

Abstract
Fragmentation is a degradation pathway ubiquitously observed in proteins despite the remarkable stability of peptide bond; proteins differ only by how much and where cleavage occurs. The goal of this review is to summarize reports regarding the non-enzymatic fragmentation of the peptide backbone of monoclonal antibodies (mAbs). The sites in the polypeptide chain susceptible to fragmentation are determined by a multitude of factors. Insights are provided on the intimate chemical mechanisms that can make some bonds prone to cleavage due to the presence of specific side-chains. In addition to primary structure, the secondary, tertiary and quaternary structures have a significant impact in modulating the distribution of cleavage sites by altering local flexibility, accessibility to solvent or bringing in close proximity side chains that are remote in sequence. This review focuses on cleavage sites observed in the constant regions of mAbs, with special emphasis on hinge fragmentation. The mechanisms responsible for backbone cleavage are strongly dependent on pH and can be catalyzed by metals or radicals. The distribution of cleavage sites are different under acidic compared to basic conditions, with fragmentation rates exhibiting a minimum in the pH range 5–6; therefore, the overall fragmentation pattern observed for a mAb is a complex result of structural and solvent conditions. A critical review of the techniques used to monitor fragmentation is also presented; usually a compromise has to be made between a highly sensitive method with good fragment separation and the capability to identify the cleavage site. The effect of fragmentation on the function of a mAb must be evaluated on a case-by-case basis depending on whether cleavage sites are observed in the variable or constant regions, and on the mechanism of action of the molecule.
Key words: fragmentation, cleavage, clipping, hinge region, peptide bond hydrolysis, IgG1, IgG2
Introduction
Fragmentation of monoclonal antibodies (mAbs) is a critical quality attribute that needs to be monitored to assess the purity and integrity of the protein. Fragmentation can be generated during protein production in the cell culture, is modulated by the purification process and will accrue during storage or circulation in the blood. Overall, the fragmentation pattern of a mAb represents a finger-print of both manufacture and stability consistency, making it a critical element in assessing comparability of materials produced at different sites.
Fragmentation is a very general term that usually pertains to disruption of a covalent bond in a protein as a result of either spontaneous or enzymatic reaction. The focus of this review is on the non-enzymatic fragmentation or cleavage of the protein backbone; any fragmentation or chemical modification of side chains or disulfide bonds that do not lead to fragmentation of the protein backbone are outside the scope of this paper. The protein backbone is extremely stable under physiological conditions, but certain sites may become prone to fragmentation as a function of amino acid sequence (presence of specific side-chains that may facilitate cleavage), flexibility of the local structure, solvent conditions (pH, temperature) and the presence of metals or radicals.
This review is structured in two parts: the first part describes different mechanisms of cleavage that may explain the fragmentation patterns observed in mAbs and the factors that have an effect on fragmentation rates; the second part presents experimental techniques developed to detect fragmentation in mAbs and summarizes fragmentation sites identified in mAbs.
It may be argued that, due to the considerable sequence similarity of mAbs, fragmentation properties are expected to be very similar for molecules belonging to the same subclass (for instance, IgG1). Although this assumption was proven correct for several molecules examined by one group,1,2 differences are to be expected when comparing data published by different groups due to the large variety of factors that can have an effect on fragmentation rates, e.g., conditions that directly influence the cleavage rates, such as pH, temperature and solvent composition, but also experimental approaches used for the detection of the fragmentation.
Fragmentation Mechanisms
The peptide bond has an intrinsic exceptional chemical stability with respect to non-enzymatic hydrolysis. The half-life of compounds containing peptide bonds may span several hundreds of years3 unless very harsh conditions like extreme pH and high temperatures are used. However, fragmentation may occur at higher rates when side-chains of few residues are involved in peptide bond cleavage via a specific degradation mechanism. In proteins, the flexibility of the backbone and of the reactive side chain is another key factor that has an effect on the peptide bond fragmentation rate; as will be illustrated below, fragmentation is observed in the solvent-exposed, flexible loops of a protein and it may not occur in rigid parts despite the cleavage propensity of a certain primary sequence. Based on the comprehensive study by Liu and coworkers4 and other reports on antibody fragmentation,1,5–7 most of the backbone fragmentation events in mAbs occur at one of the following residues: Asp, Gly, Ser, Thr, Cys or Asn. Interestingly, the side chains of these residues (with the exception of Gly) can facilitate peptide bond cleavage via specific mechanisms. It is therefore possible that many fragmentation events observed in mAb result from specific cleavage reactions involving amino acid side chains, although other mechanisms (e.g., free-radical-induced hydrolysis or direct hydrolysis) may contribute as well. A brief description of the cleavage mechanism will be given below for the most frequently observed cleavage events. A summary of the model systems, conditions and references that describe in detail the proposed reaction mechanisms involving specific residues is given in Table 1. The purpose of the chemical schematics shown in this review is to depict the chemical mechanisms and the key species involved in the fragmentation reactions; the original papers that elucidated each reaction pathways provide more details on the reaction mechanisms and complex equilibria involved.
Table 1.
Summary of residues involved in peptide bond cleavage, conditions for testing and references that discuss mechanisms of cleavage
| Residue | Peptide bond | Primary sequence | System | pH | Temperature | Reference |
| Asp | Asp-Xaa | Xaa: Gly | hexapeptide | 0.3–10 | 37°C | 14 |
| Xaa: Ser, Gly, Val | hexapeptide | 1.0, 10.0 | 70°C | 19 | ||
| Xaa: Tyr, Ser, Phe | glucagon | 1.0–2.4 | 60°C | 15 | ||
| Xaa: Pro | 18 | |||||
| Xaa:Gln, Pro, Lys, Leu | decapeptide | 4.0–5.5 | 25°C, 40°C, 60°C | 17 | ||
| Xaa: Asp | IgG2 | 5.2 | 4°C, 25°C, 45°C | 7 | ||
| Xaa-Asp | Xaa: Pro, Gly | hexapeptide | 1.0, 10.0 | 70°C | 19 | |
| Gly | Gly-Xaa | Xaa:Gly, Ala, Leu, Val | dipeptide | 12.6 | 30°C | 21 |
| Xaa-Gly | Xaa:Ala, Leu, Ser | |||||
| Thr | Gly-Thr | |||||
| Ser | Xaa-Ser | Xaa: Gly, Ala, Leu | ||||
| Xaa-Ser | alkaline | 25 | ||||
| Cys-Cys | Xaa-Cys | IgG1 | 7–10 | 45°C | 6 | |
| Asn | Asn-Xaa | Xaa: Ser, Val, Leu, Pro | hexapeptides | 7.5, 10 | 37°C | 27 |
Fragmentation of peptide bond mediated by water.
Hydrolysis of peptide bonds has been the subject of numerous experimental and computation studies due to the importance of peptide bonds for biological systems. The pH-dependence of peptide bond fragmentation was measured over a wide pH range (pH 0–14) in a model compound.8 The conclusion of the study was that the first-order rate constant is well described over the entire pH range by Schematic 1; the reaction rate is minimal around neutral pH with a half-life in the range of 175–564 years and is accelerated under acidic and basic conditions with the rate constants for specific-acid (kH3O+) and specific-base (kOH−) being almost identical. Half-lives of hundreds of years around neutral pH were reported for other peptide model compounds as well.9
Schematic 1.
