

Abstract
Purpose
We previously reported that a fibroblast growth factor (FGF) receptor (FGFR) signaling pathway drives growth of lung cancer cell lines of squamous and large cell histologies. Herein, we explored FGFR dependency in cell lines derived from the tobacco-related malignancy, head and neck squamous cell carcinoma (HNSCC).
Experimental Design
FGF and FGFR mRNA and protein expression was assessed in nine HNSCC cell lines. Dependence on secreted FGF2 for cell growth was tested with FP-1039, an FGFR1-Fc fusion protein. FGFR and EGFR-dependence was defined by sensitivity to multiple inhibitors selective for FGFRs or EGFR.
Results
FGF2 was expressed in eight of the nine HNSCC cell lines examined. Also, FGFR2 and FGFR3 were frequently expressed while only two lines expressed FGFR1. FP-1039 inhibited growth of HNSCC cell lines expressing FGF2, identifying FGF2 as an autocrine growth factor. FGFR inhibitors selectively reduced in vitro growth and ERK signaling in three HNSCC cell lines while three distinct lines exhibited responsiveness to both EGFR and FGFR inhibitors. Combinations of these drugs yielded additive growth inhibition. Finally, three cell lines were highly sensitive to EGFR TKIs with no contribution from FGFR pathways.
Conclusions
FGFR signaling was dominant or co-dominant with EGFR in six HNSCC lines while three lines exhibited little or no role for FGFRs and were highly EGFR-dependent. Thus, the HNSCC cell lines can be divided into subsets defined by sensitivity to EGFR and FGFR-specific TKIs. FGFR inhibitors may represent novel therapeutics to deploy alone or in combination with EGFR inhibitors in HNSCC.
Keywords: FGF2, FGFR, head and neck squamous cell carcinoma, receptor tyrosine kinase, tyrosine kinase inhibitor
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most frequent cancer worldwide with a cancer associated five-year survival rate less than 50% (reviewed in (1–4)). Risk factors include tobacco use, alcohol consumption, human papilloma virus exposure and genetic disorders such as Fanconi Anemia. Mutations in well-defined oncogene drivers such as KRAS and EGFR are generally rare in HNSCC (2) relative to other solid tumors such as non-small cell lung cancer (NSCLC). Yet, frequent over-expression of EGFR and EGFR ligands is seen in HNSCC tumors, with EGFR over-expression identified in up to 90% of tumors (3, 5). Based on this rationale, EGFR-specific tyrosine kinase inhibitors (TKIs) and blocking antibodies such as cetuximab have been tested in HNSCC patients. Cetuximab yielded modest increases in patient survival when used in combination with standard chemo- and radiotherapies and has been an approved treatment for HNSCC since 2006 (reviewed in (2, 6)).
The increased understanding of tumor heterogeneity dictated by different driver oncogenes dominant in distinct subsets of tumors has led to the concept of personalized therapy in NSCLC, breast cancer and colorectal cancer (7–9). By contrast, personalized treatment approaches for HNSCC lag behind, due in part to limited information on the dominant oncogenes in this cancer. In this regard, the modest impact of EGFR inhibitors observed in HNSCC may, in fact, reflect an averaged response whereby a subset of highly EGFR-dependent tumors exist within a larger set of EGFR-independent tumors. As a precedent, the initial clinical evaluation of EGFR-specific TKIs in lung cancer patients noted a rather small benefit in survival relative to the larger responses when treatment was restricted to patients with lung tumors bearing mutated EGFR (8, 10). Attempts to correlate clinical response to EGFR targeting with cetuximab in HNSCC with EGFR protein expression (2) or EGFR gene copy number (11) have proven negative. Thus, it will be necessary to stratify HNSCC into therapy-responsive subsets by a means other than the simple EGFR expression or mutation.
We hypothesize that the insensitivity of the majority of HNSCC to EGFR inhibitors is mediated by dominant activity of alternative receptor tyrosine kinase (RTK) systems in distinct subsets of tumors. Among RTKs with reported activity in HNSCC, the literature highlights MET (12), IGF-1R (13) and cooperative roles between PDGFR and VEGFR (14). Although no functionality has been explored, co-expression of FGFs and FGFRs has also been observed in HNSCC tumors compared to normal epithelia and dysplastic lesions of the head and neck (15, 16). We, and others, have demonstrated that FGF2 and FGFRs participate in autocrine signaling contributing to both intrinsic and acquired EGFR TKI resistance in NSCLC lines (17–19). A general role for FGFs and FGFRs is emerging in multiple cancers (reviewed in (20–22)) including prostate (23, 24), thyroid (25, 26), skin (27, 28), lung (reviewed in (29)), urinary bladder (30, 31), and head and neck (15, 32) cancers. Taken together, these studies show FGF and FGFR-mediated oncogenesis through gene amplification, somatic mutations, and increased expression of FGFs and/or FGFRs in human cancer. Thus, FGFs and FGFRs are likely to play roles in cancer equal to or greater in scope to that of EGFR.
