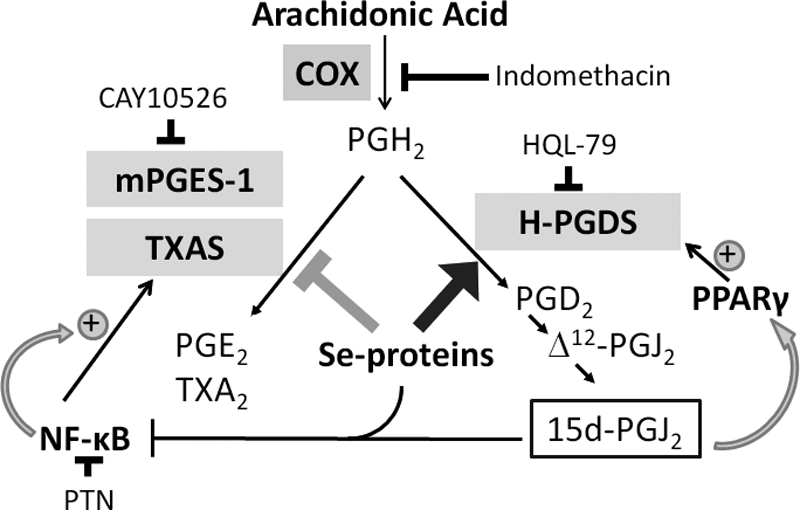
Schematic representation of the selenoprotein-dependent shunting of AA metabolism in macrophages. Treatment of macrophages with bioavailable selenium shunts the pathway of AA metabolism to produce higher levels of PGJ2 metabolites, while lowering the levels of PGE2 and TXA2 via the differential regulation of the respective synthases. CAY10526, PTN, and HQL-79 represent inhibitors of mPGES-1, NF-κB, and H-PGDS, respectively.
In summary, our studies demonstrate a critical role for selenium in anti-inflammation that also has a bearing on the protection from malignant transformation of WBCs. This is particularly relevant because macrophages are implicated in both the initiation and resolution of inflammation. Thus, it is important to understand how exactly their functions are so tightly regulated. Although the initial phases of inflammation involve increases in levels of pro-inflammatory mediators like PGE2 and TXA2, a switch toward the pro-resolving and anti-inflammatory mediators like 15d-PGJ2 during the latter stages suggests the presence of a critical regulator being involved. As seen with the increased activity of arginase in macrophages treated with selenium, shunting of the AA pathway, particularly in macrophages, may have many implications, the most notable being the switch from classically activated “M1” macrophage to alternatively activated “M2” macrophage phenotypes that have wound-healing and resolving properties in addition to affecting the progression of FV leukemia. Such studies delineating the underlying mechanisms centered on the common theme of “lipid class” switching by micronutrient selenium are being carried out in the laboratory and will be reported in the near future.
Acknowledgments
We thank the past and present members of the Prabhu laboratory for their assistance, Kayleigh McCormick for lipid extractions from cells, Dr. Shantu Amin (Penn State University College of Medicine) for providing p-XSC, and Dr. Dolph Hatfield (National Cancer Institute, National Institutes of Health) for providing us the shRNA construct for SPS2.
This work was supported, in whole or in part, by National Institutes of Health Grants DK077152 and AT004350. This work was also supported by the American Institute for Cancer Research.
- Sec
- selenocysteine
- H-PGDS
- hematopoietic prostaglandin D2 synthase
- TXAS
- thromboxane A2 synthase
- mPGES-1
- microsomal prostaglandin E2 synthase
- PGIS
- prostacyclin (prostaglandin I2) synthase
- GPX
- glutathione peroxidase
- AA
- arachidonic acid
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- SPS2
- selenophosphate synthetase 2
- NF-κB
- nuclear factor-κB
- PPAR
- peroxisome proliferator-activated receptor
- PPRE
- peroxisome proliferator response element
- MSA
- methylseleninic acid
- Se-Met
- l-selenomethionine
- p-XSC
- 1,4-phenylenebis(methylene) selenocyanate
- FV
- Friend virus
- BMDM
- bone marrow-derived macrophage
- PTN
- parthenolide
- cPPRE
- consensus PPRE
- TXA2
- thromboxane A2.