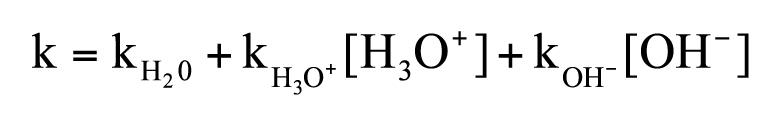
pH-dependence of peptide bond hydrolysis.8
The mechanism of peptide bond hydrolysis under acidic or basic conditions has been described in detail.10–12 In the reaction, a tetrahedral intermediate is formed and cleavage of the peptide bond follows. According to this mechanism, the side chains of the residues on both sides of the cleavage site are not involved; therefore the frequent fragmentation at Gly-containing sites (in particular Gly-Gly sequence) may be facilitated by a lack of steric hindrance to formation of the intermediate that precedes peptide bond fragmentation. The exceptionally long half-life of the peptide bond derived from model compounds would suggest that fragmentation at Gly-Gly sites is unlikely to be observed. However, as will be shown later in examples, fragmentation at two different Gly-Gly sites (one in the lower hinge/CH2 domain and another in a CH1 domain loop) was reported for mAbs under both acidic and basic conditions.4,5,13
Fragmentation involving the Asp residue.
Fragmentation occurring at the C-terminus of an Asp residue (peptide bond Asp-Xaa, where Xaa is any residue) is one of the most frequent degradation pathways of mAbs under mildly acidic conditions. Several mechanisms involving Asp side-chain have been considered3,14,15 and the currently proposed mechanism of peptide bond hydrolysis15 is described in Schematic 2. The reaction is internal to the Asp residue and begins with a nucleophilic attack of the ionized side-chain carboxylate on the protonated carbonyl carbon of the peptide bond. The peptide C-terminal to Asp residue is released. The N-terminal peptide contains an aspartic anhydride that is very reactive and further hydrolyzes to an Asp residue. When studied in a model peptide,14 hydrolysis is the dominant Asp degradation pathway below pH 3. The cleavage rate decreases with increasing pH (at pH 1 the hydrolysis rate is twice larger than at pH 3). Between pH 3 and pH 5 the peptide bond cleavage occurs at a comparable rate with Asp isomerization, which involves formation of a cyclic imide; above pH 5 the cleavage reaction becomes negligible.14
Schematic 2.

Although systematic studies addressing the effect of the primary sequence on the rate of Asp-Xaa hydrolysis are missing, several conclusions can be drawn: (1) Asp-Pro bond fragmentation is usually much faster than fragmentation of other Asp-Xaa bonds; (2) Xaa may impact the hydrolysis rate due to its charge (by altering the pKa of the Asp residue) or due to its size (by altering the local flexibility of the peptide backbone).
Compared to other Asp-Xaa bonds, the Asp-Pro bond is 8- to 20-fold more labile at pH 2,16 and about 10-fold more labile at pH 4.5,17 presumably due to the enhanced basicity of the proline nitrogen in a peptide linkage.18
Based on the proposed reaction mechanism,15 the ionized form of the Asp side chain is more reactive than the unionized form because it is a better nucleophile; the cleavage rate at an Asp-Xaa bond therefore is expected to increase when the pKa decreases. For glucagon, a 29-residue hormone containing three Asp residues, a difference of about 0.3 units in the pKa of Asp residues correlated with more than doubling of the cleavage rate.15 The pKa can vary due to the interaction with adjacent residues or with residues remote in sequence but in close proximity in the folded structure. For glucagon, it was proposed that the pKa decrease of Asp residues at sites 9 and 15 is due to hydrogen bonding with side-chains of Tyr at position 10 and Ser at position 17, respectively. A similar mechanism involving Asp pKa decrease as a result of H-bonding was proposed for the observed Asp-Asp fragmentation in one complementarity determining region (CDR) of a mAb.7
There is only a slight dependence of the Asp-Xaa cleavage rate on the type of Xaa side chain: the Asp-Gly bond hydrolyzes 1.6 times faster compared with the Asp-Ser bond and 2.3 times faster compared with the Asp-Val bond.19 A subtle, but systematic, trend was also observed for Asp-Lys sequence, which presents an increased cleavage rate (about twice larger at 25°C) compared to Asp-Leu and Asp-Gln.17
Cleavage at the N-terminal side of an Asp residue is described in Schematic 3 and occurs only under acidic conditions by a nucleophilic attack of the Asp side chain to the carbonyl of the preceding residue (position n-1), resulting in a 6-membered ring intermediate that further breaks down into two fragments.20
Schematic 3.

Fragmentation at Xaa-Asp site.3
Cleavage at the N-terminal side of an Asp residue is less frequently observed than at the C-terminal side because the formation of the five-membered ring intermediate is entropically more favored than the formation of the six-membered ring intermediate.3 As expected, the steric hindrance or local flexibility can have a significant effect on the N-terminal Asp hydrolysis. Two hexapeptides containing either Pro-Asp-Gly or Gly-Asp-Gly presented similar hydrolysis rates on the C-terminal site of Asp, but the N-terminal hydrolysis rate for the Pro-Asp-Gly was eight times slower than for the Gly-Asp-Gly.19 It is interesting to note that N-terminal hydrolysis in these model hexapeptides occurred only after hydrolysis at the C-terminal side of Asp, suggesting that hydrolysis events at an Xaa-Asp bond may be determined by the propensity to hydrolysis of the peptide bond on the C-terminal site of Asp.
Fragmentation involving Ser or Thr residues.
Fragmentation of model dipeptides studied under strong alkaline conditions and moderate temperatures (pH 12.6 and 30°C) revealed two important features of the stability of the peptide bond: (1) bulky side chains have a stabilizing effect; (2) residues containing hydroxyalkyl groups facilitate hydrolysis on the N-side based on the electron withdrawing capacity of the OH group (cleavage of Xaa-Ser is about threefold faster than for Xaa-Thr).21 It was shown that Gly-Gly and Gly-Ser bonds have comparable fragmentation rates and at least twice larger than the fragmentation rates of a dozen of other dipeptides studied, thus explaining why protein fragments produced under strong alkaline conditions have Gly, Ser or Thr residues at the N-terminal site.21
The side chain of Ser residue can facilitate fragmentation of the backbone at Xaa-Ser sites in the pH range 3–8 via a different mechanism than the one described above that occurs under strong alkaline conditions (pH larger than 11.5). The alternative mechanism is shown in Schematic 4 and involves sequential formation of two intermediates; the fragmentation is initiated by a nucleophilic addition of the serine OH group to the neighboring N-terminal peptide bond to form an oxazolidine intermediate that further rearranges to an ester intermediate that is finally hydrolysed.22
Schematic 4.