The family of FGFs is encoded by 22 distinct genes that bind and activate a family of four receptor tyrosine kinases designated FGFR1 - FGFR4 (20–22). The extracellular domain of FGFRs contains two or three immunoglobulin-like (Ig-like) loops where the two membrane-proximal loops encode the FGF binding site. Of particular importance to FGF binding specificity is the third Ig loop, the N-terminal half of which is encoded by an invariant IIIa exon with alternative usage of IIIb or IIIc exons for the C-terminal half. As a general rule, FGFRs encoding exon IIIb (FGFR IIIb) are expressed on epithelial cells while the FGFRs encoding exon IIIc (FGFR IIIc) are expressed on mesenchymal cells. By contrast, the ligands for FGFR IIIb are often expressed in mesenchymal cells while ligands for FGFR IIIc are expressed in epithelial cells. This establishes a paracrine mechanism of signaling between epithelia and mesenchyme that is critical to normal development and tissue homeostasis. Moreover, FGFs and FGFRs become involved in oncogenesis through acquisition of somatic mutations within the receptors that confer gain-of-function, over-expression of specific FGFRs or inappropriate expression of one or more FGFs to establish autocrine or paracrine signaling (20–22). In regards to the latter, FGF genes are targets of murine mammary tumor virus equal in frequency to the better known WNT genes and mediate virus-dependent murine mammary tumorigenesis (33).
In a recent study, we demonstrated an autocrine growth factor role for FGF2 or FGF9 and FGFR1 and FGFR2 in NSCLC, especially in cell lines of squamous and large cell histology (18). The demonstration of selective FGFR1 gene amplification in lung squamous cell carcinomas and lung cancer cell lines further supports the role of the FGFR signaling pathway in specific lung cancer histologies (34). In the present study, we have explored the involvement of FGF and FGFR autocrine signaling in HNSCC, also of squamous histology. The results reveal frequent co-expression of FGF2 and FGFRs in HNSCC cell lines, thereby instituting an autocrine loop that, alone or in collaboration with EGFR, drives cell growth. FGF2 and FGFRs may define a distinct subset of HNSCC tumors to targeted with emerging FGFR-specific TKIs (22).
Materials and Methods
Cell Culture
The authenticity of the HNSCC cell lines used in this study was validated by fingerprint analysis by the University of Colorado Cancer Center DNA Sequencing and Analysis Core. All cell lines were routinely cultured in DMEM (UMSCC8, UMSCC19, UMSCC25, HN4, HN31, CCL30, Detroit562) or RPMI-1640 (Ca9-22, 584-A2) growth medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) with 1% penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO) at 37°C in a humidified 5% CO2 incubator. To reduce mitogenic effects of serum components, cells were switched to HITES medium (RPMI-1640 containing 10 nM hydrocortisone, 5 μg/ml insulin, 10 μg/ml transferrin, 10 nM estradiol, 30 nM Na3SeO3 and 1% bovine serum albumin) where indicated. Primary human oral keratinocytes (HOKs; ScienCell, Carlsbad, CA), the immortalized human keratinocyte cell line, HaCaT (gift of Dr. Xiao-Jing Wang, UC Anschutz Medical Campus) and primary human gingival fibroblasts (HGF-1; ATCC, Manassas, VA) were obtained and cultured according to the suppliers.
Quantitative real-time PCR
Total RNA was purified from cells using RNeasy™ mini kits (QIAGEN, Valencia, CA) and aliquots (5 μg) were reverse transcribed in a 20 μl volume using random hexamers and MMLV reverse transcriptase. Aliquots (5 μl) of the reverse transcription reactions were submitted to PCR reactions with SYBR® green Jumpstart Taq Readymix (Sigma-Aldrich, St. Louis, MO) using an I Cycler (BioRad, Hercules, CA). Primers used for FGF2, FGFR1 and FGFR2 QPCR assays are previously described (18) and QPCR analysis of FGFR3 mRNA was performed with forward primer 5′-CCA TCG GCA TTG ACA AGG AC-3′ and reverse primer 5′-GCA TCG TCT TTC AGC ATC TTC AC-3′. Expression of the different mRNAs in samples was normalized to GAPDH mRNA levels measured by quantitative RT-PCR in replicate samples. Data are presented as “Relative Expression”.
Growth Assays
To measure the effect of inhibitors on single cell colony formation in a clonogenic assay, HNSCC cells were seeded in 6-well plates at 100 to 200 cells/well in full growth media. Twenty-four hours later, cells were treated with RO4383596 (provided by Hoffman-La Roche), AZ12908010 (provided by AstraZeneca), AG1478 (Calbiochem, San Diego, CA), Gefitinib (provided by AstraZeneca) or FP-1039 (http://www.cancer.gov/drugdictionary/?CdrID=599037; provided by FivePrime Therapeutics, San Francisco, CA) and cultured for 9 additional days. Wells were rinsed with phosphate-buffered saline (PBS) and stained and fixed with 0.5% (wt/vol) crystal violet in 6.0% (vol/vol) gluteraldehyde solution (Fisher Scientific, Fair Lawn, NJ). Following destaining with distilled water, the plates were photographed and total colony area was quantified using the MetaMorph imaging software program (Molecular Devices, Downingtown, PA).
