

Abstract
Background
A molecular characterization of Alzheimer's Disease (AD) is the key to the identification of altered gene sets that lead to AD progression. We rely on the assumption that candidate marker genes for a given disease belong to specific pathogenic pathways, and we aim at unveiling those pathways stable across tissues, treatments and measurement systems. In this context, we analyzed three heterogeneous datasets, two microarray gene expression sets and one protein abundance set, applying a recently proposed feature selection method based on regularization.
Results
For each dataset we identified a signature that was successively evaluated both from the computational and functional characterization viewpoints, estimating the classification error and retrieving the most relevant biological knowledge from different repositories. Each signature includes genes already known to be related to AD and genes that are likely to be involved in the pathogenesis or in the disease progression. The integrated analysis revealed a meaningful overlap at the functional level.
Conclusions
The identification of three gene signatures showing a relevant overlap of pathways and ontologies, increases the likelihood of finding potential marker genes for AD.
Background
Alzheimer's Disease (AD) is a common progressive brain disease generally diagnosed in individuals over 65 years of age and it is mostly characterized by cognition deterioration that causes dementia [1]. Within 3 to 9 years after diagnosis, it usually leads to death.
From the molecular point of view, AD is characterized by many different lesions: the most evident are deposits of beta amyloid and tangles of hyperphosphorylated tau proteins, together with a marked loss of neurons in the neocortex and hippocampus [2,3]. In the early stages, the most common symptom is memory loss, followed by mood swings, difficult in speech, long-memory loss and confusion. Several characteristics of AD are common to normal aging or to other neurological diseases, making its diagnosis very difficult. Usually, psycho-logical tests are used to indicate the presence of the disease, but only a post-mortem exam can confirm it. The diagnostic process is time-consuming and, by the time AD is detected, the disease has been progressing for many years, causing increased brain damages along with the deterioration of cognitive capacities. For these reasons, AD patients need constant care from their relatives or from specialized structures. Clearly, this phenomenon has a relevant economical impact on the national health systems.
Although many scientific papers are published every year, AD is still a very open research topic and its etiology is still unknown. In this context, the mainstream focus is to understand the underlying molecular mechanisms with the ultimate goal of identifying potential biomarkers to be used in the clinical practice.
The basis of our work is the assumption that candidate marker genes for a given disease belong to specific pathogenic pathways. Our aim was to uncover molecular pathways that are stable across tissues, treatments and measurement systems. The identification of these pathways or functional groups across different datasets is fundamental to unveil those that really feed the progression of the disease and that might harbor relevant genes.
We considered AD as a case study and obtained results from the supervised analysis of three publicly available datasets: one that collects the abundance of 120 signaling proteins [4] and two, retrieved from the Gene Expression Omnibus (GEO) database, that store gene expression data from DNA microarray experiments: GSE1297 [5] and GSE5281 [6,7]. The rationale behind [4] is very convincing and motivated our choice: since the brain controls many body functions through the release of signaling proteins in the blood stream, a brain disease like AD could induce unique changes of these proteins in the blood. We chose GSE1297 because it is homogeneous with the protein dataset for the Mini-Mental State Exam (MMSE) parameter (t-test, p-value < 0.01), which is a 30-points questionnaire test that is commonly used to screen for cognitive impairment. Unfortunately, for GSE5281 the MMSE parameter was not available, but we used it anyway because its platform, i.e. Affymetrix HG-U133 Plus2.0, provides a more accurate coverage of the human genome and completely includes the probesets measured with Affymetrix HG-U133A (GSE1297).
Supervised analysis of high-throughput data allows for the identification of lists of genes with good prediction ability. In the remainder of the paper we refer to such lists as signatures. Gene signature analysis is fundamental to discover the most relevant functional classes or biological pathways involved in the progression of disease.
In this work, we adopted a supervised analysis schema: l1l2FS regularization with double optimization framework, set in a nested cross-validation structure (l1l2FS). This method is inspired by [8] and it was recently proven to be robust and very effective for high-throughput data analysis [9]. The statistical accuracy of the system was measured by its prediction error that is the ability of predicting the outcome on future data (see Materials and Methods) [10].
By separately applying l1l2FS to each dataset, we obtained three AD signatures all showing high prediction performances. The small overlap between the two microarray signatures confirmed the necessity to consider more data coming from the same measuring technique and also different kind of data in order to incorporate all the genes that are significantly modulated by the disease.
The analysis was completed by a functional characterization of each signature in the Medical Literature Analysis and Retrieval System Online (Medline) [11], Gene Ontology (GO) [12] and the Kyoto Encyclopedia of Genes and Genomes (KEGG) [13]. This final step identified a functional overlap of ontologies and pathways. Even if the majority of the discriminant genes were different, they were frequently involved in the same KEGG pathways and/or shared similar GO ontologies. Moreover, the presence in each signature of some genes already known to be involved in the disease confirmed the reliability of the method in selecting relevant genes and also increased the likelihood that the remaining selected genes could be involved in the development of AD.
Results
The first purpose of this work was to define significant signatures that are gene or protein lists able to distinguish, with a certain degree of reliability, diseased from control subjects. The second purpose was to test the biological soundness of the genes selected by the adopted statistical method. The third and main goal was to characterize AD at a functional level, identifying those pathways and functions that are stable across heterogeneous data sources.
l1l2FS is a rather novel method for feature selection and classification but it has recently been applied with success in the analysis of data coming from high-throughput techniques [14-17]. We are convinced that the ability of detecting correlated features is the most relevant property of l1l2FS, since correlation is a peculiar and important property characterizing the genes. It is relevant to note that, in this context, the correlation parameter μ in l1l2FS is not a threshold value, but it is a regularization parameter within the naïve elastic net functional (see Materials and Methods section). It allows for detecting correlated genes that contribute to the final outcome in a multivariate fashion.
Our analysis was based on one protein dataset [4] and two microarray datasets [5,6]. The obtained results are presented in this order: the classification error estimated by l1l2FS, a bibliographic (Medline) characterization of the relevant variables (proteins and probesets) in the signature, the results of the WebGestalt enrichment analysis performed in KEGG and GO and the analysis of the significant gene groups identified by the k-means clustering technique. The Medline bibliographic content we considered relevant concerns: the potential role in AD, in other brain diseases, in pathways already known to be related with AD, or the specific expression in some brain regions. Additional data on the enrichment analysis is available in the Additional files section.
Protein data analysis
Results of the l1l2FS
The analysis of the protein dataset consisted in two main phases. As shown in Figure 1, we firstly trained l1l2FS on the Training Set, learning a predictive statistical model and evaluating a cross-validation error. We then assessed the generalization ability of the results on independent datasets (Test Set AD and Test Set MCI - Mild Cognitive Impairment). The algorithm distinguished AD and control samples with a 10-fold cross-validation error of 19%. The presented signature corresponds to the highest value of the correlation parameter μ and it is composed by 21 genes, reported in Table 1. The frequency score associated to each gene indicates its stability (presence) across the lists produced by l1l2FS in the cross-validation procedure.
Figure 1.
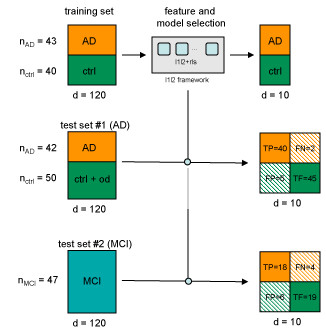
Proteomic Training and Test (AD and MCI). The Training Set is composed by 43 AD samples and 40 NDC samples. The Test Set AD is composed by 42 AD samples and 50 non-AD samples (9 NDC and 11 OD). The Test Set MCI is composed by 47 samples diagnosed with a mild cognitive impairment and with known final follow-up (2-6 years). The Training Set is fed as input to the l1l2FS framework (grey box), which splits the data in K subsplits (light blue boxes), evaluating the relevant variables and the classification error for each one. Performance of the classifier is then tested on the Test Set AD and Test Set MCI using only the 21 proteins selected in the training phase. The test error is decomposed in True Positives (TP), False Negatives (FN), False Positives (FP), True Negatives (TN).
Table 1.