REFERENCES
- 1. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., Gladyshev V. N. (2003) Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 2. Rayman M. P. (2000) Lancet 356, 233–241 [DOI] [PubMed] [Google Scholar]
- 3. Bellinger F. P., Raman A. V., Reeves M. A., Berry M. J. (2009) Biochem. J. 422, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen J., Berry M. J. (2003) J. Neurochem. 86, 1–12 [DOI] [PubMed] [Google Scholar]
- 5. McKenzie R. C., Rafferty T. S., Beckett G. J. (1998) Immunol. Today 19, 342–345 [DOI] [PubMed] [Google Scholar]
- 6. Jackson M. I., Combs G. F., Jr. (2008) Curr. Opin. Clin. Nutr. Metab. Care 11, 718–726 [DOI] [PubMed] [Google Scholar]
- 7. Gärtner R., Gasnier B. C. (2003) Biofactors 19, 165–170 [DOI] [PubMed] [Google Scholar]
- 8. Forceville X., Aouizerate P., Guizard M. (2001) Therapie 56, 653–661 [PubMed] [Google Scholar]
- 9. Gartner R., Angstwurm M. (1999) Med. Klin. 94, Suppl. 3, 54–57 [DOI] [PubMed] [Google Scholar]
- 10. Forman H. J., Torres M. (2001) Mol. Aspects Med. 22, 189–216 [DOI] [PubMed] [Google Scholar]
- 11. Ma J., Chen T., Mandelin J., Ceponis A., Miller N. E., Hukkanen M., Ma G. F., Konttinen Y. T. (2003) Cell. Mol. Life Sci. 60, 2334–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Osterud B., Bjorklid E. (2003) Physiol. Rev. 83, 1069–1112 [DOI] [PubMed] [Google Scholar]
- 13. Smith W. L., DeWitt D. L., Allen M. L. (1983) J. Biol. Chem. 258, 5922–5926 [PubMed] [Google Scholar]
- 14. Stachowska E., Dolegowska B., Dziedziejko V., Rybicka M., Kaczmarczyk M., Bober J., Rac M., Machalinski B., Chlubek D. (2009) J. Physiol. Pharmacol. 60, 77–85 [PubMed] [Google Scholar]
- 15. Catley M. C., Chivers J. E., Cambridge L. M., Holden N., Slater D. M., Staples K. J., Bergmann M. W., Loser P., Barnes P. J., Newton R. (2003) FEBS Lett. 547, 75–79 [DOI] [PubMed] [Google Scholar]
- 16. Prabhu K. S., Zamamiri-Davis F., Stewart J. B., Thompson J. T., Sordillo L. M., Reddy C. C. (2002) Biochem. J. 366, 203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zamamiri-Davis F., Lu Y., Thompson J. T., Prabhu K. S., Reddy P. V., Sordillo L. M., Reddy C. C. (2002) Free Radic. Biol. Med. 32, 890–897 [DOI] [PubMed] [Google Scholar]
- 18. Vunta H., Belda B. J., Arner R. J., Channa Reddy C., Vanden Heuvel J. P., Sandeep Prabhu K. (2008) Mol. Nutr. Food Res. 52, 1316–1323 [DOI] [PubMed] [Google Scholar]
- 19. Rajakariar R., Hilliard M., Lawrence T., Trivedi S., Colville-Nash P., Bellingan G., Fitzgerald D., Yaqoob M. M., Gilroy D. W. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 20979–20984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herlong J. L., Scott T. R. (2006) Immunol. Lett. 102, 121–131 [DOI] [PubMed] [Google Scholar]
- 21. Kim E. H., Surh Y. J. (2006) Biochem. Pharmacol. 72, 1516–1528 [DOI] [PubMed] [Google Scholar]
- 22. Maddipati K. R., Marnett L. J. (1987) J. Biol. Chem. 262, 17398–17403 [PubMed] [Google Scholar]
- 23. Poli G., Leonarduzzi G., Biasi F., Chiarpotto E. (2004) Curr. Med. Chem. 11, 1163–1182 [DOI] [PubMed] [Google Scholar]