For measurement of anchorage-independent cell growth in soft agar assay, 20,000 cells were suspended in 1.5 ml growth medium containing 10% FBS and 0.35% Difco™ agar noble (Becton, Dickinson and Co., Sparks, MD) and overlaid on base layers containing 1.5 ml growth medium containing 10% FBS and 0.5% Difco™ agar noble in 6-well plates. Wells were fed once a week with 2 mL growth medium containing inhibitors. The plates were incubated for 21 days and viable colonies were stained for 24 hrs with 200 μl of 1 mg/ml nitroblue tetrazolium. Digital photographs of wells were used to quantify colonies using MetaMorph.
FGF2 ELISA
HNSCC cell lines were cultured in HITES medium for 24 hours. Subsequently, the rinsed cell monolayers were lysed in 250 μl MAP Kinase Lysis buffer (MKLB; 0.5% Triton X-100, 50 mM β-glycerophosphate (pH 7.2), 0.1 mM Na3VO4, 2 mM MgCl2, 1 mM EGTA, 1mM DTT, 0.3 M NaCl, 2 μg/ml leupeptin and 4 μg/ml aprotinin) and supernatants from centrifuged lysates were assayed for FGF2 using a Quantikine- human FGF basic assay kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Cellular FGF2 measured by ELISA was normalized to total cellular protein and the data are presented as pg FGF2/mg cell protein.
Immunoblot Analyses
For analysis of phospho-ERK levels in HNSCC cells, 1.5 × 105 cells were plated in wells of 6-well plates. The next day, cells were placed in HITES media for 2 hours to minimize signaling from serum factors and subsequently treated with concentrations of AG1478 (0–0.3 μM) or RO4383596 (0–1 μM) for 2 hours. Cells were rinsed, lysed in 250 μl MKLB and centrifuged (5 min at 13,000 RPM). Supernatants (100 μl) were mixed with 25 μl SDS sample buffer and proteins resolved by SDS-PAGE. Following electrophoretic transfer to nitrocellulose filters and blocking in 3% bovine serum albumin (BSA; Cohn Fraction V, ICN Biomedicals, Inc., Aurora, OH) in Tris-buffered saline with 0.1% Tween 20 (TTBS), the filters were incubated for 16 hours at 4°C with rabbit polyclonal anti-phospho-ERK (Cell Signaling Technology, Inc., Danvers, MA). The filters were washed with four changes of TTBS, then incubated with alkaline phosphatase-coupled goat anti-rabbit antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 1 hour and developed with LumiPhos reagent (Pierce, Rockford, IL) according to the manufacturer’s instructions. The filters were then stripped and re-probed for total ERK1 and ERK2 using a mixture of rabbit polyclonal anti-ERK1 (sc-93) and ERK2 (sc-154) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
For immunoblot analysis of FGFR1, FGFR2, FGFR3, EGFR, and the NaK-ATPase α-subunit, MKLB extracts from HNSCC cells were submitted to SDS-PAGE using 7.5% acrylamide separating gels. Nitrocellulose filters were blocked in 3% non-fat dehydrated milk in TTBS for one hour at room temperature and incubated with antibodies to FGFR1 (sc-57132), FGFR2 (sc-122), FGFR3 (sc-13121) or NaK-ATPase α-subunit (sc-21712) from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). EGFR was detected with a rabbit polyclonal antibody (Cell Signaling Technology, Inc., cat. no. 2232). Filters were then incubated with alkaline phosphatase-coupled secondary antibodies for 1 hour and developed with Lumi-Phos reagent (Thermo Fisher Scientific, Rockford, IL).
Results
HNSCC cell lines co-express FGF2 and FGFR1-3
We previously demonstrated that a subset of NSCLC cell lines, especially of squamous and large cell histologies, co-express FGFs and FGFRs and induce autocrine signaling contributing to transformed growth (18). To explore if FGFs and FGFR signaling may play a similar role in HNSCC, previously published Affymetrix microarray data (35) from HNSCC cell lines were queried for expression of FGF and FGFR genes (Supplementary Table 1). Of the 22 FGF family members, FGF2 was expressed in the majority of these lines and FGF5 was expressed in one cell line, 584-A2. Expression of the remaining FGFs was not detected. FGFR1 expression was highly expressed in a single HNSCC cell line (584-A2), but expression of FGFR2 and FGFR3 was predicted in the majority of the cell lines. FGFR4 expression was not expressed as assessed by the Affymetrix data (Supplementary Table 1) and was not studied further. Thus, expression array data suggest that components of an FGF2-FGFR signaling pathway are generally detected in HNSCC cell lines.