Fragmentation at Xaa-Ser site.3
Although the mechanism described in Schematic 4 is pH-independent in the range pH 3–8, fragmentation adjacent to Ser may be pH-dependent due to different flexibility of protein structure to accommodate the reaction intermediates under different solvent conditions. The importance of protein structure in modulating the cleavage rates is demonstrated by the fact that fragmentation does not occur at every Ser residues in a protein, but only at selected Ser positions.23
Finally, cleavage at Xaa-Ser or Xaa-Thr residues may proceed under basic conditions via an alternative mechanism that involves beta-elimination. It was well-described in the literature that Ser residues (especially those which are phosphorylated) can undergo a beta-elimination reaction with formation of dehydroalanines;24 the formation of dehydroalanine can either lead to fragmentation (the cleavage products are amide and pyruvoyl derivatives; see Schematic 5) or to crosslinking (by reacting with side chains, for instance Lys to form lysinoalanine or with Cys side chains to form lanthionine).11,25
Schematic 5.

β-elimination at Ser residue.11
Fragmentation involving Cys or Cys-Cys sites.
The beta-elimination reaction is one of the most frequent and most important reactions of proteins in alkaline solution involving not only seryl (Schematic 5) and threonyl residues, but also cysteinyl and cystinyl residues.11 The mechanism of peptide bond cleavage for cysteinyl residues, i.e. Cys residues not involved in disulfide bonds, follows the beta-elimination pathway described in Schematic 5. The emphasis in this section will be on backbone fragmentation adjacent to cystinyl residues, i.e., Cys residues involved in disulfide bonds (Schematic 6).
Schematic 6.

Cleavage of a disulfide bond followed by fragmentation at dehydroalanine.11
Three mechanisms of backbone cleavage adjacent to disulfide bonds were considered, alpha-elimination, beta-elimination and direct hydrolysis, and among these the beta-elimination mechanism was found to be consistent with the degradation products observed in proteins.11 The beta-elimination reaction involving disulfide bonds is described in Schematic 6: the cystine bond is broken, which results in formation of dehydroalanine and a persulfide; the persulfide can further dissociate into sulfur and cysteine, and the latter can undergo beta-elimination with formation of another dehydroalanine. Overall, the β-elimination of a disulfide bond can lead to formation of two dehydroalanines, one elemental sulfur and one sulfide. The cleavage of the peptide can subsequently occur to the N-terminal site of the dehydroalanine groups (Schematic 6).
The cleavage of the peptide bond due to formation of dehydroalanines may not occur if, due to close proximity, dehydroalanine (formed at first step) reacts with Cys (formed at the second step) to produce a thioether bond, a non-reducible covalent link. It is important to note that, in mAbs, both thioether formation and backbone cleavage were observed as a result of b-elimination of the disulfide bond between the heavy chain (HC) and light chain (LC).6,26
Future studies may reveal the effect of protein structure on the asymmetric fragmentation of a disulfide bond. For instance, for a mAb of type IgG1, if dehydroalanine is preferentially formed on the HC at step 1, subsequent backbone cleavage can be easily detected because the Fab fragment is detached from the mAb molecule. Alternatively, if dehydroalanine is preferentially formed on the LC at step 1, subsequent backbone cleavage can escape detection because the HC and LC are held together via large domain-domain interfaces, and the Fab fragment will be anchored to the rest of mAb molecule via the heavy chain.
Fragmentation involving Asn residues.
Asn residues have a complex contribution to the peptide bond cleavage due to alternative degradation pathways that the side chain can undergo under different solvent conditions. Under acidic conditions, the Asn residues undergo deamidation via direct hydrolysis to yield Asp residues (iso-aspartic acid is not formed); backbone cleavage can then occur at the newly formed Asp according to the mechanisms described in Schematics 2 and 3. The hydrolysis rate that produces deamidation at acidic pH increases as pH is decreased and does not depend on the residue on the C-terminal side of Asn.27 Under alkaline conditions, the major degradation pathway of Asn is deamidation via a cyclic imide intermediate and subsequent formation of Asp and isoAsp; the deamidation rate is highly dependent on the C-terminal residue of Asn. If Asn undergoes deamidation under alkaline conditions, no subsequent peptide bond cleavage is expected to occur because, as described in Schematics 2 and 3, fragmentation of the peptide bond adjacent to an Asp residue is not observed at basic pH. If the deamidation reaction is very slow (depending on the C-terminal residue), the hydrolysis of Asn-Xaa peptide bond may occur according to the mechanism shown in Schematic 7.27 The reaction is initiated by attack of the side-chain amide nitrogen on the peptide-bond carbonyl to release the C-terminal peptide and form a cyclic imide, which can be further hydrolyzed to form the N-terminal peptide with Asn at the end.27
Schematic 7.

Fragmentation at Asn-Xaa sites.27
There are reports on fragmentation at the N-terminus of peptides with loss of the first two residues via a diketopiperazine formation,3 but this degradation pathway will not be described here because it has not been observed for mAbs.
In conclusion, Schematics 1–7 illustrate the dependence of the fragmentation mechanism on pH and on the presence of specific residues. Temperature is also a strong determinant in inducing detectable backbone cleavage; activation energies of 15–20 kcal/mol and 23 kcal/mol were reported for model peptides17 and mAbs,2 respectively. A summary depicting the main fragmentation sites as a function of pH are shown in Figure 1.
Figure 1.
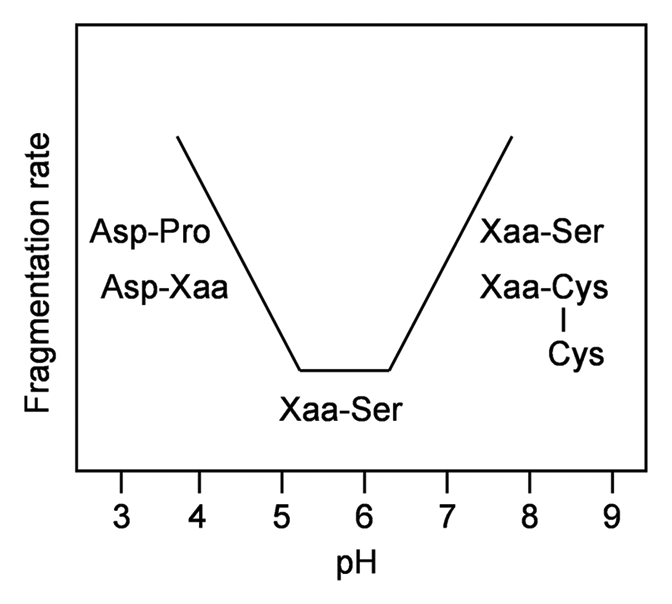
pH-dependence of the major fragmentation sites in a mAb.
In addition to pH, temperature and the chemical nature of the side chains, the rate of fragmentation is also strongly dependent on the presence of metals and formation of radicals. Contrary to the general principles illustrated above, the impact of the latter factors is more difficult to evaluate and control because there may be multiple sources (e.g., formulation composition, exposure to light or different type of surfaces during production or storage) that can modulate their contribution; therefore, the examples presented in the following section are more of qualitative than quantitative reference.
To our knowledge, there are no systematic studies on fragmentation of mAbs occurring during circulation in vivo, after the mAb is delivered to the patient as a drug. The blood represents a very crowded molecular environment at a relatively high temperature, with a strong redox system and a large variety of proteolytic enzymes. As noted in a recent review,28 the current technology has reached a level of maturation that may allow insights on mAb fragmentation events occurring in vivo.
