

Abstract
A novel cell-targeting, pH-sensitive polymeric carrier was employed in this study for delivery of the anticancer drug bortezomib (BTZ) to cancer cells. Our strategy is based on facile conjugation of BTZ to catechol-containing polymeric carriers that are designed to be taken up selectively by cancer cells through cell surface receptor-mediated mechanisms. The polymer used as a building block in this study was poly(ethylene glycol), which was chosen for its ability to reduce nonspecific interactions with proteins and cells. The catechol moiety was exploited for its ability to bind and release borate-containing therapeutics such as BTZ in a pH-dependent manner. In acidic environments, such as in cancer tissue or the subcellular endosome, BTZ dissociates from the polymer-bound catechol groups to liberate the free drug, which inhibits proteasome function. A cancer-cell-targeting ligand, biotin, was presented on the polymer carriers to facilitate targeted entry of drug-loaded polymer carriers into cancer cells. Our study demonstrated that the cancer-targeting drug–polymer conjugates dramatically enhanced cellular uptake, proteasome inhibition, and cytotoxicity toward breast carcinoma cells in comparison with nontargeting drug–polymer conjugates. The pH-sensitive catechol–boronate binding mechanism provides a chemoselective approach for controlling the release of BTZ in targeted cancer cells, establishing a concept that may be applied in the future toward other boronic acid-containing therapeutics to treat a broad range of diseases.
Targeted drug delivery systems aim to improve the efficacy and reduce the toxicity of potent therapeutics in treatments for a variety of diseases. For example, chemotherapy is one of the major systemic treatments for cancer, but it is linked to serious side effects because of its toxicity to normal proliferating cells. Cancer-targeting delivery systems have been developed to alter the biodistribution of drugs, aiming to achieve drug accumulation in cancer tissue through the enhanced permeability and retention (EPR) effect and/or targeting of the cancer cell surface.(1) Advanced delivery systems for anticancer drugs often utilize environmentally triggered mechanisms.2−4 For example, the extracellular pH in tumor tissue is slightly lower than that in normal tissue, and this has been exploited to accomplish pH-triggered drug release in tumor tissue.(5) In addition, the significantly increased acidity in subcellular compartments such as the endosome also offers a complementary route to increases in the efficacy of anticancer drug delivery via pH-initiated release of drugs from endocytosed drug carriers.5,6
Here we describe a novel polymer conjugate of the anticancer drug bortezomib (BTZ) that can be used for pH-sensitive delivery to specific cancer cells. BTZ is a dipeptide boronic acid analogue that inhibits cancer cell proteasome through direct binding between its boronic acid group and threonine residues in the active sites of several proteases.7,8 This drug is currently marketed as Velcade (Millenium Pharmaceuticals) and has been approved for multiple myeloma and mantle cell lymphoma treatment; however, BTZ is less active against many solid tumors and has significant dose-limiting toxicity.9,10 In addition to the possibility that different cancer cells may have distinct proteasome sensitivity to BTZ,11,12 the unfavorable pharmacokinetic properties of BTZ, including nonspecific binding to proteins and rapid hepatic clearance from blood, may also contribute to its limited efficacy against solid tumors.13,14 This suggests that polymeric delivery vehicles capable of enhancing the pharmacokinetic properties of BTZ may improve its activity against solid tumors. Moreover, strategies to direct BTZ to cancer cells by specific cell surface recognition epitopes and/or trigger pH-regulated drug release may facilitate drug delivery and release in tumors, thereby potentially increasing the efficacy and reducing the toxicity of BTZ.
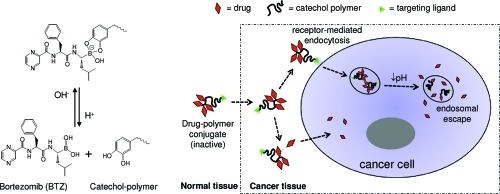
It has been previously reported that polyphenols can chemically block the proteasome-inhibiting activity of BTZ through conjugation to the boronic acid active site and that the formed conjugates cannot permeate freely through the cell membrane.15−18 Our design takes advantage of the facile conjugation of BTZ to the 1,2-benzenediol (catechol) moiety to form a membrane-impermeable compound in order to reduce nonselective cellular uptake of the drug. An important characteristic of the boronic acid–catechol conjugate is that it is formed through dynamic covalent chemistry that is reversible in a pH-sensitive manner:(19) at neutral or alkaline pH, BTZ and catechol form a stable boronate ester, which deactivates the cytotoxicity of BTZ; in a low-pH environment, the BTZ–catechol ester conjugate readily dissociates to release free BTZ and catechol groups. This mechanism has the potential to be exploited in two contexts: (1) extracellularly for localized drug release within the mildly acidic tumor interstitium and (2) intracellularly in the more acidic endosomes following cell surface receptor-mediated endocytosis. In both cases, the acidic environment would result in dissociation of BTZ from the polymer-bound catechol groups, activating its proteasome-inhibiting function (Figure 1).