A panel of nine HNSCC cell lines was analyzed for FGF2 and FGFR function. The clinical characteristics of the tumors from which the cell lines were derived are summarized in Supplementary Table 2. Among these, HN4, UMSCC8 and HN31 have previously been defined as sensitive to the EGFR-specific TKI, gefitinib, while 584-A2 was resistant (35). With the exception of 584-A2 and CCL30, all of the HNSCC cell lines express EGFR protein (see Figure 2A). Quantitative RT-PCR analysis of FGF2 mRNA (Figure 1A) and ELISA analysis of cellular FGF2 protein (Figure 1B) revealed significant expression in six of the nine HNSCC cell lines (584-A2, HN31, HN4, Detroit562, CCL30 and UMSCC8). FGF2 expression was reproducibly detected, albeit at a lower level, in Ca9-22 and UMSCC19 while UMSCC25 was negative for FGF2. Consistent with the microarray data (Supplementary Table 1), FGF7 and FGF9 mRNAs were uniformly low or undetectable in HNSCC cell lines as measured by quantitative RT-PCR (data not shown). Measurement of FGFR expression status in the nine HNSCC cell lines revealed high levels of FGFR1 mRNA (Fig. 1C) and protein (Fig. 2A) in 584-A2 with HN31 and CCL30 expressing lower, but detectable levels. By contrast, six of the HNSCC cell lines (UMSCC8, UMSCC25, Detroit562, Ca9-22, HN4 and HN31) expressed detectable FGFR2 mRNA and protein (Figs. 1C and 2A) and four of the lines (UMSCC8, UMSCC25 CCL30 and Ca9-22) expressed FGFR3 mRNA and/or protein. Thus, the data in Figures 1 and 2 provide evidence for general expression of FGF2, FGFR2 and FGFR3 and restricted expression of FGFR1 in a panel of nine HNSCC cell lines.
Figure 2. Expression of FGFRs in HNSCC cell lines and normal cellular constituents of oral tissue.

A, Cell lysates from the indicated HNSCC cell lines were immunoblotted for FGFR1, FGFR2, FGFR3, EGFR and the NaK-ATPase α-subunit as a loading control. B, Total RNA prepared from HNSCC cell lines (UMSCC8 and 584-A2), primary human oral keratinocytes (HOK), immortalized human keratinocytes (HaCaT cells) and human gingival fibroblasts (HGF-1) was reversed transcribed and submitted to RT-PCR for FGF2 mRNA. The values are normalized to GAPDH mRNA as described in Figure 1A. C, Cell lysates from the indicated cell lines were immunoblotted for FGFR1, FGFR2, FGFR3 and the NaK-ATPase α-subunit as a loading control.
Figure 1. Expression of FGF2 and FGFRs in HNSCC cell lines.

A, Total RNA prepared from a panel of nine HNSCC cell lines was reverse transcribed and submitted to quantitative real-time PCR for FGF2 mRNA. The values are normalized to GAPDH mRNA measured in replicate samples and presented as relative expression. B, Cellular FGF2 protein was measured in the nine HNSCC cell lines by ELISA (R&D Systems). The data are the means and SEM of 3 independent experiments. C, Quantitative RT-PCR assays for FGFR1, FGFR2 and FGFR3 mRNAs were performed on the nine HNSCC cell lines and normalized for GAPDH mRNA levels. The data are the means and SEM of 3 or more replicate experiments.
FGF2 mRNA levels in primary human oral keratinocytes (HOK) and immortalized human keratinocytes (HaCaT) were markedly lower than expression levels in the HNSCC cell lines and human gingival fibroblasts (HGF-1) (Fig. 3A). Likewise, the level of FGFR1 protein expression in HOK and HaCaT cells was undetectable compared to the level measured in 584-A2 cells and the gingival fibroblasts (Fig. 3B). By contrast, the relatively high levels of FGFR2 and FGFR3 in UMSCC8 cells (relative to the other 8 HNSCC cell lines) were similar to expression observed in HaCaT and HOK cells (Fig. 3B). A simple interpretation of the findings is that FGF2 and FGFR1 levels are increased in HNSCC cell lines, but FGFR2 and FGFR3 are retained at levels similar to non-transformed oral epithelia modeled by HaCaT and HOK cells.
Figure 3. Effect of the FGF ligand trap, FP-1039, on growth of HNSCC cells.