Effect of Metals and Radicals on the Fragmentation Rate
Historically, copper ions were the first reported to significantly increase the fragmentation rate of IgG1 molecules in the hinge region: the reaction is accelerated by increased concentrations of cupric ion and inhibited by ethylenediaminetetraacetic acid (EDTA).29 It is interesting to note that specific hinge cleavage by CuII is significantly higher compared to other metals such as MgII, MnII, ZnII, FeIII and NiII under the same solvent conditions (phosphate-buffered saline at 37°C). The copper-catalyzed cleavage rate may differ between IgG1 molecules depending on their affinity to bind copper ions, and between IgG isotypes depending on the sequence and conformation of their hinge region.
The detailed mechanism of cleavage in the presence of copper ions is not completely understood: the fragmentation rate is minimal around pH 5 and increases with pH. In particular for IgG1 molecules, it was demonstrated29 that the main copper cleavage site is in the hinge region sequence S219CDK222T223HTC between Lys222-Thr223, presumably supported30 by the His224 residue.
Iron atoms in the presence of histidine buffer were found to catalyze cleavage of the hinge region of IgG1 molecules containing lambda LCs; no fragmentation catalyzed by iron atoms was observed in the absence of histidine buffer or for IgG1 molecules with kappa LCs.31 The dominant cleavage sites catalyzed by iron/histidine are on the HC between Ser-Cys and on the LC between Glu-Cys, suggesting that cleavage via disruption of HC-LC disulfide is enhanced for lambda LC-containing mAbs.
Recently, a 3-step, radical-induced hinge fragmentation was described for an IgG1 mAb under strong oxidizing conditions (prolonged exposure to H2O2): first, the reaction is initiated by the formation of a hydroxyl radical via a Fenton-like reaction of a transition metal with H2O2; second, the hydroxyl radical disrupts the HC-HC disulfide at Cys226, yielding the formation of a thiyl radical (Cys226-S) on one cysteine and sulfenic acid (Cys226-SO3H) on the other cysteine; finally, the thiyl radical induces an electron transfer to an upper hinge residue where a cleavage occurs either via diamide or α-amidation pathway.32,33 The radical-induced fragmentation mechanism was supported by the following observations: the fragmentation rate is significantly increased in the presence of H2O2 and can be further enhanced by the presence of iron or copper; very little fragmentation is observed in the absence of H2O2 with or without iron, or with H2O2 but in the presence of a metal chelator (e.g., EDTA). The involvement of a hydroxyl radical in the fragmentation mechanism was demonstrated by complete inhibition of H2O2-induced fragmentation in the presence of catalase. The initial radical formation site on the peptide backbone was identified by peptide mapping upon formation of a covalent adduct with 5,5-dimethyl-1-pyrroline N-oxide.32 Mutagenesis studies suggested that fragmentation induced by the electron transfer along the hinge sequence SCDKTHT is facilitated by the His residue and attenuated by the Lys residue.33
Methods for Detection of Fragmentation
Cleavage of a peptide bond can significantly alter protein properties (particularly if followed by dissociation of the two protein fragments) and, as such, can be detected by many different analytical methods. The analytical methods can be divided into two groups according to their mode of separation: (1) Those where the separation is based primarily on the size of the molecule, such as size-exclusion chromatography (SEC), sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and capillary electrophoresis with SDS (CE-SDS); and (2) those where the separation also involves the chemistry of amino acid side chains, which mainly include various types of chromatography. While the above methods are typically used for monitoring and quantitation of protein fragmentation, the identification of the exact cleavage site is accomplished using mass spectrometry (MS) or N-terminal sequencing, although the latter method is used less frequently now than in the past.29,34,35
Size-based methods provide straightforward data interpretation because other chemical degradations of amino acid side chains, which occur usually at higher rates compared to peptide bond cleavage, are not detected. If separation is based on the size of the molecule, peptide bond cleavage is only detectable when followed by spatial separation of the two fragments. That is often not the case under native conditions where non-covalent interactions or disulfide bridges prevent separation of the two fragments.6,7 This simple mechanistic principle is illustrated when comparing cleavages in the antibody hinge region with cleavages within the immunoglobulin domains. The upper hinge region36 is unique in the sense that it links independent structural units—Fab and Fc fragments—and, when this link is severed by cleavage of the peptide backbone, the two fragments can freely dissociate in solution. Thus, the cleavage in the hinge region is readily detectable by SEC run under native conditions. In contrast, cleavages in folded immunoglobulin domains are typically not detectable without denaturation and often also reduction of disulfide bridges. In fact, due to the significant cross-linking of the antibody molecule by disulfides, their reduction is required to detect peptide bond cleavage in more than a half of the antibody sequence. As illustrated in Figure 2, when the monomeric fraction of a stressed antibody is purified by native SEC and then analyzed by reducing SDS-PAGE, a number of fragment bands can be typically identified, despite the apparent intactness of the monomer by native SEC. Cleavage sites corresponding to these bands are localized within the folded immunoglobulin domains.
Figure 2.

Reducing SDS PAGE analysis of a stressed mAb (2 weeks at 45°C, pH 9) and its SEC-purified monomer. Compared to the whole sample, the monomeric fraction lacks bands corresponding to the hinge cleavage (Fc HC and Fab HC) but contains similar bands corresponding to several cleavages within the immunoglobulin domains. Cleavage in the CH1 domain loop K133STSGGT yields two fragments of approximately 35 and 15 kDa.
SEC is a critical method to monitor antibody aggregation, and it also provides information about fragmentation, but mainly in the hinge region. Two fragment peaks, corresponding to Fab and Fc-Fab, are usually detected when antibodies subjected to various forced-degradation conditions are analyzed by SEC.1,6,13,29,31–33,37 In some relatively rare cases, cleavages outside of the hinge region may be resolved as well by SEC, in particular for highly degraded samples where multiple cleavages in one antibody molecule can aid dissociation of the fragments without the need for denaturation.38 One interesting example was reported for a cleavage in the lower hinge/CH2 domain between G236 and G237;13 after storage at pH 4, the SEC peak that would typically consist of the Fc-Fab fragment (when stored at pH 5–9) contained the N-terminal portion of the antibody with both HCs ending at G236 (a similar Fab2 is obtained by pepsin digestion). Additionally, this cleavage was significantly enhanced in the deglycosylated antibody. Because the G236G237 sequence is within the lower hinge adjacent to the CH2 domain,36 its cleavage in both HCs seems to allow dissociation of the two antibody fragments under native conditions. The formation of the two fragments could be further facilitated by the different, less stable conformation, that the CH2 domain assumes at pH 4.39
SEC is usually the method of choice to quantify the extent of hinge fragmentation, although it is not without complications. Poor resolution between the monomeric peak and the Fc-Fab peak hinders accurate integration, especially for mildly degraded samples. An alternative approach that has been used in our laboratory is to calculate the percentage of the fragmented IgG by multiplying the fraction of the well-resolved Fab-fragment peak by a factor of three (the Fab fragment is approximately 1/3 of the molecule weight).
SEC can also be run under denaturing conditions, for instance, in the presence of guanidine hydrochloride,7 SDS,40,41 or an organic solvent.42 When combined with reduction of the sample, denaturing SEC (dSEC) should, theoretically, detect the same fragments as reducing SDS PAGE, although with significantly lower resolution.