Figure 1.

pH-sensitive polymer–drug conjugates for delivering BTZ selectively into cancer cells. (A) The catechol and the boronic acid structure in BTZ form a stable, covalently-bonded, inactive conjugate at neutral and alkaline pH, but this structure dissociates in acidic environments to release the free active drug. (B) The catechol polymer–BTZ conjugate may dissociate in response to a mildly acidic cancer tissue microenvironment to liberate the free drug, which can be taken up by cancer cells. Alternatively, through the use of a targeting ligand, the catechol polymer–BTZ conjugate may be transported intact into the cell via receptor-mediated endocytosis, whereupon the acidic environment of the endosome induces intracellular drug release.
We first characterized the pH-dependent reversible binding between BTZ and catechol compounds using 1H NMR spectroscopy (Figure 2A). At pH 5.5, a mixture of BTZ and dopamine (DA) produced a spectrum similar to that of the two individual compounds superimposed upon each other, indicating the presence of uncomplexed BTZ and DA. However, the same mixture at pH 7.4 gave a spectrum with striking changes in chemical shifts and peak splittings, revealing the formation of a BTZ–DA conjugate. To investigate further the pH sensitivity of BTZ–catechol dissociation, preformed BTZ–DA conjugate [0.1 M in deuterated dimethyl sulfoxide (DMSO-d6)] was diluted to 1 mM in deuterated phosphate buffer at pH 5.5–8.5 and then analyzed by NMR spectroscopy (Figure SI 1 in the Supporting Information). The peak integrals in the ranges 7.0–7.2, 6.6–6.8, and 5.5–6.5 ppm, corresponding to the H atoms on the phenyl ring of BTZ, the benzene ring of free DA, and the benzene of the BTZ–DA complex, respectively, were used to estimate the degree of BTZ–DA complexation. As shown in Figure 2B, dissociation of the BTZ–DA complex increased as the pH decreased from 8.5 to 6.5, and the BTZ–DA complex was undetectable at pH 5.5.
Figure 2.

pH-dependent interactions between BTZ and DA. (A) 1H NMR spectra of BTZ and DA in deuterated PBS at pH 5.5 and 7.4. DA was chosen as a representative catechol-containing model compound whose conjugate with BTZ is fully soluble in aqueous solutions. Circled in red are proton signals resulting from the formation of a stable BTZ–DA conjugate at pH 7.4, which are not present at pH 5.5. (B) Peak integrals in the pH ranges 7.0–7.2, 6.6–6.8, and 5.5–6.5 ppm were used to estimate the degree of BTZ–DA binding at pH 5.5–8.5.
The pH dependence of the BTZ–catechol complex suggested that polymer constructs containing catechols may be useful for pH-triggered release of BTZ. We therefore synthesized several BTZ–polymer constructs as pH-responsive carriers of BTZ to cancer cells (Scheme 1). We employed a modular heterobifunctional polymer design consisting of a poly(ethylene glycol) (PEG) polymer derivatized with one or more catechols and a cell-targeting moiety. PEG is well-known to reduce nonspecific interactions of therapeutic molecules with proteins and cells and therefore has been widely applied in the form of drug–PEG conjugates that exhibit reduced drug degradation and extended drug circulation time.(20) Here we used biotin as a cell-targeting ligand because previous reports have shown that biotinylated polymers can be selectively taken up by cancer cells.21,22 Although the biotin receptors/transporters on cancer cell surfaces have not been well-characterized, biotin has a relatively simple structure and thus can also be used as a tag for cellular tracking of polymer drug carriers, making this molecule an attractive choice for demonstrating cancer cell targeting. We used fluorescence microscopy to examine the uptake of fluorescein isothiocyanate (FITC)-labeled biotinylated PEG (biotin–PEG–FITC) into two breast cancer cell lines, MDA-MB-231 cells and MCF-10A-H-RasV12 cells, as well as the nontransformed or noncancerous breast epithelial cell line MCF-10A-Vector.(23) Figure 3 shows that biotin–PEG–FITC was rapidly taken up by cells, whereas the cellular uptake of PEG–FITC without the biotin moiety was minimal. The uptake of biotinlylated polymers was inhibited by free biotin in a dose-dependent manner (Figure SI 3), demonstrating that polymer uptake by cells is mediated by the cell surface receptor for biotin. Notably, the inhibition of uptake of biotinylated polymer by free biotin was less robust in cancer cell lines (MDA-MB-231 and MCF-10A-H-RasV12) than in noncancer MCF-10A-Vector cells, suggesting greater expression levels of biotin receptors in cancer cells than in noncancer cells.