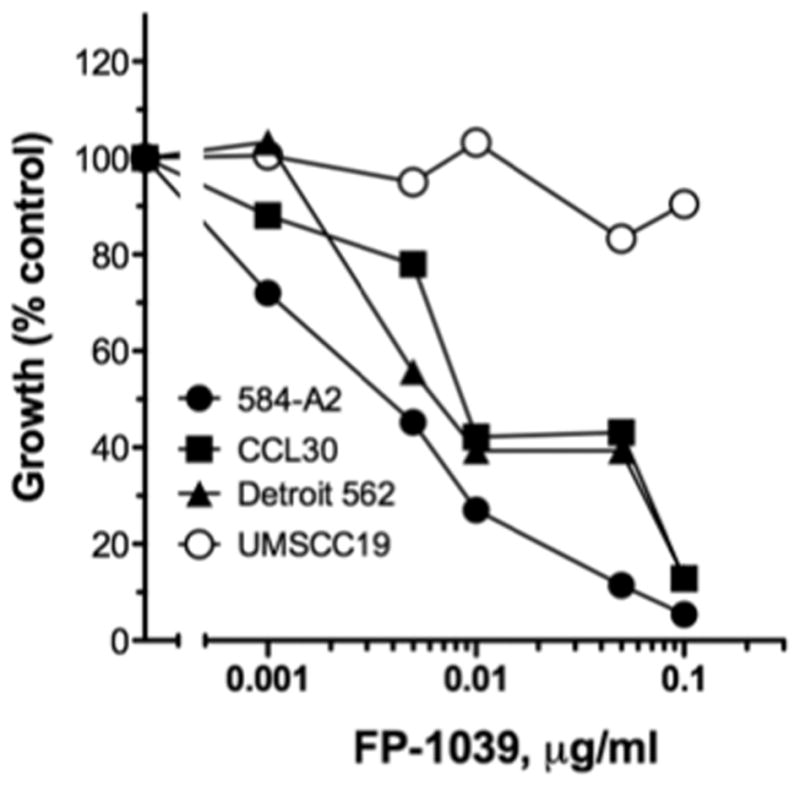
The indicated HNSCC cell lines were submitted to anchorage-independent growth assays in the presence of the indicated concentrations of FP-1039 as described in the Materials and Methods. As shown in Figure 1, 584-A2, CCL30 and Detroit562, but not UMSCC19, express FGF2 mRNA and protein.
FGF2 functions as an autocrine growth factor in HNSCC cell lines
To test the role of FGF2 as an autocrine growth factor in HNSCC cell lines, FP-1039, a soluble Fc fusion protein encoding the extracellular ligand-binding domain of FGFR1c (see Materials and Methods), was used as a ligand trap to sequester FGF2 released into the medium by HNSCC cells. UMSCC19 cells produce little FGF2 (see Figure 1) and cell growth is not inhibited by FP-1039 (Figure 3). By contrast, FP-1039 elicited dose-dependent inhibition of anchorage independent growth of 584-A2, CCL30 and Detroit562 cells that express abundant FGF2. These findings indicate that FGF2 functions as an autocrine growth factor in distinct HNSCC cell lines.
Differential sensitivity of HNSCC cell lines to EGFR and FGFR TKI treatment
As a distinct means to test if the FGF2 and FGFRs constitute a signal pathway contributing to HNSCC growth and transformation, cells were treated with increasing concentrations of the FGFR inhibitor, RO4383596 (18, 36), or an EGFR specific inhibitor, AG1478, and submitted to immunoblot analyses for basal ERK phosphorylation as a measure of proximal RTK signaling. As shown in Figure 4, CCL30 and 584-A2 responded to RO4383596 with a complete, dose-dependent inhibition of basal ERK phosphorylation while Detroit562 cells exhibited partial reduction of phospho-ERK levels. Moreover, AG1478 was without effect on phospho-ERK levels in these cells. By contrast, UMSCC19, HN4, and HN31 exhibited potent inhibition of ERK signaling in response to AG1478, while RO4383596 was without effect. Finally, UMSCC8, Ca9-22 and UMSCC25 cells exhibited partial reduction of phospho-ERK levels in response to both RO4383596 and AG1478. Moreover, a chemically-distinct FGFR-specific TKI, AZ12908010 (19) reduced phospho-ERK levels similarly to RO4383596 (Supplementary Figure 1), supporting the interpretation that FGFRs are driving signaling of the ERK pathway in these cell lines. Based on this signaling endpoint, some cell lines (584-A2, CCL30 and Detroit 562) exhibit dependence solely on FGFR signaling, while others are completely dependent on EGFR signaling (UMSCC19, HN4, HN31). Finally, Ca9-22, UMSCC25 and UMSCC8 responded to both TKIs with inhibition of basal ERK phosphorylation.
Figure 4. Inhibition of basal ERK phosphorylation by EGFR and FGFR-specific TKIs in HNSCC cell lines.

HNSCC cell lines were incubated in serum-free HITES media for 2 hours and then treated with DMSO (control), the FGFR inhibitor, RO4383596, or the EGFR inhibitor, AG1478, at the indicated concentrations for 2 hours. Cell-free extracts were prepared as described in the Materials and Methods and immunoblotted for phospho-ERK. The filters were subsequently stripped and re-probed for total ERK1 and ERK2 to verify equal loading. The data are representative of at least two independent experiments for each cell line.
To determine if TKI-induced inhibition of ERK signaling correlated with effects on HNSCC cell growth, the effect of AG1478 or RO4383596 was tested on anchorage-independent or clonogenic growth. As predicted from the findings in Figure 4, growth of CCL30, Detroit562, and 584-A2 was inhibited by RO4383596, but not AG1478 (Fig. 5A and B). By contrast, growth of cell lines exhibiting dominant EGFR signaling (UMSCC19, HN4 and HN31) was reduced by AG1478, but not RO4383596 (Fig. 5C and D). Finally, UMSCC8, UMSCC25 and Ca9-22 cells exhibit growth inhibition in response to either TKI (Fig. 5E and F), indicating that both EGFR and FGFR contribute to growth and transformation in these cell lines.