SDS-PAGE6,29,35,43 or its capillary counterpart CE-SDS31,44–49 provides excellent resolution of fragments, and these methods are widely used to monitor overall fragmentation in mAbs. CE-SDS is now commonly used in the pharmaceutical industry due to the straightforward quantification, often better resolution compared with the traditional slab gel SDS PAGE and improved sensitivity with fluorescence detection.45,46 Identification of the cleavage sites, however, is hindered by the difficulty of fraction collection, and consequently, very little31,34 has been published regarding the identity of the observed fragments. Further development is needed in methods that would allow identification of gel bands or CE-SDS peaks. One approach is elution of full-length fragments from the gel and subsequent analysis by mass spectrometry.50 This approach has some advantages over more traditional methods used for band identification, such as N-terminal Edman sequencing and in-gel digestion. As discussed below, cleavage sites are usually clustered in loops resulting in a ladder of fragments with varying N- or C-termini. The N-terminal heterogeneity can significantly complicate data interpretation for N-terminal sequencing and prevent detection of minor fragments. Elution of the full-length fragment avoids these complications, but encounters different challenges related to sensitivity (i.e., faint bands) and intractability (i.e., bands that resist elution). In-gel digestion, although likely more amenable and sensitive than whole-band elution, relies on the detection of the terminal peptide, which may not always be feasible.
Separation methods with contributions from the side-chain chemistry.
Most of the information regarding cleavage sites within antibody immunoglobulin domains come from reversedphase HPLC with in-line MS detection.4–7,40,51,52 Reversed-phase HPLC is a very powerful tool to identify the sites of peptide bond cleavage; however, quantitation can be hindered by low resolution and co-elution with variants resulting from amino acid side-chain degradation. Consequently, this method may not be optimal to monitor overall fragmentation in the molecule. As a technical comment, it is worth noting that due to the low pH and high temperature of reversed-phase HPLC, on-column acid hydrolysis of the D270-P271 bond in the CH2 domain typically occurs to a small degree.53 Liu and coworkers4 have characterized the fragmentation of an entire antibody molecule using reversedphase HPLC; to our knowledge, this is the most comprehensive study of antibody fragmentation. The work is a good example of the power of LC/MS to detect fragments and identify cleavage sites, but it also shows the difficulty of applying this approach to quantitation of individual fragments.
Other methods such as hydrophobic-interaction HPLC49,54 or cation-exchange HPLC55 could detect peptide bond cleavage as well, but are mainly used to detect degradations of the amino acid side chains.
To control the quality of mAb drugs, it is important to accurately quantify degradation processes. For certain sites of fragmentation, “dedicated” methods like SEC for hinge fragmentation or reversed-phase HPLC for particular, well-resolved fragments7 may be the most suitable. On the other hand, reducing SDS PAGE or CE-SDS seem to be the most appropriate for capturing and quantifying the overall fragmentation profile of a molecule.
Cleavage Sites Observed in mAbs
To date, a number of cleavage sites have been reported for mAbs. The cleavages are often clustered in small regions, typically loops connecting individual β-strands, presumably because of the lack of structural constraints and enhanced flexibility. The cleavage sites discussed in this section are sites that were consistently reported by different labs (including ours) and they are likely to be observed by other investigators as well. Nevertheless, the kinetics are very sensitive to pH and other factors, and the relative abundance of different fragments depends also on the separation technique. Often, cleavage sites are indentified in samples exposed to significant stress and for milder conditions, especially the pH range of 5–6 where fragmentation is very slow, there is little information about the dominant fragmentation sites.
When presenting the hot spots of peptide bond cleavage in mAbs, it is helpful to divide the antibody molecule into three regions: (1) the hinge region, which is unique both in terms of its flexibility/solvent exposure and in terms of detection of peptide bond cleavage by native SEC; (2) constant immunoglobulin domains and (3) variable domains, in particular the CDRs, where cleavage-susceptible sites may or may not be present depending on antibody sequence.
Hinge region.
Cleavages in the IgG1 upper hinge region (Fig. 3) have received significant attention, in our opinion, largely due to the ease of detection by native SEC. Multiple mechanisms of scission in the hinge have been proposed, including direct hydrolysis,1,6 β-elimination,6 copper-mediated cleavage,29 and free-radical catalysis.32,33 Hinge region fragmentation has mostly been studied on fragments resolved by native SEC. Cleavages were found across a portion of the upper hinge region spanning the HC-LC and the N-terminal HC-HC disulfide (Fig. 4A). In this region, a single polypeptide connects the Fab fragment with the rest of the molecule and, thus, a cleavage of the backbone is followed by dissociation of the Fab fragment. One additional site located N-terminally of the HC-LC disulfide, between S219 and C220, was also identified. This cleavage is associated with disruption of the HC-LC disulfide via β-elimination, which explains why such fragment is detected by native SEC.6
Figure 3.

Frequently observed cleavage sites in mAbs. Only one heavy chain and one light chain are shown. Dotted lines-disulfide bridges, shaded boxes-cleavage sites. Glycosylated Asn297 is indicated.
Figure 4.

(A) Schematic drawing of a portion of IgG1 and IgG2 hinge region indicating sites of fragmentation. (B) pH-dependence of IgG1 hinge fragmentation monitored by SEC. Fragmentation between S219 and C220 in IgG1 that proceeds via β-elimination is shown as a solid line. The rest of the cleavages are shown as dotted lines. Disulfide bridges are indicated. The samples in (B) were incubated for approximately two weeks at 45°C. To identify the cleavage sites, the Fab fragments were purified by SEC and then analyzed by LC/MS. Considering the relatively wide pH range of 4–10, the cleavage sites can be divided into three groups. (1) Ser-Cys bond, where cleavage occurs via beta elimination, becomes dominant at higher pH. (2) Asp-Lys bond, where cleavage dramatically increases at pH < 5, presumably involving Asp side chain. (3) The rest of the cleavage sites in the hinge, where pH dependence is not as remarkable.
Cleavage in the hinge region has significant pH dependence, both in terms of the magnitude of the fragmentation rate and in terms of the predominant cleavage sites (Fig. 4B).4,6,13 Typically minimal around pH 6, the cleavage significantly increases both at basic and acidic pH. At acidic pH (≤5), the predominant cleavage site is usually at the Asp-Lys bond, which can be explained by the pH-driven mechanism of Asp-Xaa cleavage. At pH > 8, cleavage of the S219-C220 via β-elimination becomes dominant because this reaction is accelerated at alkaline pH. In addition to accelerating particular chemical reactions, pH can also influence antibody structure, which, in turn, may have an effect on the cleavage pattern.4,13 Comparison of antibody fragmentation with fragmentation of a peptide containing the hinge sequence suggests that antibody structure can indeed influence both the rate and the pattern of the cleavage.37 At pH between 5 and 7, where most antibodies are formulated, the cleavages are more equally distributed across the upper hinge SCDKTHTC sequence, although typically showing stronger cleavage between Asp-Lys and His-Thr.1,4,37 Factors other than solution pH can also influence the rate and cleavage sites. A noteworthy example is a specific cleavage between Lys-Thr mediated by copper.29
The hinge region of IgG2 is substantially different from that of IgG1 (Fig. 4A) and seems to be less prone to cleavage.56 One cleavage site (K/CCVECPPC) in the IgG2 hinge was detected by hydrophobic-interaction chromatography.49
Constant immunoglobulin domains.