Scheme 1. Chemical Structures of Polymers 1–5.

Figure 3.

Selective uptake of fluorescent FITC-terminated PEG molecules by cancer cells (MDA-MB-231 and MCF-10A-H-RasV12) and noncancerous cells (MCF-10A-Vector). All cells showed robust uptake of biotin–PEG–FITC but little uptake of PEG–FITC. Free biotin (0.1 μM) in the culture medium inhibited uptake of biotin–PEG–FITC by noncancerous cells and, to a lesser extent, by the two cancer cell lines, indicating that the cellular entry of FITC-terminated polymers is mediated by biotin receptors on the cell surface and that the expression of these receptors on cancer cell surfaces is very likely higher than on noncancerous cell surfaces.
After confirming the cancer-targeting function of the biotin ligand, we synthesized PEG polymers containing four catechol groups each (BPC and PC; Scheme 1) in order to take advantage of multiple catechol moieties to increase the drug-loading capacity of the polymeric carrier. Time-dependent release of BTZ from these catechol polymers was carried out in buffered solutions at pH 7.4, 6.5, and 5.0, mimicking the physiological pH in normal tissue and blood, the tumor extracellular environment, and subcellular endosome, respectively. Figure 4 shows that at pH 7.4, ∼30% of the BTZ was released from both BPC and PC over a 12 h period, while >80% of the BTZ was released from these catechol polymers at pH 5.0. This further confirmed the pH-dependent BTZ dissociation from the catechol-presenting polymeric delivery vehicles, which could render BTZ non-cell-permeable and inactive in normal tissues but allow the BTZ activity to be recovered in cancer tissue, where the acidity increases.
Figure 4.
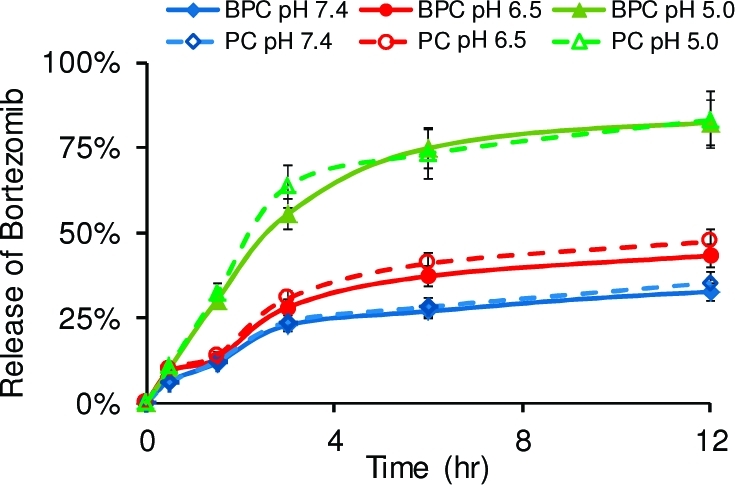
Time-dependent release of BTZ from the catechol polymers BPC and PC at pH 5.0, 6.5, and 7.4. The fraction of BTZ released was 30% from both polymers at pH 7.4 over 12 h, while >80% of the BTZ was released at pH 5.0 in the same period of time. Drug–polymer conjugates formed between 0.2 mM BTZ and 0.05 mM BPC or PC were used for all measurements of release in 10 mM PBS buffers at 37 °C.