Figure 5. Effect of EGFR and FGFR TKIs on growth of HNSCC cell lines.

HNSCC cell lines were submitted to clonogenic assays (UMSCC8, UMSCC25, Ca9-22, HN31) or anchorage-independent growth assays (UMSCC19, CCL30, Detroit562, HN4, 584-A2) in the absence (DMSO, control) or presence of indicated concentrations of RO4383596 or the EGFR-specific TKI, AG1478, as described in the Materials and Methods. Cell lines exhibiting sensitivity to RO4383596, but not AG1478 (A and B) sensitivity to AG1478, but not RO4383596 (C and D), or sensitivity to both RO4383596 and AG1478 (E and F) are grouped accordingly.
The growth inhibitory response of UMSCC8, UMSCC25 and Ca9-22 to RO4383596 and AG1478 suggests that both EGFR and FGFRs are active in these cell lines. Therefore, additive inhibition of growth would be predicted in response to treatment with combinations of TKIs targeting EGFR and FGFRs. UMSCC8, UMSCC25 and Ca9-22 cells were treated with single concentrations of AG1478 (100 nM) or RO4383596 (300 nM) or combinations of both drugs and clonogenic growth was measured. Growth of UMSCC25 cells was inhibited by ~50% with AG1478 or RO4383596 and a further inhibition was observed in response to combined treatment with these TKIs (Fig. 6A). Growth of UMSCC8 and Ca9-22 cells was significantly reduced by AG1478 alone, but not by RO4383596 alone. However, treatment with combined AG1478 and RO4383596 induced significantly greater growth inhibition than AG1478 alone (Fig. 6A). Nearly identical results were obtained when the three HNSCC cell lines were treated with chemically-distinct EGFR and FGFR-specific TKIs, gefitinib and AZ12908010 (19), respectively (Fig. 6B). Moreover, the EGFR-specific TKIs can be replaced with cetuximab, an anti-EGFR antibody inhibitor with similar results (Fig. 6C). These results indicate that a subset of HNSCC lines exist in which both FGFR and EGFR pathways are simultaneously engaged to drive maximal growth. These results are consistent with the functioning of coactivation networks whereby multiple distinct RTKs simultaneously engage in oncogenic signaling rather than single dominant receptors (37).
Figure 6. Inhibition of HNSCC growth by combinations of FGFR and EGFR-specific inhibitors.

A, The indicated HNSCC cell lines shown to be responsive to both EGFR and FGFR-specific TKIs (Fig. 5E and F) were submitted to a clonogenic growth assay in the absence (DMSO, control) or presence of the FGFR TKI, RO4383596 (300 nM), the EGFR-specific TKI, AG1478 (100 nM) or both drugs (AG + RO) at the same concentrations. Clonogenic growth assays were performed with B, EGFR-specific TKI, gefitinib (100 nM), and/or the FGFR- specific TKI, AZ12908010 (300 nM) or C, cetuximab (1 μg/ml) in the presence or absence of AZ12908010 (300 nM). After 2 weeks, colonies were stained and quantified as described in the Materials and Methods. The data are the means and SEM where * indicates p value < 0.05 by unpaired student t test relative to EGFR inhibitor treatment alone.
Discussion
Our study provides evidence for functioning of an FGF2-FGFR autocrine pathway, either alone or in combination with EGFR, to drive HNSCC growth and transformation. Although there is little prior published evidence for activity of FGFs and FGFRs in growth of HNSCC, previous reports support our observation of co-expression of FGF2 and distinct FGFRs. For example, analysis of tissue samples reflecting the progression of HNSCC from normal oral mucosa to carcinomas showed increased FGF2, FGFR2 and FGFR3 expression compared to normal surrounding mucosa (38). Also, FGF2 was detected in conditioned medium from HNSCC cell lines (39) and in urine and serum obtained from head and neck cancer patients (40). Thus, similar to our study showing autocrine growth activity of FGFR signaling pathways in NSCLC of squamous and large cell histologies (18), the present study highlights the FGFR pathway as a novel target for therapeutic intervention in HNSCC. These findings also support and extend our previously published results showing sensitivity of a subset of HNSCC cell lines to the EGFR-specific TKI, gefitinib (35) and indicate HNSCC cell lines can be more precisely classified into subsets that are dependent on 1) EGFR signaling, 2) FGFR signaling or 3) both EGFR and FGFRs.