Table of protein signature
| Gene symbol (d) | Official Gene Symbol | Entrez ID | Frequency(%) |
|---|---|---|---|
| EGF_1 | EGF | 1950 | 100 |
| PDGF-BB_1 | PDGFB | 5155 | 100 |
| RANTES_1 | CCL5 | 6352 | 100 |
| TNFα | TNFα | 7124 | 100 |
| GCSF_1 | CSF3 | 1440 | 100 |
| ICAM-1_1 | ICAM1 | 3383 | 100 |
| IL-1α | IL-1α | 3552 | 90 |
| M-CSF_1 | CSF1 | 1435 | 90 |
| PARC_1 | CCL18 | 6362 | 80 |
| Acrp30_1 | ADIPOQ | 9370 | 70 |
| ANG-2_1 | ANGPT2 | 285 | 70 |
| IL-8_1 | IL8 | 3576 | 60 |
| IL-3_1 | IL3 | 3562 | 60 |
| IL-11_1 | IL11 | 3589 | 60 |
| IL-6 R_1 | IL6R | 3570 | 50 |
| IGFBP-6_1 | IGFBP6 | 3489 | 50 |
| MSP-a_1 | MST1 | 4485 | 50 |
| TRAIL R3_1 | TNFRSF10C | 8794 | 40 |
| ANG_1 | ANGTP1 | 284 | 40 |
| AgRP(ART)_1 | AGRP | 181 | 40 |
| TRAIL R4_1 | TNFRSF10D | 8793 | 40 |
The 21 gene signature identified by l1l2FS. The Gene symbol column reports the corresponding genes following the same nomenclature used by [4]. The Official Gene Symbol column shows the nomenclature of Entrez-Gene and the Frequency column shows the cross-validation frequency scores.
Following Ray et al. [4] and Ravetti and Moscato [18], we used the Test Set AD and the Test Set MCI to verify the predicting ability of our signature.
Ray et al. adopted a shrunken centroid algorithm and identified 18 predictors characterizing AD status. Similarly, Ravetti and Moscato considered the dataset and applied more than 20 different classifiers to achieve a highly predictive 5-protein signature.
After the feature selection step, for each test set the test phase consisted in extracting the sub-matrix corresponding to the 21 relevant variables identified in the training phase and in applying the learned model.
The Test Set AD is composed by samples affected by either AD or other dementia and by controls. In this case, our model scored a 7/92 error (see Figure 1), while Ray and co-authors obtained a 10/92 error and Ravetti and Moscato an error of ~ 6/92, averaged over all the methods they applied.
The Test Set MCI is composed by 47 samples corresponding to subjects with MCI as illustrated in Figure 1. In this case, we used the statistical model as a predictor of outcome, considering the conversion to AD as benchmark status (follow-up: 2-6 years from MCI diagnosis). The statistical model scored a 10/47 error, while Ray and co-authors obtained a 9/47 overall error and Ravetti and Moscato achieved an average error of ~ 16/47.
Literature characterization
Table 1 reports the 21 relevant genes identified by l1l2FS, ranked according their stability in terms of the frequency score. Thirteen genes are meaning-fully associated to AD, to other brain diseases or to brain-related processes. The signature completely includes the one of Ravetti and Moscato [18] and almost completely the one presented in [4]. Some genes uniquely belong to our signature: ADIPOQ, MST1, TNFRSF10C, ANGTP1, AGRP and IL6; with the exception of the latter, the other proteins have never been associated to AD. ADIPOQ encodes for the adiponectin protein that circulates in the plasma and it is involved in the metabolic and hormonal processes. This protein is unable to cross the blood-brain barrier but it is able to modify cytokine expression in the brain endothelial cells [19]; the cytokines are known to be involved in AD. ADIPOQ also characterizes the pathogenesis of the insulin resistance [20] that is a common trait of AD patients.
AGRP encodes for a protein homolog to agouti, a murine protein that regulates the hypothalamic control of feeding behavior via melanocortin receptor and/or intracellular calcium regulation. It therefore influences the weight homeostasis. Kim et al. [21] note that AGRP stimulates insulin secretion through calcium release in pancreatic beta cells. Imbalances in insulin and calcium are well-known risk factors for AD.
ANGPT1 and ANGPT2 contribute to the glucose metabolism by interacting with VEGF [22] and they are both indicated to have a prognostic value in adult forms of malignant brain tumors [23].
TNFR10C encodes for a member of the tumor necrosis factor that protects the cells from TRAIL-induced apoptosis. It is regulated by p53 and it is inducible by DNA damage. It is not constitutively expressed in the human brain but its apoptosis-mediating and apoptosis-blocking receptors are found on neurons, astrocytes and oligodendrocytes [24].
The same holds for TNFRSF10D, which is involved in the same KEGG pathways of TNFRSF10C (i.e. apoptosis, cytokine-cytokine receptor interaction, natural killer cell mediated cytotoxicity).
MST1 encodes for the macrophage stimulating 1 factor. MST1, interacting with FOXO1, induces its accumulation in the nucleus leading to cell death, upon withdrawal of growth factors and neuronal activity [25].
Functional analysis of the signature
Table 2 shows the results of gene set enrichment analysis of the signature using KEGG [26].
Table 2.
Table of the functional analysis made in KEGG for protein signature
| KEGG pathway | KEGG category | n. | P |
|---|---|---|---|
| Cytokine-cytokine receptor interaction | 14 | 9.69e-26 | |
| Cell adhesion molecules (CAMs) | Signaling Molecules and Interaction | 1 | 7.59e-2 |
| Neuroactive ligand-receptor interaction | 1 | 1.67e-1 | |
| Hematopoietic cell lineage | 7 | 6.00e-14 | |
| Natural killer cell mediated cytotoxicity | 4 | 9.12e-7 | |
| Toll-like receptor signaling pathway | Immune System | 3 | 2.19e-5 |
| Fc epsilon RI signaling pathway | 2 | 1.01e-3 | |
| Leukocyte transendothelial migration | 2 | 2.29e-3 | |
| T cell receptor signaling pathway | 1 | 5.57e-2 | |
| Apoptosis | Cell Growth and Death | 5 | 1.54e-9 |
| MAPK signaling pathway | 4 | 2.21e-5 | |
| Jak-STAT signaling pathway | Signal Transduction | 4 | 1.90e-6 |
| TGF-beta signaling pathway | 1 | 4.96e-2 | |
| Adipocytokine signaling pathway | Endocrine System | 3 | 1.13e-5 |
| PPAR signaling pathway | 1 | 4.17e-2 | |
| Epithelial cell signaling in Helicobacter pylori infection | Infectious Diseases | 2 | 8.05e-4 |
| Gap junction | Cell Communication | 2 | 1.33e-3 |
| Focal adhesion | 2 | 6.43e-3 | |
| Glioma | Cancers | 2 | 6.66e-4 |
| Pancreatic cancer | 1 | 4.42e-2 | |
| Type I diabetes mellitus | Metabolic Diseases | 2 | 2.96e-4 |
| Type II diabetes mellitus | 2 | 3.26e-4 | |
| Regulation of actin cytoskeleton | Cell Motility | 2 | 6.89e-3 |
Enriched pathways of the protein signature resulting from the WebGestalt functional analysis with KEGG. For each pathway, the table reports the pathway name, its KEGG category, the number of selected genes (n.) and the p-value. The enriched pathways are displayed in boldface.
The selected proteins are especially involved in the Signaling Molecules and Interaction and Immune System categories, but also in processes related to the cell (Cell Growth and Death, Cell Communication, Signal Transduction). These results underline the role of the selected genes within pathways already linked to AD: cytokine-cytokine receptor interaction [2,3,27], hematopoietic cell lineage [2,3], apoptosis [2,3,27], pathways involved in the immune and inflammatory response [2] and pathways related to Metabolic Diseases [28]. We also identified pathways not previously associated with AD: adipocytokine, PPAR signaling pathway, glioma and pancreatic cancer. We extended the functional analysis of our signature applying the gene set enrichment procedure on GO. The results are presented in Additional file 1. The heatmap plot in Figure 2 visualizes the structured signature obtained by l1l2FS and postprocessed by k-means clustering. Such structured representation confirmed that the genes belonging to the same clusters, having highly correlated abundance profiles, are indeed grouped in the same ontologies or biological pathways or they are known to interact. For instance, gene set enrichment in GO showed two gene pairs: GCSF/IL3 in the positive biological processes and IL8/TNFRSF10D in the negative biological processes. The heatmap plot in Figure 2 shows them into two different clusters. The enrichment in KEGG provided additional examples. For instance, EGF and PDGFB were clustered together and they are both involved in several pathways: cytokine-cytokine receptor interaction, MAPK signaling pathway, gap junction, focal adhesion, glioma and regulation of actin cytoskeleton. TNFα and CSF1 show similar abundance profiles and they are included in the hematopoietic cell lineage and cytokine-cytokine signaling pathways. These proteins are also known to interact. Similar examples are: IL3/IL-1α, CSF3/IL-1α, ADIPOQ/AGRP, TNFRSF10C/TNFRS10D.
Figure 2.

Heatmap for the proteomic signature. Heatmap representing the abundance of the 21 selected proteins by l1l2FS. The thick black lines show the gene groups identified with the k-means clustering technique. The samples are divided in two classes: AD and control. The red and green colors represent high and low abundance respectively. For visualization purposes the expressions have been scaled between 0 and 1.
Microarray data analysis - GSE1297
Results of the l1l2FS
The l1l2FS procedure applied to the GSE1297 dataset provided a signature of 12 probesets associated to 11 genes, reported in Table 3. The list corresponds to the highest value of the correlation parameter μ. In this case the 10-fold cross-validation error was 17%. Nine of the 11 genes are related to AD, other neurological disorders or brain functions. It is not straightforward to compare our results to those in [5], because they applied a Pearson correlation test, without providing a classification error.