The effects of polymer–BTZ conjugates on proteasome inhibition and cell viability were first evaluated using MDA-MB-231 breast cancer cells (Figure 5 A,B). The untargeted (no biotin ligand) PC–BTZ conjugate exhibited dramatically decreased proteasome inhibition in comparison to free BTZ, indicating deactivation of BTZ due to formation of the BTZ–catechol polymer conjugate. This result is consistent with the previously reported inhibition of BTZ activity by polyphenols,15,16,18 which we speculate may be due to the inability of BTZ–catechol complexes to enter cells. In stark contrast, the biotinylated conjugate BPC–BTZ retained high proteasome-inhibiting activity. Cell viability assays 48 h after treatment of cells with the polymer–BTZ conjugates demonstrated a similar pattern: the biotin-receptor-targeting BPC–BTZ conjugate maintained fairly high cytotoxicity of BTZ, whereas catechol-containing polymers (BPC and PC) without loaded drug exhibited little proteasome inhibition activity or cytotoxicity. These results suggest that cellular uptake of the polymer–BTZ conjugate through biotin-receptor-mediated transport can restore the BTZ activity within the cells, possibly via internalization of the polymer–BTZ conjugate followed by low-pH-triggered intracellular dissociation of BTZ from the polymer in the endosomes. A detailed investigation of the intracellular events responsible for this effect is in progress. Some preliminary evidence for self-assembly of the polymer–BTZ construct into nanostructures was observed by cryogenic transmission election microscopy (cryo-TEM) (Figure SI 2). However, the significance of these structures in relation to the observed cytotoxic effects requires further study.
Figure 5.

Cytotoxicity of catechol polymer–BTZ conjugates toward breast cancer cells. (A) Proteasome inhibition assays 6 h after treatment of MDA-MB-231 cells with (I) free BTZ, (II) BPC–BTZ, (III) PC–BTZ, (IV) BPC without BTZ, and (V) PC without BTZ. (B) Cell death as determined by fluorescence imaging 48 h after treatment using a commercial cell viability assay kit (calcein AM for live cells and ethidium homodimer-1 for dead cells). (C) IC50 values of BPC–BTZ estimated from MTS assays indicate the enhanced cytotoxicity of BPC–BTZ toward cancer cells relative to noncancer cells.
To evaluate further the selectivity of the polymer–drug conjugates for cancer cells over normal cells, we treated immortalized human mammary epithelial cells (MCF-10A-Vector cells, a noncancer control) and transformed MCF-10A-H-RasV12 cells with BPC–BTZ (0, 10, and 25 nM). We found that BPC loaded with BTZ at 25 nM concentration induced 1.7-fold increased cell death in the cancer cell line relative to the control cells (Figure SI 5). Calculated IC50 values for cell growth inhibition by BPC–BTZ and free BTZ revealed that cancer cells were 2.2-fold more sensitive to BPC–BTZ than noncancer cells, while both cell types were equally sensitive to free BTZ (Figure 5C). This result suggests that cancer cells are more susceptible to BPC–BTZ toxicity than noncancer cells, a major potential therapeutic advantage of our targeted polymeric carrier platform.
This work has demonstrated targeted delivery of an anticancer drug to cancer cells using catechol-presenting polymers. These polymeric drug carriers have a chemically defined mechanism for drug loading and release through the pH-sensitive catechol–boronic acid interaction. With the use of biotin to target cancer cells, the proteasome inhibitor bortezomib delivered by such polymeric carriers showed enhanced cytotoxicity against breast cancer cells compared to noncancer cells in vitro. Variation of the catechol polymer architecture and composition and the choice of boronic acid-containing therapeutic can potentially provide great tailorability of catechol polymer–boronic acid complexes for a variety of drug delivery applications. For instance, alternative targeting ligands can be exploited to alter the tissue biodistribution, and modifications of the chemical structure of the catechol (or, more generally, diol groups) can be exploited to fine-tune the pH sensitivity of the diol–boronic acid complex for drug release in different tissue environments.24,25 Since the boronic acid structure exisits in many potent therapeutics,26−28 this pH-sensitive strategy may provide a versatile, chemoselective approach for targeted drug delivery to diseased tissues resulting from a broad spectrum of disorders.
Acknowledgments
This work was supported by NIH Grant DE014193, the NCI Center of Cancer Nanotechnology Excellence at Northwestern University, and the Breast Cancer Research Foundation. We are grateful to Dr. Jinsong Wu of the Electron Probe Instrumentation Center at Northwestern University for help with cryo-TEM analysis.