An important question is how the FGFR autocrine pathway may become engaged or activated during progression of HNSCC. In this regard, cancer research presently focuses on increased activity of specific RTKs through somatic mutations to induce gain-of-function phenotypes or through marked over-expression of specific RTKs, often through gene amplification. In this regard, fluorescence in situ hybridization (FISH) analysis of primary oral squamous cell carcinomas revealed amplification of the FGFR1 gene in ~17% of oral squamous cell tumors and FGFR1 protein over-expression in ~12% (41). Of note, a recent report has shown that FGFR1 is amplified in approximately 20% of NSCLC of the squamous histology, but is rare in lung adenocarcinomas (34). Our own inspection of Affymetrix 6.0 SNP array findings deposited in the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/CellLines/) suggests that 2 of the 22 HNSCC cell lines analyzed (HN; sample ID 907059) and (SCC25; sample ID 910701)) may have increased copy number at the chromosome 8p12 locus containing the fgfr1 gene. While these two particular cell lines were not tested in our study, we did analyze 584-A2 cells that express abundant FGFR1 mRNA and protein (Fig. 2). Analysis of fgfr1 gene copy number by FISH in 584-A2 cells revealed no evidence for amplification (data not shown). Regarding a role for somatic gain-of-function mutations in specific FGFRs, a published report revealed frequent (62%) mutation of FGFR3 (G697C) in primary HNSCC derived from Japanese patients (42). However, an independent screening of a French head and neck cancer patient population revealed no evidence for this FGFR3 mutation (43). Also, the COSMIC database reveals no identified FGFR3 mutations in any of the 22 HNSCC cell lines that were sequenced. Thus, it is possible that FGFR3 mutations may be highly restricted to HNSCC derived from specific ethnic populations. Increased frequency of EGFR mutations in patients of Asian ethnicity provides precedent for this possibility (44).
As an alternative mechanism for the observed FGF2 and FGFR2/FGFR3 co-expression in HNSCC cell lines, we propose that FGFR2 and FGFR3 are expressed in normal epithelial cell precursors exemplified by HOK and HaCaT cells (see Fig. 2) and simply retained and co-opted for a role in transformed growth of HNSCC cells. In support, FGFR2 has been established as a key RTK mediating proliferation and maintenance of normal oral keratinocytes (45–47). The simple retention of FGFR2 and FGFR3 expression as normal mucosa advances to squamous cell carcinoma may be sufficient to contribute to progression of this disease. In this manner, FGFR signaling pathway may contribute to HNSCC transformation through the establishment of an autocrine loop without frequent amplification or mutation of any of the key FGFR components. By contrast, a general induction of FGF2 expression may occur in the transition from early lesions to carcinomas. In support, FGF2 expression is low in HOK and HaCaT cells relative to HNSCC cell lines and gingival fibroblasts (Fig. 2). Interestingly, inspection of two GEO Dataset expression arrays of HNSCC tumor and normal mucosa samples (Supplementary Figure 2) reveals no significant differences in FGF2, FGFR2 and FGFR3 mRNA expression in HNSCC tumors compared to normal tissues. In fact, FGFR3 mRNA levels are somewhat lower in tumors relative to normal tissues. The contributions of FGF2 and FGFR mRNA from non-epithelial stromal cell types present in both the normal and tumor specimens likely accounts for the discrepancy between the findings in Figure 2 and Supplemental Figure 2.
Despite over-expression in ~90% of HNSCC tumors, EGFR inhibitors have not exerted a major therapeutic impact (reviewed in (1–4)). In fact, our previous study (35) and present experiments (see Fig. 5) indicate that only a subset of HNSCC cell lines exhibit a significant growth inhibitory response to EGFR-specific TKIs as single agents. Currently, the only EGFR targeted therapy approved for treatment of locally advanced HNSCC tumors is cetuximab, which has shown modest benefit in patients when used in conjunction with chemotherapy or radiotherapy (2, 6). Clearly, expression of EGFR protein is not an accurate indicator of EGFR dominance in primary HNSCC tumors or in HNSCC cell lines (see Fig. 2A, Supplementary Table 2 and (2, 11)). Importantly, the efficacy of EGFR-specific TKIs for treatment of lung cancer was largely underestimated in early trials that failed to appreciate that benefit was largely restricted to those lung adenocarcinomas bearing mutations in EGFR (8). Thus, the relatively weak clinical efficacy of EGFR inhibitors in HNSCC is predicted from the absence of somatic mutations in EGFR (48) and the failure to selectively target a subset of HNSCC tumors in which EGFR is dominant.
Mutations in defined oncogene drivers are rare in HNSCC relative to NSCLC where subsets of lung cancers bearing mutations in KRAS, PIK3CA, EGFR, ERBB2, ALK and BRAF have emerged (48, 49). Because targeted therapy strategies depend on matching inhibitors with tumors bearing their mutated oncogene targets, the deployment of targeted therapies in HNSCC is problematic. Importantly, this issue is not limited to HNSCC as at least 50% of NSCLC tumors cannot be presently assigned a defined oncogenic driver mutation (49). Yet, we propose that RTKs may still serve as useful targets for therapeutics in HNSCC. Besides EGFR and FGFRs in HNSCC growth, published studies indicate roles for IGF-1R, Met, PDGFR and VEGFR (12–14). In fact, our study and others (48), provide support for the concept of RTK coactivation networks (37) as the more accurate representation of oncogenic growth factor inputs in HNSCC. Our present study demonstrates potent growth inhibition by an FGFR TKI in a subset of HNSCC cell lines while EGFR-specific TKIs exert strong growth inhibition in a distinct subset of cell lines. Finally, a third subset of HNSCC cell lines exhibited additive growth inhibition upon treatment with combined EGFR and FGFR-specific TKIs (Fig. 6). Additional RTKs including IGF-1R and Met are also likely to be actively engaged in growth signaling in this panel of nine HNSCC cell lines. Therefore, identifying the minimal set of RTKs that provide dominant or co-dominant growth inputs in HNSCC as well as discovering biomarkers that define specific RTK networks represent important goals for future investigations. If successful, combinations of TKIs that block the operative RTK coactivation network within a given HNSCC tumor, despite lack of somatic mutations, may prove to be an effective therapeutic strategy for HNSCC.