Table 3.
Table of gene expression signature (GSE1297)
| Gene symbol (d) | Official Gene Symbol | Entrez ID | Frequency(%) |
|---|---|---|---|
| 221728_x_at | XIST | 7503 | 100 |
| 221729_at | COL5A2 | 1290 | 100 |
| 221730_at | COL5A2 | 1290 | 100 |
| 206552_s_at | TAC1 | 6863 | 90 |
| 200800_s_at | HSPA1A | 3303 | 60 |
| 200664_s_at | DNAJB1 | 3337 | 50 |
| 201645_at | TNC | 3371 | 50 |
| 204337_at | RGS4 | 5999 | 50 |
| 212063_at | CD44 | 960 | 50 |
| 213436_at | CNR1 | 1268 | 50 |
| 202018_s_at | LTF | 4057 | 40 |
| 220122_at | MCTP1 | 79772 | 40 |
The 12 probesets signature identified by l1l2FS. The Gene symbol refers to the Affymetrix probeset ID, the Official Gene Symbol shows the nomenclature of Entrez-Gene and the Frequency column shows the cross-validation frequency score.
Literature characterization
XIST encodes for a transcript not translated in a protein. Its main role is the X chromosome inactivation during the early development in mammal females. More recently, one transcription variant of XIST has been found expressed in a subset of neurons as part of a group of gender-specific genes differentially expressed in some brain regions [29].
The literature describes TAC1 as the encoder for many hormones that may function as neurotransmitters, interacting with nerve receptors and smooth muscle cells.
HSPA1A is a heat shock protein of the Hsp70 family. It prevents the subsequent aggregation of misfolded proteins and it is instrumental in targeting them for degradation when the above mechanism fails [30]. The misfolding and the aggregation of proteins are common characteristics of several neurodegenerative diseases including AD and Parkinson's disease, therefore it is very likely that HSPA1A may also characterize AD.
Heat shock protein DNAJB1 has an increased expression in the lymphoblastoid cell lines from patients with bipolar I and II disorders and schizophrenia [31]. It is also known to modulate the activity of the Hsp70 family that leads to the translocation of proteins into the mitochondria and endoplasmatic reticulum thus affecting the functions of these organelles [32].
TNC encodes tenascin-C that contributes to the invasive nature of glioblastoma and the majority of high-grade gliomas [33], to the adhesion of medulloblastoma [34] and to the malignant transformation of plexiform neurofibromas [35].
RGS4 belongs to the regulators of G protein signaling family. A recent paper [36] reports that RGS4 contributes to the regional differences in the coupling of muscarinic M1 receptors in AD. The expression of this gene in the human brain might be spatially and temporally regulated by alternative promoters, resulting in several previously unknown isoforms [37]. This may have implications for the physiological role of RGS4 in some pathologies of the brain.
Besides being involved in the cannabinoid-induced central nervous system (CNS) effects, cannabinoid receptor 1 (CNR1) participates in the development of insulin resistance in the human skeletal muscle and its expression is markedly decreased in AD brains [38].
LTF is a major iron-binding protein in milk and body secretions with an antimicrobial activity. It has several functions, including regulation of iron homeostasis, host defense against a broad range of microbial infections, anti-inflammatory activity, regulation of cellular growth and differentiation. This protein seems to be up-regulated in AD patients [39].
MCTP1 encodes for a protein integral to the cell membrane that is involved in the calcium-mediated signaling process. Indeed, the imbalance of calcium is one of the major AD risk factors.
Functional analysis of the signature
Table 4 and Additional file 2 present the results of the functional characterization performed in KEGG and GO.
Table 4.
Table of the functional analysis made in KEGG for GSE1297 signature
| KEGG pathway | KEGG Category | n. | P |
|---|---|---|---|
| ECM-receptor interaction | Signaling Molecules and Interaction | 3 | 2.39e-6 |
| Neuroactive ligand-receptor interaction | 2 | 3.93e-3 | |
| Cell Communication | Cell Communication | 2 | 5.40e-4 |
| Focal adhesion | 2 | 1.75e-3 | |
| MAPK signaling pathway | Signal Transduction | 1 | 8.51e-2 |
| Antigen processing and presentation | Immune System | 1 | 2.37e-2 |
| Hematopoietic cell lineage | 1 | 2.76e-2 | |
Enriched pathways of the GSE1297 signature resulting from the WebGestalt functional analysis with KEGG. For each pathway, the table reports the pathway name, its KEGG category, the number of selected genes (n.) and the p-value. The enriched pathways are displayed in boldface.
KEGG analysis indicated that these genes are especially involved in the Signaling Molecules and Interaction KEGG category (ECM-receptor-interaction and neuroactive ligand-receptor interaction)and in Cell Communication and Immune System (cell communication, focal adhesion, antigen processing and presentation and hematopoietic cell lineage).
Again, we considered the functional results together with the structured heatmap, identifying those groups of probesets with similar expression profiles also involved in the same pathways or in the same ontologies, see Figure 3. For instance, CNR1, TAC1 and RGS4 are involved in the G-protein coupled receptor protein signaling pathway GO process; CNR1 and TAC1 are involved in the neuroactive ligand-receptor interaction KEGG pathway. CD44 and TNC share the ECM-receptor interaction pathway. The two probesets referring to the same gene, COL5A2, were nicely clustered together in the same group and showed very similar expression profiles. The last pair is composed by HSPA1 and DNAJB1 which have similar expression profiles and belong to the same biological process, i.e. response to stimulus. These proteins are also known to interact [40].
Figure 3.

Heatmap for the GSE1297 signature. Heatmap representing the gene expression of the 12 selected probestes by l1l2FS in the GSE1297 experiment. The thick black lines show the gene groups identified with the k-means clustering technique. The samples are divided in two classes: AD and control. The red and green colors represent high and low expression respectively. For visualization purposes the expressions have been scaled between 0 and 1.
Microarray data analysis - GSE5281
Results of the l1l2FS
Table 5 reports the 39 probesets that formed the second microarray-based signature, associated with the highest value of the correlation parameter μ. In this case, the 5-fold cross-validation error was 5%. Nine of the 39 identified genes are already associated to AD, other brain diseases or brain-related processes. As opposed to the other datasets, the number of well-characterized genes was low compared to the number of selected genes. A comparison of the classification results was not feasible because in [6] the authors used a t-test filtering method, which is not directly associated to a classification error.
Table 5.
Table of gene expression signature (GSE5281)
| Gene symbol (d) | Official Gene Symbol | Entrez ID | Frequency(%) |
|---|---|---|---|
| 202234_s_at | SLC16A1 | 6566 | 100 |
| 205048_s_at | PSPH | 5723 | 100 |
| 212063_at | CD44 | 960 | 100 |
| 213921_at | SST | 6750 | 100 |
| 223380_s_at | LATS2 | 26524 | 100 |
| 230629_s_at | EP400 | 57634 | 100 |
| 231735_s_at | MALAT1 | 378938 | 100 |
| 235987_at | PRKXP1 | 441733 | 100 |
| 204142_at | ENOSF1 | 55556 | 80 |
| 212417_at | SCAMP1 | 9522 | 80 |
| 213872_at | C6orf62 | 81688 | 80 |
| 214246_x_at | MINK1 | 50488 | 80 |
| 217028_at | CXCR4 | 7852 | 80 |
| 220122_at | MCTP1 | 79772 | 80 |
| 220182_at | SLC25A23 | 79085 | 80 |
| 221646_s_at | ZDHHC11 | 79844 | 80 |
| 227413_at | UBLCP1 | 134510 | 80 |
| 228697_at | HINT3 | 135114 | 80 |
| 209116_x_at | HBB | 3043 | 60 |
| 212451_at | SECISBP2L | 9728 | 60 |
| 216834_at | RGS1 | 5996 | 60 |
| 224588_at | XIST | 7503 | 60 |
| 228946_at | INTU | 27152 | 60 |
| 229120_s_at | CDC42SE1 | 56882 | 60 |
| 230748_at | SLC16A6 | 9120 | 60 |
| 1554447_at | LOC554203 | 554203 | 60 |
| 1569110_x_at | LOC728613 | 728613 | 60 |
| 202436_s_at | CYP1B1 | 1545 | 40 |
| 203540_at | GFAP | 2670 | 40 |
| 204338_s_at | RGS4 | 5999 | 40 |
| 206826_at | PMP2 | 5375 | 40 |
| 211959_at | IGFBP5 | 3488 | 40 |
| 213274_s_at | CTSB | 1508 | 40 |
| 213791_at | PENK | 5179 | 40 |
| 214980_at | UBE3A | 7337 | 40 |
| 227062_at | NEAT1 | 283131 | 40 |
| 229676_at | MTPAP | 55149 | 40 |
| 229793_at | ASAH2B | 653308 | 40 |
| 235060_at | LOC100190986 | 100190986 | 40 |
The 39 probesets signature identified by l1l2FS. The Gene symbol refers to the Affymetrix probeset ID, the Official Gene Symbol shows the nomenclature of Entrez-Gene and the Frequency column shows the cross-validation frequency score.