Supporting Information Available
Synthesis of catechol-presenting polymers, preparation of BTZ–polymer conjugates, NMR study, drug release measurement, and cytotoxicity assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kwon G. S.; Kataoka K. Adv. Drug Delivery Rev. 1995, 16, 295–309. [Google Scholar]
- Duncan R. Nat. Rev. Cancer 2006, 6, 688–701. [DOI] [PubMed] [Google Scholar]
- Rapoport N. Prog. Polym. Sci. 2007, 32, 962–990. [Google Scholar]
- MacEwan S. R.; Callahan D. J.; Chilkoti A. Nanomedicine (London) 2010, 5, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. S.; Gao Z.; Bae Y. H. J. Controlled Release 2008, 132, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P.; Lukyanov A. N. Drug Discovery Today 2003, 8, 259–266. [DOI] [PubMed] [Google Scholar]
- Adams J.; Behnke M.; Chen S.; Cruickshank A. A.; Dick L. R.; Grenier L.; Klunder J. M.; Ma Y.-T.; Plamondon L.; Stein R. L. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [DOI] [PubMed] [Google Scholar]
- Groll M.; Berkers C. R.; Ploegh H. L.; Ovaa H. Structure 2006, 14, 451–456. [DOI] [PubMed] [Google Scholar]
- Kane R. C.; Farrell A. T.; Sridhara R.; Pazdur R. Clin. Cancer Res. 2006, 12, 2955–2960. [DOI] [PubMed] [Google Scholar]
- Russo A.; Bronte G.; Fulfaro F.; Cicero G.; Adamo V.; Gebbia N.; Rizzo S. Curr. Cancer Drug Targets 2010, 10, 55–67. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Ricker J. L.; Malhotra P. S.; Nottingham L.; Bagain L.; Lee T. L.; Yeh N. T.; Van Waes C. Mol. Cancer Ther. 2008, 7, 1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri B.; Miknyoczki S.; Dorsey B.; Hui A.-M. Adv. Pharmacol. 2009, 57, 91–135. [DOI] [PubMed] [Google Scholar]
- Voorhees P. M.; Dees E. C.; O’Neil B.; Orlowski R. Z. Clin. Cancer Res. 2003, 9, 6316–6325. [PubMed] [Google Scholar]
- Pekol T.; Daniels J. S.; Labutti J.; Parsons I.; Nix D.; Baronas E.; Hsieh F.; Gan L.-S.; Miwa G. Drug Metab. Dispos. 2005, 33, 771–777. [DOI] [PubMed] [Google Scholar]
- Fernández Y.; Miller T. P.; Denoyelle C.; Esteban J. A.; Tang W.-H.; Bengston A. L.; Soengas M. S. J. Biol. Chem. 2006, 281, 1107–1118. [DOI] [PubMed] [Google Scholar]
- Liu F. T.; Agrawal S. G.; Movasaghi Z.; Wyatt P. B.; Rehman I. U.; Gribben J. G.; Newland A. C.; Jia L. Blood 2008, 112, 3835–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W.; Yue P.; Lin N.; He M.; Zhou Z. M.; Lonial S.; Khuri F. R.; Wang B. H.; Sun S. Y. Clin. Cancer Res. 2006, 12, 273–280. [DOI] [PubMed] [Google Scholar]
- Golden E. B.; Lam P. Y.; Kardosh A.; Gaffney K. J.; Cadenas E.; Louie S. G.; Petasis N. A.; Chen T. C.; Schonthal A. H. Blood 2009, 113, 5927–5937. [DOI] [PubMed] [Google Scholar]
- Rowan S. J.; Cantrill S. J.; Cousins G. R. L.; Sanders J. K. M.; Stoddart J. F. Angew. Chem., Int. Ed. 2002, 41, 898–952. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Pasut G. Drug Discovery Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- Yang W.; Cheng Y.; Xu T.; Wang X.; Wen L.-P. Eur. J. Med. Chem. 2009, 44, 862–868. [DOI] [PubMed] [Google Scholar]
- Yellepeddi V. K.; Kumar A.; Palakurthi S. Anticancer Res. 2009, 29, 2933–2943. [PubMed] [Google Scholar]
- Moyano J. V.; Evans J. R.; Chen F.; Lu M. L.; Werner M. E.; Yehiely F.; Diaz L. K.; Turbin D.; Karaca G.; Wiley E.; Nielsen T. O.; Perou C. M.; Cryns V. L. J. Clin. Invest. 2006, 116, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M. C.; Hanson M. C.; Massey A. P.; Karren E. A.; Kiser P. F. Adv. Mater. 2007, 19, 2503–2507. [Google Scholar]
- Yan J.; Springsteen G.; Deeter S.; Wang B. H. Tetrahedron 2004, 60, 11205–11209. [Google Scholar]
- Yang W.; Gao X.; Wang B. Med. Res. Rev. 2003, 23, 346–368. [DOI] [PubMed] [Google Scholar]
- Minkkilä A.; Saario S. M.; Käsnänen H.; Leppänen J.; Poso A.; Nevalainen T. J. Med. Chem. 2008, 51, 7057–7060. [DOI] [PubMed] [Google Scholar]
- Kong Y.; Grembecka J.; Edler M. C.; Hamel E.; Mooberry S. L.; Sabat M.; Rieger J.; Brown M. L. Chem. Biol. 2005, 12, 1007–1014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.