- 24. Cao Y. Z., Reddy C. C., Sordillo L. M. (2000) Free Radic. Biol. Med. 28, 381–389 [DOI] [PubMed] [Google Scholar]
- 25. Vunta H., Davis F., Palempalli U. D., Bhat D., Arner R. J., Thompson J. T., Peterson D. G., Reddy C. C., Prabhu K. S. (2007) J. Biol. Chem. 282, 17964–17973 [DOI] [PubMed] [Google Scholar]
- 26. Lee C. R., Cervi D., Truong A. H., Li Y. J., Sarkar A., Ben-David Y. (2003) Anticancer Res. 23, 2159–2166 [PubMed] [Google Scholar]
- 27. Subramanian A., Hegde S., Porayette P., Yon M., Hankey P., Paulson R. F. (2008) J. Virol. 82, 382–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Persons D. A., Paulson R. F., Loyd M. R., Herley M. T., Bodner S. M., Bernstein A., Correll P. H., Ney P. A. (1999) Nat. Genet. 23, 159–165 [DOI] [PubMed] [Google Scholar]
- 29. Hashimoto K., Sheller J. R., Morrow J. D., Collins R. D., Goleniewska K., O'Neal J., Zhou W., Ji S., Mitchell D. B., Graham B. S., Peebles R. S., Jr. (2005) J. Immunol. 174, 525–532 [DOI] [PubMed] [Google Scholar]
- 30. Paglia D. E., Valentine W. N. (1967) J. Lab. Clin. Med. 70, 158–169 [PubMed] [Google Scholar]
- 31. Corraliza I. M., Campo M. L., Soler G., Modolell M. (1994) J. Immunol. Methods 174, 231–235 [DOI] [PubMed] [Google Scholar]
- 32. Livak K. J., Schmittgen T. D. (2001) Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 33. Small-Howard A. L., Berry M. J. (2005) Biochem. Soc. Trans. 33, 1493–1497 [DOI] [PubMed] [Google Scholar]
- 34. Nakamura Y., Feng Q., Kumagai T., Torikai K., Ohigashi H., Osawa T., Noguchi N., Niki E., Uchida K. (2002) J. Biol. Chem. 277, 2687–2694 [DOI] [PubMed] [Google Scholar]
- 35. Gallardo-Soler A., Gómez-Nieto C., Campo M. L., Marathe C., Tontonoz P., Castrillo A., Corraliza I. (2008) Mol. Endocrinol. 22, 1394–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rouzer C. A., Ivanova P. T., Byrne M. O., Milne S. B., Marnett L. J., Brown H. A. (2006) Biochemistry 45, 14795–14808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maxey K. M., Hessler E., MacDonald J., Hitchingham L. (2000) Prostaglandins Other Lipid Mediat. 62, 15–21 [DOI] [PubMed] [Google Scholar]
- 38. Yamaguchi S., Aldini G., Ito S., Morishita N., Shibata T., Vistoli G., Carini M., Uchida K. (2010) J. Am. Chem. Soc. 132, 824–832 [DOI] [PubMed] [Google Scholar]
- 39. Brigelius-Flohé R. (2008) Chem. Biodivers. 5, 389–395 [DOI] [PubMed] [Google Scholar]
- 40. Ip C. (1998) J. Nutr. 128, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 41. Lee C. H., Chawla A., Urbiztondo N., Liao D., Boisvert W. A., Evans R. M., Curtiss L. K. (2003) Science 302, 453–457 [DOI] [PubMed] [Google Scholar]
- 42. Coll T., Rodrïguez-Calvo R., Barroso E., Serrano L., Eyre E., Palomer X., Vázquez-Carrera M. (2009) Curr. Mol. Pharmacol. 2, 46–55 [DOI] [PubMed] [Google Scholar]
- 43. Straus D. S., Pascual G., Li M., Welch J. S., Ricote M., Hsiang C. H., Sengchanthalangsy L. L., Ghosh G., Glass C. K. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 4844–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]