Literature characterization
The somatostatin hormone (SST) is expressed in the body and a ects the rates of neurotransmission [41].
CXCR4, the chemokine receptor 4, is specific for stromal cell-derived factor-1. This protein is known to be highly expressed in the neural precursor cells [42], to be implicated in the inflammation affecting the brain [43] and, like several others chemokines and their receptors, to be implicated in AD [44]. This gene is also critical to the progression of various brain malignancies [45,46].
GFAP, the glial fibrillary acidic protein, encodes one of the major intermediate filament proteins of mature astrocytes. Besides its use as a marker to distinguish astrocytes from other glial cells during development [47,48], GFAP is expressed in the neurons of hippocampus of AD patients [49-51].
CTSB, the lysosomal cysteine proteinase, is known as an amyloid precursor protein (APP) secretase and it is involved in the proteolytic processing of APP. Indeed, the incomplete proteolytic processing of APP is the most known and important causative factor in AD [52].
Enkephalin precursor PENK is a neuropeptide hormone that, together with protachykinin A the precursor of SP, is altered in both dementia and acute neuroinflammatory disorders [53].
Finally, the ubiquintin protein ligase E3A (UBE3A) can be implicated not only in the pathogenesis of Angelman syndrome but also in the neurodegenerative disorders involving protein aggregation [54].
Functional analysis of the signature
Table 6 reports the results of the enrichment analysis in KEGG. We did not find any enriched pathway because the majority of the selected genes are not yet functionally characterized. Nevertheless, some of the involved pathways had a significant p-value (p-value < 0.05) and, among them, those in Neurodegenerative Diseases, Signaling Molecules and Interaction and Immune System are the most significantly connected with AD. The results of GO enrichment analysis are reported in Additional file 3.
Table 6.
Table of the functional analysis made in KEGG for GSE5281 signature
| KEGG pathway | KEGG Category | n. | P |
|---|---|---|---|
| Glycine, serine and thereonine | Amino Acid Metabolism | 1 | 3.20e-2 |
| Tryptophan metabolism | 1 | 5.88e-2 | |
| Metabolism of xenobiotics by cytochrome P450 | Xenobiotics Biodegradation and Metabolism | 1 | 4.23e-2 |
| Neurodegenerative Diseases | Neurodegenerative Diseases | 1 | 2.54e-2 |
| Prion disease | 1 | 9.04e-3 | |
| MAPK signaling pathway | 1 | 1.83e-1 | |
| Cytokine-cytokine receptor interaction | 1 | 1.68e-1 | |
| ECM-receptor interaction | Signaling Molecules and Interaction | 1 | 6.02e-2 |
| Neuroactive ligand-receptor migration | 1 | 1.96e-1 | |
| Axon guidance | Development | 1 | 8.77e-2 |
| Antigen processing and presentation | 1 | 5.31e-2 | |
| Hematopoietic cell lineage | Immune System | 1 | 6.17e-2 |
| Leukocyte transendothelial migration | 1 | 8.14e-2 | |
Enriched pathways of the GSE5281 signature resulting from the WebGestalt functional analysis with KEGG. For each pathway, the table reports the pathway name, its KEGG category, the number of selected genes (n.) and the p-value. The enriched pathways are displayed in boldface.
Integrated functional analysis of the three signatures
First, we proceeded by simply intersecting the three gene signatures. This procedure identified RGS4, MCTP1, CD44 and XIST as common between the two microarray signatures.
Taking a step towards an integrated functional characterization, we compared our results at the functional level, in order to identify common pathways and/or ontologies. The integration followed a late integration schema [55].
The comparison at the pathway level for the KEGG results is shown in Table 7 and Figure 4. Table 7 presents three common pathways and several pathways common between two of the three signatures. Figure 4 illustrates the selected genes and their relation to the aforementioned pathways. This figure summarizes the main result of the integrated analysis, highlighting the common pathways between two signatures, the pathways shared by all signatures and also those known to be related to AD or other brain diseases. The shared pathways were: hematopietic cell lineage, neuroactive ligand-receptor interaction and MAPK signaling pathways.
Table 7.
Table of the integrated functional analysis for the three signatures
| KEGG pathway | KEGG Category | Protein | GSE1297 | GSE5281 |
|---|---|---|---|---|
| Cytokine-cytokine receptor interaction | ● | o | ||
| Neuroactive ligand-receptor migration | Signaling Molecules and Interaction | o | o | o |
| ECM-receptor interaction | ● | o | ||
| Antigen processing and presentation | o | o | ||
| Hematopoietic cell lineage | Immune System | ● | o | o |
| Leukocyte transendothelial migration | o | o | ||
| MAPK signaling pathway | Signal Transduction | ● | o | o |
| Focal adhesion | Cell Communication | o | o | o |
KEGG pathways common between two or three signatures. The table reports the pathway name, its KEGG category and the signatures IDs. The enriched pathways are identified with filled circles, whereas empty circles are associated to not enriched pathways.
Figure 4.

Integrated functional analysis for the three signatures. Relevant KEGG pathways, including: the enriched pathways, those common between two or more signatures and also those related to AD or other brain diseases (in boldface the pathways common to all the signatures). Within each pathway we report the selected genes color-coded according to the signature: purple for the protein dataset, pink for the GSE1297 dataset and dark green for GSE5281 dataset. KEGG categories are displayed in uppercase.
In the GO integrated analysis (see Additional files) it is clear that the annotation for the protein dataset is richer than the other two datasets, hence the overlap takes place at a somewhat high level in the GO hierarchy. For instance, in the BP domain, the overlap reaches down to the cellular process node. If we consider two signatures at a time, we note slightly more specific common nodes. In both the protein and GSE1297 signatures, the following ontologies are enriched: extracellular region and extracellular region part in the CC domain and response to stimulus and response to biotic stimulus for the BP domain. If we consider the protein and GSE5281 signatures, the common enriched nodes are negative regulation of biological process and negative regulation of cellular process.
Discussions
In this section we will clarify the reasons behind the choice of the datasets and we will illustrate more extensively the results in the context of identifying common functional pathways possibly harboring marker genes.
We primarily chose to study heterogeneous datasets to deal with the multifactorial nature of AD. Indeed, AD affects different brain areas [7] with various lesions: the most evident are deposits of beta amyloid and tangles of hyperphosphorylated tau proteins, together with a marked loss of neurons mostly in the neocortex and hippocampus [2,3]. Moreover, AD impacts many molecular levels causing the depletion of neurotrophins and neurotransmitters, dysfunctions affecting the mitochondria, disturbances affecting the metabolism of cholesterol and insulin, inflammation and loss of calcium regulation [3]. Hence, we analyzed data measuring the effects of AD at different molecular levels, i.e. DNA and proteins.
The protein dataset mainly measures the abundance of a specific class of signaling proteins while the two microarray datasets quantify the expression of nearly the entire genome. Nevertheless, we deemed the integrated functional analysis feasible because there exists an overlapping set of measured genes across the three datasets. This assumption was also validated a posteriori, because the number of common pathways between the microarray datasets was comparable with the number of common pathways between the protein dataset and each microarray set (see Table 7). Despite the blood protein data are heavily shifted towards cytokine measurements, the overall results were unbiased.
The integration of results from different datasets corresponds to an in silico validation phase. A consistency assessment of the results across datasets is fundamental in verifying their reliability and in deciding for a further biological validation step [56].
The integrated functional analysis led to very promising results: most of the significant identified genes and pathways are likely related to AD and worth further investigation.
The classification performances of the protein dataset were equivalent or slightly better than those achieved by [4], who provided the dataset, and by [18], who later analyzed it. The l1l2FS protein signature was comparable with those in [4,18], completely including the latter and differing on only three genes with the former. Seven genes were uniquely selected by l1l2FS, namely: IL6R, MST1, TNFRSF10C, ANGTP1, ADIPOQ and AGRP. Aside for IL6R, they have never been associated to AD.