Compared to the fragmentation of the hinge region, our understanding of peptide bond hydrolysis within immunoglobulin domains is limited. Detailed work on this subject was published by Liu and co-workers4 who studied by LC/MS the dependence of IgG1 fragmentation on pH. A large number of cleavage sites were described; however, from the data it is difficult to compare their relative rates and thus highlight those that are dominant.
When considering the Liu et al. publication with other published work, some general conclusions can be made. Several reports,4,5 as well as our own experience (see Figs. 2 and 5), suggest that, depending on the conditions, cleavages in the constant immunoglobulin domains can occur at comparable or larger rates compared to cleavages in the hinge. Similar to hinge fragmentation, pH is an important factor both in terms of the magnitude and localization of the cleavages. At pH 7 and above, a CH1 domain loop K133STSGGT (Fig. 3) can become the most prevalent cleavage site in the entire antibody (including the hinge region).4 The cleavage in the K133STSGGT loop yields two fragments of approximately 35 and 15 kDa (Fig. 2). While this cleavage is accelerated at higher pH it also occurs at pH 5–7,4 and it is commonly observed in formulated antibodies (our unpublished results and ref. 5). The homologous loop (with a different sequence—R133STSEST) in IgG2 is also prone to cleavage and, interestingly, the rates differ between the IgG2-A and IgG2-B disulfide isoforms.41 Starting slightly at pH 5 and increasing dramatically at lower pH, two specific sites within the CH2 domain (Fig. 3), G236-G237,4,13 and D270-P271,34,40,53 undergo cleavage rapidly. The D270-P271 bond is subject to acid hydrolysis involving the Asp side chain, and therefore, is specifically accelerated under low pH conditions. The susceptibility of the G236-G237 bond may be related to conformational changes of the CH2 domain at low pH.39 The new conformation may lower constraints that the structure typically imposes on degradation rates or may even specifically accelerate the degradation.
Figure 5.

Comparison of the cleavage pattern in three monoclonal antibodies. The three antibodies were stored in PBS at pH 7.0 for three months at 45°C. All antibodies show comparable cleavage in the hinge (Fc HC band, the Fab HC band is not apparent in Ab3 due to co-migration with the light chain), comparable cleavage in the K133STSGGT loop (in agreement with the sequence of the variable domain the N-terminal fragment (CH1 band) showed different molecular weight), comparable cleavage in the CL domain. Ab3 has two additional bands that correspond to two cleavages (seen as a doublet on the small N-terminal fragment) in heavy chain CDR3.
A number of other cleavage sites both in the HC and LC,4,35,49,51 particularly around domain-domain interfaces4,51 have been described, but they seem to be less significant in terms of the abundance of their products or, based on the data, it is not possible to assess their abundance. In light of this, we propose to use the cleavage rate in the hinge region as an internal reference to compare the rates of cleavage at other sites in the mAb.
Variable domains.
Because the variable domains, in particular the CDRs, are unique to each antibody, it can be expected that the susceptibility to peptide bond hydrolysis will also differ greatly. Nevertheless, there are so far no reports showing significant rates of peptide bond hydrolysis in the CDR, and therefore, this degradation is of a lesser concern than degradations of amino acid side chains such as deamidation, isomerization or oxidation. Several examples of cleavage sites in CDRs have been reported in references 4, 7, 49 and 52. We have compared three IgG1κ antibodies in their susceptibility to fragmentation in phosphate buffered saline at pH 7.0. While all showed the same bands corresponding to cleavages in the hinge and in the constant domains, one antibody had an additional cleavage site in heavy chain CDR3 (Fig. 5).
Effect of Fragmentation on the Function of mAbs
Fragmentation in the CDRs is likely to have an effect on the binding of a mAb to the target and, consequently, have an effect on its potency. We are not aware of any reports in the literature to describe the altered binding affinity to a target for a mAb with fragmentation occurring in the CDR; it is probable that, unless the CDR-clipped species is purified and tested, the low levels of clipping in CDRs may preclude any detectable changes by potency assays.
Fragmentation in the hinge region may have more implications on the function of a mAb molecule: the Fab fragment generated will be devoid of any Fc-mediated effector function and have a reduced circulation half-time; the Fc-Fab fragment may not be potent at all if interaction with the target receptor requires both Fab arms. For these reasons, the effect of the hinge fragmentation on the potency of a mAb has to be evaluated as a function of the mechanism of action.
Depending on the cleavage site, fragmentation in the constant regions of mAbs may have an effect on either the Fc-mediated effector function or on the circulation half-time. To our knowledge, there are currently no reports regarding the effect of fragmentation in the constant regions of mAbs on potency.
Finally, fragmentation may have an effect on the quality of mAb materials by altering the aggregation rates. Aggregation is an important degradation pathway due to potential immunogenic response that aggregates can trigger and, consequently, only very low levels of aggregates are considered acceptable in a mAb formulation. There may be multiple mechanisms that lead to formation of aggregates, and the contribution of each pathway to the overall population of aggregated species will depend significantly on pH, temperature and other solvent conditions.40,41 Whether fragmentation leads to aggregation or fragmentation is increased in aggregates is still up for debate; studies of IgG2 mAbs in the pH range 4 to 6 propose that either fragmentation is facilitated by aggregation40 or that aggregation is induced by fragmentation.41 It is interesting to note that, at pH 4, only aggregated species but not the monomeric form had a cleavage at the Asp-Pro bond in the CH2 domain.40 At pH 5, differences in aggregation rates between IgG2 A and B isoforms were attributed to increased fragmentation of a loop in the CH1 domain for the IgG2A isoform (the IgG2 isoforms A and B differ in the disulfide connectivity in the hinge region, hence subtle conformational differences in the loop flexibility may have an effect on the fragmentation rate).41 The impact of CDR fragmentation on the aggregation rate has to be evaluated caseby- case; the very limited observations published so far suggest that, in a particular case, cleavage in HC CDR3 does not trigger aggregation.7
Acknowledgments
The authors are grateful to Dr. Steve L. Cohen for insightful discussions and comments on the manuscript.