Both ADIPOQ and AGRP are involved in the same KEGG pathway of TNFα, which is a protein considered to be a probable prognostic factor of AD and recently mentioned in several works [2,4,18]. This pathway, namely adipocytokine signaling, also comprises PGC-1α, a protein that is a potential target for treating type II diabetes (it is indeed involved in the regulation of glucose metabolism) and that has been shown to decrease the hyperglycemic-mediated production of beta-amyloid [57]. In addition, the work of Gavrila et al. [58] suggested the use of AGRP as useful peripheral marker of metabolism change. ANGPT1 as well as ANGPT2 encodes for angiopoietins, relevant for vascular development, angiogenesis and lymphogenesis. Both proteins were studied in patients with type II diabetes mellitus and found to be related to VEGF, glycemic control, endothelial damage/dysfunction and atherosclerosis [22]. In particular, ANGPT1 has a role in the pathological vascularization of malignant astrocytomas [59] and the balance between ANGPT1 and ANGPT2 has prognostic value in patients with primary glioblastoma multiforme [23]. The involvement of these proteins in the glucose metabolism as well as in the pathogenesis of brain tumors makes them interesting for further investigation with in vitro techniques.
The functional analysis revealed pathways previously unrelated to AD, e.g. adipocytokine, PPAR signaling pathway and other related to different diseases, such as glioma and pancreatic cancer. Despite having a significant p-value, these three pathways were not enriched, because of the small number of genes belonging to the signature, see Table 2.
The analysis of GSE1297 led to the identification of TAC1. TAC1 encodes for substance P (SP), neurokinin A, neuropeptide K and neuropeptide gamma. SP stimulates human peripheral blood monocytes to produce in ammatory cytokines including IL12, IL6, IL1 and TNFα, all belonging to the protein signature except for the former two. Recently, [53] highlighted the alteration of the highly correlated PENK and SP in both dementia and acute neuroinflammatory disorders. SP is a neuropeptide that is widely distributed in the central and peripheral nervous systems and it has a well-established role as neurotransmitter and as neuroimmunoregulator [60]. SP and its receptor NK1R are involved in inflammation and neurological disorders within the CNS [61,62]. PENK also belonged to the GSE5281 signature. At the functional level, the analysis selected the ECM-receptor-interaction and the neuroactive ligand-receptor interaction pathways, both belonging to the Signaling Molecules and Interaction KEGG category, which is related to AD [4].
In the GSE5281 signature, SST and UBE3A are relevant for further studies. SST is an important regulator of the endocrine system and it is likely to have a role in the regulation of ADIPOQ, AGRP and TNFα, that l1l2FS selected in the protein signature and that were assigned to enriched KEGG pathways within the Endocrine system category. SST also affects the rates of neurotransmission in the CNS and proliferation of both normal and tumorigenic cells. Associations of this gene with AD are already suggested [63-66], even if its exact role in the disease is not clear yet. SST is also known to be co-transported with SP, one of the products of TAC1 mentioned above.
UBE3A functions as a cellular quality control ubiquitin ligase. Since AD is characterized by the accumulation of amyloid-beta and tau peptides, this protein could be a good candidate for investigation because it participates to the ubiquitin protein degradation system, with the main function of breaking down the non-functional proteins.
The l1l2FS analysis performed separately on the microarray datasets scored low cross-validation errors and revealed 4 common significant genes: RGS4, MCTP1, CD44 and XIST. Besides RGS4 [36], the other genes have never been associated with AD but, as noted in literature characterization, they are very likely to play a role in the disease.
The integrated functional analysis showed pathways that are common across two or three of the signatures, see Table 7 and Figure 4. Such pathways are already related to AD and they belong to the Signaling Molecules and Interaction, Immune System, Signal Transduction and Cell Communication KEGG categories. In particular, these pathways are involved in the immune system and inflammation (cytokine-cytokine receptor interaction, antigen processing and presentation, hematopoietic cell lineage, leukocyte transendothelial migration), in the nervous system and related diseases (neurodegenerative disorders, neuroactive ligand-receptor interaction, prion disease and axon guidance), in cell proliferation, differentiation and migration and also in the maintenance of the structure and function of one cell line or tissue (MAPK signaling and ECM-receptor interaction).
The combined analysis of the three signatures led to the identification of common probesets between the two microarray signatures and of a meaningful functional overlap among the three signatures. The statistical method adopted for the analysis allowed for the selection of genes that are already known to be related with AD or to be expressed in the brain. This strengthened the hypothesis that the remaining selected genes might be relevant for further studies, especially because the results were obtained without using any prior knowledge on the subject. Despite the very convincing results, we are aware of some limitations that affected our work and that should be overcome in the future. From the data viewpoint, we coarsely divided the available samples in two broad classes (healthy and diseased) but it would be more accurate to separately consider the three AD stages (incipient, moderate, severe) versus controls. More-over, when dealing with the GSE5281 data, we could discard those samples extracted from the visual cortex that seems to be spared by AD [6]. On the computational side, the l1l2FS framework has the advantage of being correlation-aware and multivariate. Unfortunately, the algorithm itself and the double loop of nested cross-validation procedures heavily demand computational power, in contrast to more standard techniques, such as t-test, that are easier to run even by non-specialists. Nonetheless, the computational burden can be significantly reduced by running the statistical framework on distributed computing facilities, such as clusters, grids or cloud computing.
Conclusion
Gene signatures are the first indispensable step towards the identification of genes and proteins highly related with disease and belonging to pathogenic pathways. Indeed, in the context of AD, there is a urgent need to improve its molecular characterization to establish novel therapeutic targets and reliable biomarkers.
By applying l1l2FS on three AD datasets, we defined gene signatures with good discriminative properties for the classification of diseased and healthy subjects. Our work, focused on the functional characterization of potentially meaningful AD genes, revealed 21 genes in the protein dataset [4], 11 and 39 genes in the GSE1297 [5] and GSE5281 [6] microarray datasets respectively. Each signature was robust from the statistical viewpoint and it was associated to a low cross-validation error. Some of the selected genes are already known to be involved in AD; others have never been associated with the disease but the current biological knowledge suggests their possible correlation with AD. Specifically, the first group comprises TNFα, IL6R, IL-1α, GFAP, CXCR4, CTSB, SST, LTF, CNR1, RGS4 and the second is formed by ADIPOQ, AGRP, HSPA1A, PENK, UBE3A, TAC1, TNFRSF10C and ANGPT1.
The functional analysis of the signatures confirmed the validity of the results, identifying pathways that are biologically meaningful and related with AD, see Figure 4.
The integrated functional analysis revealed three overlapping pathways: hematopoietic cell lineage, neuroactive ligand-receptor interaction and MAPK signaling pathways. The validity of our procedure was also confirmed at the functional level, in fact the common pathways are already mentioned in the literature as important for AD [2,3].
The use of l1l2FS as core algorithm for feature selection allowed for the identification of relevant and correlated genes. Therefore those genes not included in common pathways and still unmentioned in the literature could play a role in the pathogenesis of the disease.
In the near future, we plan to extend the analysis in order to include more available domain knowledge. We are currently working on designing methods that explicitly use prior knowledge on the subject, to drive the feature selection step for example adopting filtering techniques [15] or designing appropriate kernel functions [56,67]. We also plan to integrate heterogeneous data from different techniques (Chip-on-Chip, SNP, GWAS and miRNA) and possibly from different-omics domains.
Materials and methods
5.1 Data
The protein dataset of Ray and co-authors [4] is a collection of 259 plasma samples from individuals with presymptomatic to late-stage AD and from various controls. The abundance of 120 well-known signaling proteins was measured for each sample with the filtered-based arrayed sandwich ELISA. The data-set is available online, already normalized and subdivided in four groups: the Training Set, composed by 43 AD samples and 40 nondemented control (NDC) samples; the Test Set AD, composed by 42 AD, 39 NDC and 11 other dementia (OD) samples; the Test Set MCI, composed by 47 MCI samples with follow-up (2-6 years from MCI diagnosis); the OND and RA patient set, composed by 21 other neurological disease (OND) and 16 rheumatoid arthrithis (RA) samples. The latter dataset was excluded from our analysis because it did not contain any AD data. The remaning test sets that presented more than two classes were considered as binary problems: AD versus not-AD.
The other datasets are stored in the GEO repository: GSE1297 of Blalock et al. [5] and GSE5281 of Liang et al. [6]. The first consisted of 22 AD samples and 9 controls retrieved from the hippocampal brain region. The gene expressions were measured on the Affymetrix HG-U133A platform. The second dataset was measured on the Affymetrix HG-U133 Plus2.0 platform and it was composed by 87 AD late onset samples and 74 controls on six different brain regions originating from the same subject: entorhinal cortex, hippocampus, medial temporal gyrus, posterior cingulate, superior frontal gyrus and primary visual cortex. Some of the samples originated from the same subject, but we did not take explicitly into account this information. The statistical framework treats those samples as if they belonged to different subjects. For both microarray datasets, the gene expressions were extracted from the. CEL files and normalized using the Robust Multichip Average method [68] by running an R [69] script, based on the aroma package.affymetrix [70] http://www.aroma-project.org.
Feature selection framework
l1l2FS is a regularization method capable to select subsets of discriminative genes. The algorithm can be tuned to give a minimal set of discriminative genes or larger sets including correlated genes. The method is based on the optimization principle presented in [8] and further developed and studied in [9,71]. First we fix some notation and then we explain the idea behind the algorithm. Assume we are given a collection of n subjects, each represented by a d-dimensional vector x of measurements (e.g. the gene expression or the protein abundance vector). Each sample is also associated with a binary label y, assigning it to a class (e.g. AD or control). The dataset is therefore represented by a n × d matrix X, where d >>n and Y is the n-dimensional labels vector. The l1l2FS with double optimization algorithm looks for a linear model f(x) = βx, whose sign gives the classification rule that can be used to associate a new sample to one of the two classes. The offset is zero, since we normalize the X matrix to have zero mean. Note that the vector of weights β is forced to be a sparse vector, that is some of its entries are zero, then some variables (probesets or proteins) will not contribute in building the estimator f(x). The weight vector β is found by minimizing the naïve elastic net functional:
where the least square error is penalized with the l1 and l2 norm of the coefficient vector β. The least square term ensures fitting of the data whereas adding the two penalties allows to avoid over-fitting. The role of the two penalties is different, the l1 term (sum of absolute values) enforces the solution to be sparse, the l2 term (sum of the squares) preserves correlation among the variables. The solution of this step, computed through the simple iterative softthresholding [72], is followed by a second optimization, namely regularized least squares (RLS):
to estimate the classifier on the selected features and to improve the classification performance [73]. The training for selection and classification requires the choice of the regularization parameters for both l1l2FS regularization and RLS denoted with τ* and λ*, respectively. In fact, the model selection and statistical significance assessment are performed within two nested K-fold cross-validation loops, similarly to [14,74]. When a separate set of new data, i.e. a test set, is difficult to gather because of cost or time issues, cross-validation represents the most common solution to achieve an accurate estimate of prediction error. In K-fold cross-validation, the available data are split into K roughly equal-sized parts. For each k-th part, a model is trained on the other K - 1 (training) and a prediction error is evaluated using the fitted model to predict the outcome on the k-th part (test). The estimated prediction error is the combination (usually the average) of the K estimated errors. Specifically, we emploied stratified cross-validation, where the folds are selected so that the mean response value is approximately equal in all the folds. In the case of binary classification, this means that each fold contains roughly the same proportions of the two types of class labels. Stratified cross-validation is particularly useful to avoid unbalances between Type I (False Positive - FP) and Type II (False Negative - FN) errors.
Being interested in a comprehensive list of relevant variables, we fixed our attention on the lists obtained with the highest values for the correlation parameter μ (μ = 1). The framework was implemented in Python, based on the L1L2Py library http://slipguru.disi.unige.it/Research/L1L2Py.
The statistical framework described above provides a set of K lists of selected variables, therefore it it is necessary to choose an appropriate criterion [75] in order to assess a common list of relevant variables (probesets or proteins, in our case). We based ours on the absolute frequency, i.e. we decided to promote as relevant variables the most stable probesets across the lists. The threshold we used to select the final lists was chosen according to the slope variation of the number of selected genes vs. frequency (plot not shown), its value being 40%. In this way we managed to cut out those variables that were not stable across the cross-validation lists, similarly to the procedure adopted in [14].
We also visualized the signatures in heatmap plots in order to devise the modules of correlated variables within the signatures by applying a k-means clustering procedure, with the aim of enhancing the genes with correlating expressions [76]. We used the correlation distance and set the number of clusters according to the dimensionality of the minimal list that is the list selected by l1l2FS when the correlation parameter is set to the minimum value of μ.
Functional Analysis
For the functional analysis of the signatures we used the online gene set analysis toolkit WebGestalt [77] http://bioinfo.vanderbilt.edu/webgestalt/.
This online toolkit performs the gene set enrichment in KEGG and GO and identifies the most relevant pathways and ontologies in the signatures. GO is a database of controlled vocabularies (ontologies) that describes gene products in terms of their associated domains, that are biological process (BP), cellular component (CC) and molecular function (MF), in a species independent manner. GO is structured as a directed acyclic graph where each term has a defined relationship to one or more terms in the same domain and sometimes to other domains. The most common visual representation of GO is a graph where the relations among the ontologies (nodes) are represented by connecting lines (arcs). KEGG is a repository that stores the higher-order systemic behaviors of the cell and the organism from genomic and molecular information. It is an integrated database resource consisting of 16 main databases, broadly categorized into systems information, genomic information, and chemical information. All the available KEGG pathways have been biologically validated before publishing. Both for KEGG and GO, we selected the WebGestalt human genome as reference set, p-value ≤ 0.05 as level of significance, 3 as the minimum number of genes and the default Hypergeometric test as statistical method. Medline [11] was used to retrieve the available prior knowledge on the genes from the current literature.
List of Abbreviations
AD: Alzheimer's Disease; GEO: Gene Expression Omnibus; MMSE: Mini-Mental State Exam; l1l2FS: l1l2FS regularization with double optimization feature selection; Medline: Medical Literature Analysis and Retrieval System Online; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; MCI: Mild Cognitive Impairment; CNS: Central Nervous System; NDC: Non Demented Controls; OD: Other Dementia; OND: Other Neurological Disease; RA: Rheumathoid Arthritis; RLS: Regularized Least Squares FP: False Positive; FN: False Negative; BP: Biological Process; CC: Cellular Component; MF: Molecular Function.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MS contributed to the functional analysis, the biological interpretation of the results and the writing of the paper. AB curated the data normalization and analysis and the visualization of the results. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Ontologies for the protein signature. Results of WebGestalt analysis of the gene set enrichment made for the 21 protein signature in GO. The enriched ontologies are marked in red color. The most significant ontologies in MF are associated to the selected cytokines: some have chemokines activity, some belong to the hematopoietin/interferon class, some have a growth factor activity and others are coupled with the G-proteins. The most enriched process in the BP domain is cell communication, that is connected to signal transduction. More general processes follow, like regulation of cellular processes, of physiological processes and of cellular physiological processes. It is interesting to observe that these general biological processes present both positive and negative regulation, having only IL3 as common gene. IL3 is a potent growth promoting cytokine involved in several activities like cell growth, differentiation and apoptosis. IL3 possesses neurotrophic activity and it is associated with neurological disorders like schizophrenia.
{kind=link}
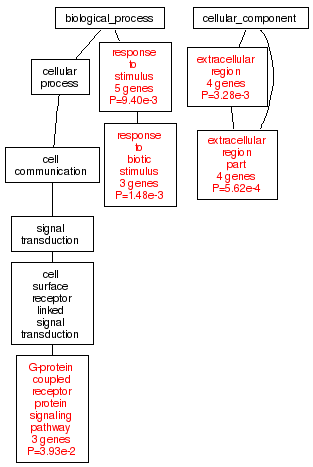
Ontologies for the GSE1297 signature. Results of WebGestalt analysis of the gene set enrichment made for the GSE1297 signature in GO. The enriched ontologies are marked in red color. The GO enrichment is not associated to a large subgraph, as in the protein signature analysis, because not all the genes are functionally characterized. The CC domain shows that the gene products of this signature are mainly located in the extracellular region, probably because they are involved in the response to stimulus process, that it is subsequently propagated inside the cell trough the G-protein coupled receptor protein signaling pathway.
{kind=link}
Ontologies for the GSE5281 signature. Results of WebGestalt analysis of the gene set enrichment made for the GSE5281 signature in GO. The enriched ontologies are marked in red color. Similarly to the GSE1297 analysis, the GO subgraph associated to this signature is not large because of the scarce functionally characterizion of its genes.
{kind=link}
Contributor Information
Margherita Squillario, Email: margherita.squillario@unige.it.
Annalisa Barla, Email: annalisa.barla@unige.it.
Acknowledgements
The authors would like to thank Alessandro Verri, Curzio Basso, Andrea Cornero and Paolo Fardin for many useful discussions and the reviewers for their helpful comments.
The study was partially supported by the EU Integrated Project Health-e-Child IST-2004-027749.
References
- Gandhi S, Wood NW. Genome-wide association studies: the key to unlocking neurodegeneration? Nat Neurosci. 2010;13(7):789–94. doi: 10.1038/nn.2584. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12(9):1005–15. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362(4):329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman L, Galasko D, Jutel M, Karydas A. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA. 2004;101(7):2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette C, Schmechel D, Alexander GE, Reiman EM, Rogers J, Stephan DA. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiological Genomics. 2007;28(3):311–22. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli RJ, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. PNAS. 2008;105(11):4441–6. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society, Series B. 2005.
- De Mol C, Mosci S, Traskine M, Verri A. A Regularized Method for Selecting Nested Groups of Relevant Genes from Microarray Data. Journal of Computational Biology. 2009;16:1–15. doi: 10.1089/cmb.2008.0137. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. An introduction to the bootstrap. 1993.