Abbreviations
- HC
heavy chain
- LC
light chain
- SEC
size-exclusion chromatography
- CE
capillary electrophoresis
- CDR
complementarity determining regions
References
- 1.Cordoba AJ, Shyong BJ, Breen D, Harris RJ. Nonenzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818:115–121. doi: 10.1016/j.jchromb.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 2.Kamerzell TJ, Li M, Arora S, Ji JA, Wang YJ. The relative rate of immunoglobulin gamma 1 fragmentation. J Pharm Sci. 2010;100:1341–1349. doi: 10.1002/jps.22389. [DOI] [PubMed] [Google Scholar]
- 3.Bernard Testa, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism. Verlag Helvetica Chimica Acta and Wiley-VCH; 2003. [Google Scholar]
- 4.Liu H, Gaza-Bulseco G, Lundell E. Assessment of antibody fragmentation by reversed-phase liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:13–23. doi: 10.1016/j.jchromb.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 5.Dillon TM, Bondarenko PV, Rehder DS, Pipes GD, Kleemann GR, Ricci MS. Optimization of a reversedphase high-performance liquid chromatography/mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. J Chromatogr A. 2006;1120:112–120. doi: 10.1016/j.chroma.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Cohen SL, Price C, Vlasak J. Beta-elimination and peptide bond hydrolysis: two distinct mechanisms of human IgG1 hinge fragmentation upon storage. J Am Chem Soc. 2007;129:6976–6977. doi: 10.1021/ja0705994. [DOI] [PubMed] [Google Scholar]
- 7.Xiao G, Bondarenko PV. Identification and quantification of degradations in the Asp-Asp motifs of a recombinant monoclonal antibody. J Pharm Biomed Anal. 2008;47:23–30. doi: 10.1016/j.jpba.2007.11.050. [DOI] [PubMed] [Google Scholar]
- 8.Smith RM, Hansen DE. The pH-Rate Profile for the Hydrolysis of a Peptide Bond. J Am Chem Soc. 1998;120:8910–8913. [Google Scholar]
- 9.Radzicka A, Wolfenden R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J Am Chem Soc. 1996;118:6105–6109. [Google Scholar]
- 10.Brown RS, Bennet AJ, Sleboka-Tilk H. Recent perspectives concerning the mechanism of H3O+ and OH− promoted amide hydrolysis. Acc Chem Res. 1992;25:481–488. [Google Scholar]
- 11.Whitaker JR. Chemical Deterioration of Proteins. Washington, DC: American Chemical Society; 1980. Changes cccuring in proteins in alkaline solution; pp. 145–164. [Google Scholar]
- 12.Pan B, Ricci MS, Trout BL. Molecular mechanism of acid-catalyzed hydrolysis of peptide bonds using a model compound. J Phys Chem B. 2010;114:4389–4399. doi: 10.1021/jp905411n. [DOI] [PubMed] [Google Scholar]
- 13.Gaza-Bulseco G, Liu H. Fragmentation of a recombinant monoclonal antibody at various pH. Pharm Res. 2008;25:1881–1890. doi: 10.1007/s11095-008-9606-3. [DOI] [PubMed] [Google Scholar]
- 14.Oliyai C, Borchardt RT. Chemical pathways of peptide degradation. IV. Pathways, kinetics and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm Res. 1993;10:95–102. doi: 10.1023/a:1018981231468. [DOI] [PubMed] [Google Scholar]
- 15.Joshi AB, Sawai M, Kearney WR, Kirsch LE. Studies on the mechanism of aspartic acid cleavage and glutamine deamidation in the acidic degradation of glucagon. J Pharm Sci. 2005;94:1912–1927. doi: 10.1002/jps.20405. [DOI] [PubMed] [Google Scholar]
- 16.Marcus F. Preferential cleavage at aspartyl-prolyl peptide bonds in dilute acid. Int J Pept Protein Res. 1985;25:542–546. doi: 10.1111/j.1399-3011.1985.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 17.Li N, Fort F, Kessler K, Wang W. Factors affecting cleavage at aspartic residues in model decapeptides. J Pharm Biomed Anal. 2009;50:73–78. doi: 10.1016/j.jpba.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Piszkiewicz D, Landon M, Smith EL. Anomalous cleavage of aspartyl-proline peptide bonds during amino acid sequence determinations. Biochem Biophys Res Commun. 1970;40:1173–1178. doi: 10.1016/0006-291x(70)90918-6. [DOI] [PubMed] [Google Scholar]
- 19.Oliyai C, Borchardt RT. Chemical pathways of peptide degradation. VI. Effect of the primary sequence on the pathways of degradation of aspartyl residues in model hexapeptides. Pharm Res. 1994;11:751–758. doi: 10.1023/a:1018944800691. [DOI] [PubMed] [Google Scholar]
- 20.Inglis AS. Cleavage at aspartic acid. Methods Enzymol. 1983;91:324–332. doi: 10.1016/s0076-6879(83)91030-3. [DOI] [PubMed] [Google Scholar]
- 21.Noll BW, Jarboe CJ, Hass LF. Kinetic studies on the alkali-catalyzed hydrolysis and epimerization of model alkyl and hydroxyalkyl di- and tripeptides. Biochemistry. 1974;13:5164–5169. doi: 10.1021/bi00722a018. [DOI] [PubMed] [Google Scholar]
- 22.Strickley RG, Brandl M, Chan KW, Straub K, Gu L. High-performance liquid chromatographic (HPLC) and HPLC-mass spectrometric (MS) analysis of the degradation of the luteinizing hormone-releasing hormone (LH-RH) antagonist RS-26306 in aqueous solution. Pharm Res. 1990;7:530–536. doi: 10.1023/a:1015829119270. [DOI] [PubMed] [Google Scholar]
- 23.Windisch V, DeLuccia F, Duhau L, Herman F, Mencel JJ, Tang SY, et al. Degradation pathways of salmon calcitonin in aqueous solution. J Pharm Sci. 1997;86:359–364. doi: 10.1021/js9602305. [DOI] [PubMed] [Google Scholar]
- 24.Patchornik A, Sokolovsky M. Nonenzymatic cleavages of peptide chains at the cysteine and serine residues through their conversion into dehydroalanine I. hydrolytic and oxidative cleavage of dehydroalanine residues. J Am Chem Soc. 1964;86:1206–1212. [Google Scholar]
- 25.Correia JJ, Lipscomb LD, Lobert S. Nondisulfide crosslinking and chemical cleavage of tubulin subunits: pH and temperature dependence. Arch Biochem Biophys. 1993;300:105–114. doi: 10.1006/abbi.1993.1015. [DOI] [PubMed] [Google Scholar]
- 26.Tous GI, Wei Z, Feng J, Bilbulian S, Bowen S, Smith J, et al. Characterization of a novel modification to monoclonal antibodies: thioether cross-link of heavy and light chains. Anal Chem. 2005;77:2675–2682. doi: 10.1021/ac0500582. [DOI] [PubMed] [Google Scholar]
- 27.Patel K, Borchardt RT. Chemical pathways of peptide degradation. III. Effect of primary sequence on the pathways of deamidation of asparaginyl residues in hexapeptides. Pharm Res. 1990;7:787–793. doi: 10.1023/a:1015999012852. [DOI] [PubMed] [Google Scholar]
- 28.Correia IR. Stability of IgG isotypes in serum. mAbs. 2010;2:221–232. doi: 10.4161/mabs.2.3.11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith MA, Easton M, Everett P, Lewis G, Payne M, Riveros-Moreno V, et al. Specific cleavage of immunoglobulin G by copper ions. Int J Pept Protein Res. 1996;48:48–55. doi: 10.1111/j.1399-3011.1996.tb01105.x. [DOI] [PubMed] [Google Scholar]