- Medline. http://www.ncbi.nlm.nih.gov/pubmed/
- The Gene Ontology database http://www.geneontology.org. http://www.geneontology.org
- KEGG PATHWAY Database. http://www.genome.ad.jp/kegg/pathway.html
- Fardin P, Barla A, Mosci S, Rosasco L, Verri A, Varesio L. The l1-l2 regularization framework unmasks the hypoxia signature hidden in the transcriptome of a set of heterogeneous neuroblastoma cell lines. BMC Genomics. 2009;10:474. doi: 10.1186/1471-2164-10-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardin P, Cornero A, Barla A, Mosci S, Acquaviva M, Rosasco L, Gambini C, Verri A, Varesio L. Identification of multiple hypoxia signatures in neuroblastoma cell lines by l1-l2 regularization and data reduction. Journal of Biomedicine and Biotechnology. 2010. [DOI] [PMC free article] [PubMed]
- Fardin P, Barla A, Mosci S, Rosasco L, Verri A, Versteeg R, Caron HN, Molenaar JJ, Ora I, Eva A, Puppo M, Varesio L. A biology-driven approach identifies the hypoxia gene signature as a predictor of the outcome of neuroblastoma patients. Molecular Cancer. 2010. [DOI] [PMC free article] [PubMed]
- Guzzetta G, Jurman G, Furlanello C. A machine learning pipeline for quantitative phenotype prediction from genotype data. BMC Bioinformatics. 2010. [DOI] [PMC free article] [PubMed]
- Ravetti MG, Moscato P. Identification of a 5-protein biomarker molecular signature for predicting Alzheimer's disease. PLoS ONE. 2008;3(9):e3111. doi: 10.1371/journal.pone.0003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger J, Verma S, Göhring I, Bobbert T, Seifert J, Sindler AL, Pfeiffer A, Hileman SM, Tschöp M, Banks WA. Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells. Diabetes. 2006;55:141–7. doi: 10.2337/diabetes.55.01.06.db05-1077. [DOI] [PubMed] [Google Scholar]
- Yang H, yuan Li Y, qiang Nie Y, jian Zhou Y, lei DU Y, hong Sha W, Hong Y. The relationship between insulin resistance and adiponectin gene expression in nonalcoholic fatty liver disease. Zhonghua Gan Zang Bing Za Zhi. 2007;15(7):525–8. [PubMed] [Google Scholar]
- Kim HS, Yumkham S, Lee HY, Cho JH, Kim MH, Koh DS, Ryu SH, Suh PG. C-terminal part of AgRP stimulates insulin secretion through calcium release in pancreatic beta Rin5mf cells. Neuropeptides. 2005;39(4):385–93. doi: 10.1016/j.npep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Lim HS, Lip GYH, Blann AD. Angiopoietin-1 and angiopoietin-2 in diabetes mellitus: relationship to VEGF, glycaemic control, endothelial damage/dysfunction and atherosclerosis. Atherosclerosis. 2005;180:113–8. doi: 10.1016/j.atherosclerosis.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Sie M, Wagemakers M, Molema G, Mooij JJA, de Bont ESJM, den Dunnen WFA. The angiopoietin 1/angiopoietin 2 balance as a prognostic marker in primary glioblastoma multiforme. J Neurosurg. 2009;110:147–55. doi: 10.3171/2008.6.17612. [DOI] [PubMed] [Google Scholar]
- Dörr J, Bechmann I, Waiczies S, Aktas O, Walczak H, Krammer PH, Nitsch R, Zipp F. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J Neurosci. 2002;22(4):RC209. doi: 10.1523/JNEUROSCI.22-04-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Lehtinen MK, Merlo P, Villén J, Gygi S, Bonni A. Regulation of neuronal cell death by MST1FOXO1 signaling. J Biol Chem. 2009;284(17):11285–92. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010. pp. D355–60. [DOI] [PMC free article] [PubMed]
- Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inammatory signaling. J Neurosci Res. 2002;70(3):462–73. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- Kroner Z. The relationship between Alzheimer's disease and diabetes: Type 3 diabetes? Altern Med Rev. 2009;14(4):373–9. [PubMed] [Google Scholar]
- Vawter MP, Evans S, Choudary P, Tomita H, Meador-Woodruff J, Molnar M, Li J, Lopez JF, Myers R, Cox D, Watson SJ, Akil H, Jones EG, Bunney WE. Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsy-chopharmacology. 2004;29(2):373–84. doi: 10.1038/sj.npp.1300337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Mills IP, Trojanowski JQ, Lee VMY. Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry. 2008;47(47):12614–25. doi: 10.1021/bi801475r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Washizuka S, Kakiuchi C, Kato T. Expression of HSPF1 and LIM in the lymphoblastoid cells derived from patients with bipolar disorder and schizophrenia. J Hum Genet. 2004;49(5):227–31. doi: 10.1007/s10038-004-0136-5. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004;9(4):406–16. doi: 10.1038/sj.mp.4001437. [DOI] [PubMed] [Google Scholar]
- Hirata E, Arakawa Y, Shirahata M, Yamaguchi M, Kishi Y, Okada T, Takahashi JA, Matsuda M, Hashimoto N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Science. 2009;100(8):1451–9. doi: 10.1111/j.1349-7006.2009.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann D, Dupanloup I, Excoffier L. Width of gene expression profile drives alternative splicing. PLoS ONE. 2008;3(10):e3587. doi: 10.1371/journal.pone.0003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy P, Ripoche H, Laurendeau I, Lazar V, Ortonne N, Parfait B, Leroy K, Wechsler J, Salmon I, Wolkenstein P, Dessen P, Vidaud M, Vidaud D, Bièche I. Microarray-based identification of tenascin C and tenascin XB, genes possibly involved in tumorigenesis associated with neurofibromatosis type 1. Clin Cancer Res. 2007;13(2 Pt 1):398–407. doi: 10.1158/1078-0432.CCR-06-0182. [DOI] [PubMed] [Google Scholar]
- Muma NA, Mariyappa R, Williams K, Lee JM. Differences in regional and subcellular localization of G(q/11) and RGS4 protein levels in Alzheimer's disease: correlation with muscarinic M1 receptor binding parameters. Synapse. 2003;47:58–65. doi: 10.1002/syn.10153. [DOI] [PubMed] [Google Scholar]
- Ding L, Mychaleckyj JC, Hegde AN. Full length cloning and expression analysis of splice variants of regulator of G-protein signaling RGS4 in human and murine brain. Gene. 2007;401(1-2):46–60. doi: 10.1016/j.gene.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Ramírez BG, Blázquez C, del Pulgar TG, Guzmán M, de Ceballos ML. Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25(8):1904–13. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An L, Sato H, Konishi Y, Walker DG, Beach TG, Rogers J, Tooyama I. Expression and localization of lactotransferrin messenger RNA in the cortex of Alzheimer's disease. Neurosci Lett. 2009;452(3):277–80. doi: 10.1016/j.neulet.2009.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels AA, Kanon B, Bensaude O, Kampinga HH. Heat shock protein (Hsp) 40 mutants inhibit Hsp70 in mammalian cells. J Biol Chem. 1999;274(51):36757–63. doi: 10.1074/jbc.274.51.36757. [DOI] [PubMed] [Google Scholar]
- Samson WK, Zhang JV, Avsian-Kretchmer O, Cui K, Yosten GLC, Klein C, Lyu RM, Wang YX, Chen XQ, Yang J, Price CJ, Hoyda TD, Ferguson AV, bin Yuan X, Chang JK, Hsueh AJW. Neuronostatin encoded by the somatostatin gene regulates neuronal, cardiovascular, and metabolic functions. J Biol Chem. 2008;283(46):31949–59. doi: 10.1074/jbc.M804784200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HT, Hu S, Sheng WS, Olson JM, Cheeran MCJ, Chan ASH, Lokensgard JR, Peterson PK. High-level expression of functional chemokine receptor CXCR4 on human neural precursor cells. Brain Res Dev Brain Res. 2004;152(2):159–69. doi: 10.1016/j.devbrainres.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Chvapil M, Peng YM. Oxygen and lung fibrosis. Arch Environ Health. 1975;30(11):528–32. doi: 10.1080/00039896.1975.10666770. [DOI] [PubMed] [Google Scholar]
- Weeraratna AT, Kalehua A, Deleon I, Bertak D, Maher G, Wade MS, Lustig A, Becker KG, Wood W, Walker DG, Beach TG, Taub DD. Alterations in immunological and neurological gene expression patterns in Alzheimer's disease tissues. Exp Cell Res. 2007;313(3):450–61. doi: 10.1016/j.yexcr.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero S, Bajetto A, Bonavia R, Porcile C, Piccioli P, Pirani P, Ravetti JL, Zona G, Spaziante R, Florio T, Schettini G. Expression of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1 in human brain tumors and their involvement in glial proliferation in vitro. Ann N Y Acad Sci. 2002;973:60–9. doi: 10.1111/j.1749-6632.2002.tb04607.x. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. PNAS. 2003;100(23):13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouser PE, Head E, Ha KH, Rohn TT. Caspasemediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer's disease brain. Am J Pathol. 2006;168(3):936–46. doi: 10.2353/ajpath.2006.050798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton SB, O'Callaghan JP, Savva GM, Nicoll JAR, Matthews F, Simpson JE, Forster G, Shaw PJ, Brayne C, Ince PG, Function MC. Group ANS. Population variation in glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord. 2009;27(5):465–73. doi: 10.1159/000217729. [DOI] [PubMed] [Google Scholar]
- Korolainen MA, Auriola S, Nyman TA, Alafuzoff I, Pirttilä T. Proteomic analysis of glial fibrillary acidic protein in Alzheimer's disease and aging brain. Neurobiol Dis. 2005;20(3):858–70. doi: 10.1016/j.nbd.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Hol EM, Roelofs RF, Moraal E, Sonnemans MAF, Sluijs JA, Proper EA, de Graan PNE, Fischer DF, van Leeuwen FW. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Mol Psychiatry. 2003;8(9):786–96. doi: 10.1038/sj.mp.4001379. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, van den Berge SA, Aronica E, Speijer D, Hol EM. Specific human astrocyte subtype revealed by affinity purified GFAP antibody; unpurified serum cross-reacts with neurofilament-L in Alzheimer. PLoS ONE. 2009;4(11):e7663. doi: 10.1371/journal.pone.0007663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa K, Kunishita T, Maruyama K, Yoshikawa K, Kominami E, Tsuchiya T, Suzuki K, Tabira T, Sugita H, Ishiura S. Alzheimer's disease amyloid beta-clipping enzyme (APP secretase): identification, purification, and characterization of the enzyme. Biochem Biophys Res Commun. 1991;177:377–87. doi: 10.1016/0006-291X(91)91994-N. [DOI] [PubMed] [Google Scholar]
- Ernst A, Buerger K, Hartmann O, Dodel R, Noelker C, Sommer N, Schwarz M, Köhrle J, Bergmann A, Hampel H. Midregional Proenkephalin A and N-terminal Protachykinin A are decreased in the cerebrospinal fluid of patients with dementia disorders and acute neuroinammation. J Neuroimmunol. 2010;221(1-2):62–7. doi: 10.1016/j.jneuroim.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Mishra A, Dikshit P, Purkayastha S, Sharma J, Nukina N, Jana NR. E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J Biol Chem. 2008;283(12):7648–56. doi: 10.1074/jbc.M706620200. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Weston J, Cai J, Noble Grundy W. Gene functional classification from heterogeneous data. Proceedings of the fifth annual international conference on Computational biology. 2001.
- Hamid JS, Hu P, Roslin NM, Ling V, Greenwood CMT, Beyene J. Data Integration in Genetics and Genomics: Methods and Challenges. Human Genomics and Proteomics. 2009;2009:1–13. doi: 10.4061/2009/869093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66(3):352–61. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrila A, Chan JL, Miller LC, Heist K, Yiannakouris N, Mantzoros CS. Circulating melanin-concentrating hormone, agouti-related protein, and alpha-melanocyte-stimulating hormone levels in relation to body composition: alterations in response to food deprivation and recombinant human leptin administration. J Clin Endocrinol Metab. 2005;90(2):1047–54. doi: 10.1210/jc.2004-1124. [DOI] [PubMed] [Google Scholar]
- Zadeh G, Reti R, Koushan K, Baoping Q, Shannon P, Guha A. Regulation of the pathological vasculature of malignant astrocytomas by angiopoietin-1. Neoplasia (New York, NY) 2005;7(12):1081–90. doi: 10.1593/neo.05424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Douglas SD, Pleasure DE, Lai J, Guo C, Bannerman P, Williams M, Ho W. Human neuronal cells (NT2-N) express functional substance P and neurokinin-1 receptor coupled to MIP-1 beta expression. J Neurosci Res. 2003;71(4):559–66. doi: 10.1002/jnr.10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, Johnson DJ, Boehmer CG, Catton MD, Vinters HV, Maggio JE, Too HP, Vigna SR. Substance P receptor binding sites are expressed by glia in vivo after neuronal injury. PNAS. 1989;86(13):5193–7. doi: 10.1073/pnas.86.13.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyk SK, Kowall NW, Hauser SL. Substance P immunoreactive astrocytes are present in multiple sclerosis plaques. Brain Res. 1989;504(2):284–8. doi: 10.1016/0006-8993(89)91369-3. [DOI] [PubMed] [Google Scholar]
- Saiz-Sanchez D, Ubeda-Bañon I, de la Rosa-Prieto C, Argandoña-Palacios L, Garcia-Muñozguren S, Insausti R, Martinez-Marcos A. Somatostatin, tau, and beta-amyloid within the anterior olfactory nucleus in Alzheimer disease. Exp Neurol. 2010;223(2):347–50. doi: 10.1016/j.expneurol.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Xue S, Jia L, Jia J. Association between somatostatin gene polymorphisms and sporadic Alzheimer's disease in Chinese population. Neurosci Lett. 2009;465(2):181–3. doi: 10.1016/j.neulet.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Vepsäläinen S, Helisalmi S, Koivisto AM, Tapaninen T, Hiltunen M, Soininen H. Somatostatin genetic variants modify the risk for Alzheimer's disease among Finnish patients. J Neurol. 2007;254(11):1504–8. doi: 10.1007/s00415-007-0539-2. [DOI] [PubMed] [Google Scholar]
- van de Nes JAP, Konermann S, Nafe R, Swaab DF. Beta-protein/A4 deposits are not associated with hyperphosphorylated tau in somatostatin neurons in the hypothalamus of Alzheimer's disease patients. Acta Neuropathol. 2006;111(2):126–38. doi: 10.1007/s00401-005-0018-8. [DOI] [PubMed] [Google Scholar]
- Lanckriet GRG, Bie TD, Cristianini N, Jordan MI, Noble WS. A statistical framework for genomic data fusion. Bioinformatics. 2004;20(16):2626–35. doi: 10.1093/bioinformatics/bth294. [DOI] [PubMed] [Google Scholar]
- Irizarry R, Bolstad B, Collin F, Cope L, Hobbs B, Speed T. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2004.
- Hansen K, Bullard J, Simpson K, Bengtsson H. aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bunded memory. Tech Report #745 Department of Statistics, University of California, Berkeley, February 2008.
- De Mol C, De Vito E, Rosasco L. Elastic Net Regularization in Learning Theory. Journal of Complexity. 2009.
- Daubechies I, Defrise M, De Mol C. An iterative thresholding algorithm for linear inverse problems with a sparsity constraint. arXiv. 2003. math.FA.
- Candes E, Tao T. The Dantzig selector: statistical estimation when p is much larger than n. Annals of Statistics. 2007.
- Barla A, Mosci S, Rosasco L, Verri A. A method for robust variable selection with significance assessment. Proceedings of ESANN 2008. 2008.
- Jurman G, Merler S, Barla A, Paoli S, Galea A, Furlanello C. Algebraic stability indicators for ranked lists in molecular profiling. Bioinformatics. 2008;24(2):258–264. doi: 10.1093/bioinformatics/btm550. [DOI] [PubMed] [Google Scholar]
- Mosci S, Barla A, Verri A, Rosasco L. Finding structured gene signatures. IEEE Proceedings BIBM 2008. 2008. p. 8.
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005. pp. W741–8. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ontologies for the protein signature. Results of WebGestalt analysis of the gene set enrichment made for the 21 protein signature in GO. The enriched ontologies are marked in red color. The most significant ontologies in MF are associated to the selected cytokines: some have chemokines activity, some belong to the hematopoietin/interferon class, some have a growth factor activity and others are coupled with the G-proteins. The most enriched process in the BP domain is cell communication, that is connected to signal transduction. More general processes follow, like regulation of cellular processes, of physiological processes and of cellular physiological processes. It is interesting to observe that these general biological processes present both positive and negative regulation, having only IL3 as common gene. IL3 is a potent growth promoting cytokine involved in several activities like cell growth, differentiation and apoptosis. IL3 possesses neurotrophic activity and it is associated with neurological disorders like schizophrenia.
Ontologies for the GSE1297 signature. Results of WebGestalt analysis of the gene set enrichment made for the GSE1297 signature in GO. The enriched ontologies are marked in red color. The GO enrichment is not associated to a large subgraph, as in the protein signature analysis, because not all the genes are functionally characterized. The CC domain shows that the gene products of this signature are mainly located in the extracellular region, probably because they are involved in the response to stimulus process, that it is subsequently propagated inside the cell trough the G-protein coupled receptor protein signaling pathway.
Ontologies for the GSE5281 signature. Results of WebGestalt analysis of the gene set enrichment made for the GSE5281 signature in GO. The enriched ontologies are marked in red color. Similarly to the GSE1297 analysis, the GO subgraph associated to this signature is not large because of the scarce functionally characterizion of its genes.