- 30.Allen G, Campbell RO. Specific cleavage of histidine-containing peptides by copper(II) Int J Pept Protein Res. 1996;48:265–273. doi: 10.1111/j.1399-3011.1996.tb00840.x. [DOI] [PubMed] [Google Scholar]
- 31.Ouellette D, Alessandri L, Piparia R, Aikhoje A, Chin A, Radziejewski C, et al. Elevated cleavage of human immunoglobulin gamma molecules containing a lambda light chain mediated by iron and histidine. Anal Biochem. 2009;389:107–117. doi: 10.1016/j.ab.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Yan B, Yates Z, Balland A, Kleemann GR. Human IgG1 hinge fragmentation as the result of H2O2-mediated radical cleavage. J Biol Chem. 2009;284:35390–35402. doi: 10.1074/jbc.M109.064147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates Z, Gunasekaran K, Zhou H, Hu Z, Liu Z, Ketchem RR, et al. Histidine residue mediates radicalinduced hinge cleavage of human IgG1. J Biol Chem. 2010;285:18662–18671. doi: 10.1074/jbc.M110.108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davagnino J, Wong C, Shelton L, Mankarious S. Acid hydrolysis of monoclonal antibodies. J Immunol Methods. 1995;185:177–180. doi: 10.1016/0022-1759(95)00110-v. [DOI] [PubMed] [Google Scholar]
- 35.Rao PE, Kroon DJ. Orthoclone OKT3. Chemical mechanisms and functional effects of degradation of a therapeutic monoclonal antibody. Pharm Biotechnol. 1993;5:135–158. [PubMed] [Google Scholar]
- 36.Brekke OH, Michaelsen TE, Sandlie I. The structural requirements for complement activation by IgG: does it hinge on the hinge? Immunol Today. 1995;16:85–90. doi: 10.1016/0167-5699(95)80094-8. [DOI] [PubMed] [Google Scholar]
- 37.Xiang T, Lundell E, Sun Z, Liu H. Structural effect of a recombinant monoclonal antibody on hinge region peptide bond hydrolysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858:254–262. doi: 10.1016/j.jchromb.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 38.Gao SX, Zhang Y, Stansberry-Perkins K, Buko A, Bai S, Nguyen V, et al. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol Bioeng. 2011;108:977–982. doi: 10.1002/bit.22982. [DOI] [PubMed] [Google Scholar]
- 39.Wang S, Ionescu R, Peekhaus N, Leung JY, Ha S, Vlasak J. Separation of post-translational modifications in monoclonal antibodies by exploiting subtle conformational changes under mildly acidic conditions. J Chromatogr A. 2010;1217:6496–6502. doi: 10.1016/j.chroma.2010.08.044. [DOI] [PubMed] [Google Scholar]
- 40.Van Buren N, Rehder D, Gadgil H, Matsumura M, Jacob J. Elucidation of two major aggregation pathways in an IgG2 antibody. J Pharm Sci. 2009;98:3013–3030. doi: 10.1002/jps.21514. [DOI] [PubMed] [Google Scholar]
- 41.Perico N, Purtell J, Dillon TM, Ricci MS. Conformational implications of an inversed pH-dependent antibody aggregation. J Pharm Sci. 2009;98:3031–3042. doi: 10.1002/jps.21539. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Gaza-Bulseco G, Chumsae C. Analysis of reduced monoclonal antibodies using size exclusion chromatography coupled with mass spectrometry. J Am Soc Mass Spectrom. 2009;20:2258–2264. doi: 10.1016/j.jasms.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Jiskoot W, Beuvery EC, De Koning AA, Herron JN, Crommelin DJ. Analytical approaches to the study of monoclonal antibody stability. Pharm Res. 1990;7:1234–1241. doi: 10.1023/a:1015925519154. [DOI] [PubMed] [Google Scholar]
- 44.Hunt G, Moorhouse KG, Chen AB. Capillary isoelectric focusing and sodium dodecyl sulfate-capillary gel electrophoresis of recombinant humanized monoclonal antibody HER2. J Chromatogr A. 1996;744:295–301. doi: 10.1016/0021-9673(96)00437-2. [DOI] [PubMed] [Google Scholar]
- 45.Hunt G, Nashabeh W. Capillary electrophoresis sodium dodecyl sulfate nongel sieving analysis of a therapeutic recombinant monoclonal antibody: a biotechnology perspective. Anal Chem. 1999;71:2390–2397. doi: 10.1021/ac981209m. [DOI] [PubMed] [Google Scholar]
- 46.Salas-Solano O, Tomlinson B, Du S, Parker M, Strahan A, Ma S. Optimization and validation of a quantitative capillary electrophoresis sodium dodecyl sulfate method for quality control and stability monitoring of monoclonal antibodies. Anal Chem. 2006;78:6583–6594. doi: 10.1021/ac060828p. [DOI] [PubMed] [Google Scholar]
- 47.Michels DA, Brady LJ, Guo A, Balland A. Fluorescent derivatization method of proteins for characterization by capillary electrophoresis-sodium dodecyl sulfate with laser-induced fluorescence detection. Anal Chem. 2007;79:5963–5971. doi: 10.1021/ac0705521. [DOI] [PubMed] [Google Scholar]
- 48.Rustandi RR, Washabaugh MW, Wang Y. Applications of CE SDS gel in development of biopharmaceutical antibody-based products. Electrophoresis. 2008;29:3612–3620. doi: 10.1002/elps.200700958. [DOI] [PubMed] [Google Scholar]
- 49.Valliere-Douglass J, Jones L, Shpektor D, Kodama P, Wallace A, Balland A, et al. Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal Chem. 2008;80:3168–3174. doi: 10.1021/ac702245c. [DOI] [PubMed] [Google Scholar]
- 50.Cohen SL, Chait BT. Mass spectrometry of whole proteins eluted from sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. Anal Biochem. 1997;247:257–267. doi: 10.1006/abio.1997.2072. [DOI] [PubMed] [Google Scholar]
- 51.Liu H, Gaza-Bulseco G, Sun J. Characterization of the stability of a fully human monoclonal IgG after prolonged incubation at elevated temperature. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;837:35–43. doi: 10.1016/j.jchromb.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 52.Zhao H, Graf O, Milovic N, Luan X, Bluemel M, Smolny M, et al. Formulation development of antibodies using robotic system and high-throughput laboratory (HTL) J Pharm Sci. 2010;99:2279–2294. doi: 10.1002/jps.22008. [DOI] [PubMed] [Google Scholar]
- 53.Rehder DS, Dillon TM, Pipes GD, Bondarenko PV. Reversed-phase liquid chromatography/mass spectrometry analysis of reduced monoclonal antibodies in pharmaceutics. J Chromatogr A. 2006;1102:164–175. doi: 10.1016/j.chroma.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 54.Valliere-Douglass J, Wallace A, Balland A. Separation of populations of antibody variants by fine tuning of hydrophobic-interaction chromatography operating conditions. J Chromatogr A. 2008;1214:81–89. doi: 10.1016/j.chroma.2008.10.078. [DOI] [PubMed] [Google Scholar]
- 55.Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol. 2008;9:468–481. doi: 10.2174/138920108786786402. [DOI] [PubMed] [Google Scholar]
- 56.Ishikawa T, Ito T, Endo R, Nakagawa K, Sawa E, Wakamatsu K. Influence of pH on heat-induced aggregation and degradation of therapeutic monoclonal antibodies. Biol Pharm Bull. 2010;33:1413–1417. doi: 10.1248/bpb.33.1413. [DOI] [PubMed] [Google Scholar]