Supplementary Material
Translational Relevance.
Frequent over-expression of epidermal growth factor receptor (EGFR) is observed in head and neck squamous cell carcinomas (HNSCC). Yet, EGFR inhibitors have failed to exert a major impact on HNSCC patient survival. In this article, we provide evidence that fibroblast growth factor (FGF) 2 and FGF receptors (FGFRs) comprise an important growth factor pathway that functions alone, or in combination with EGFR, in distinct subsets of HNSCC cell lines. Deployment of FGFR-specific tyrosine kinase inhibitors as single agents or in combination with EGFR inhibitors may be effective therapeutic strategies in HNSCC.
Acknowledgments
Grant Support: The studies were supported by NIH grant R01 CA127105.
Abbreviations
- HNSCC
Head and Neck Squamous Cell Carcinoma
- NSCLC
non-small cell lung cancer
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- PDGF
platelet-derived growth factor
- PDGFR
platelet-derived growth factor receptor
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- IGF
insulin-like growth factor
- IGF1-R
insulin-like growth factor 1 receptor
- RTK
receptor tyrosine kinase
- TKI
tyrosine kinase inhibitor
- ERK
extracellular signal regulated kinase
- RT-PCR
real-time polymerase chain reaction
References
- 1.Bozec A, Peyrade F, Fischel JL, Milano G. Emerging molecular targeted therapies in the treatment of head and neck cancer. Expert Opin Emerg Drugs. 2009;14:299–310. doi: 10.1517/14728210902997947. [DOI] [PubMed] [Google Scholar]
- 2.Egloff AM, Grandis JR. Improving Response Rates to EGFR-Targeted Therapies for Head and Neck Squamous Cell Carcinoma: Candidate Predictive Biomarkers and Combination Treatment with Src Inhibitors. J Oncol. 2009;2009:896407. doi: 10.1155/2009/896407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–54. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 4.Murdoch D. Standard, and novel cytotoxic and molecular-targeted, therapies for HNSCC: an evidence-based review. Curr Opin Oncol. 2007;19:216–21. doi: 10.1097/01.cco.0000264952.98166.99. [DOI] [PubMed] [Google Scholar]
- 5.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–72. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 6.Specenier P, Vermorken JB. Cetuximab in the treatment of squamous cell carcinoma of the head and neck. Expert Rev Anticancer Ther. 2011;11:511–24. doi: 10.1586/era.11.20. [DOI] [PubMed] [Google Scholar]
- 7.Duffy MJ, O’Donovan N, Crown J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat Rev. 2011;37:151–9. doi: 10.1016/j.ctrv.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28 (Suppl 1):S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308–24. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non- small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 11.Licitra L, Mesia R, Rivera F, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol. 2011;22:1078–87. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seiwert TY, Jagadeeswaran R, Faoro L, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69:3021–31. doi: 10.1158/0008-5472.CAN-08-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes CJ, Ohshiro K, Rayala SK, El-Naggar AK, Kumar R. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res. 2007;13:4291–9. doi: 10.1158/1078-0432.CCR-06-2040. [DOI] [PubMed] [Google Scholar]
- 14.Bran B, Bran G, Hormann K, Riedel F. The platelet-derived growth factor receptor as a target for vascular endothelial growth factor-mediated anti-angiogenetic therapy in head and neck cancer. Int J Oncol. 2009;34:255–61. [PubMed] [Google Scholar]
- 15.Hasina R, Whipple ME, Martin LE, Kuo WP, Ohno-Machado L, Lingen MW. Angiogenic heterogeneity in head and neck squamous cell carcinoma: biological and therapeutic implications. Lab Invest. 2008;88:342–53. doi: 10.1038/labinvest.2008.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsui IF, Poh CF, Garnis C, Rosin MP, Zhang L, Lam WL. Multiple pathways in the FGF signaling network are frequently deregulated by gene amplification in oral dysplasias. Int J Cancer. 2009;125:2219–28. doi: 10.1002/ijc.24611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer H, Taylor N, Allerstorfer S, et al. Fibroblast growth factor receptor-mediated signals contribute to the malignant phenotype of non-small cell lung cancer cells: therapeutic implications and synergism with epidermal growth factor receptor inhibition. Mol Cancer Ther. 2008;7:3408–19. doi: 10.1158/1535-7163.MCT-08-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marek L, Ware KE, Fritzsche A, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ware KE, Marshall ME, Heasley LR, et al. Rapidly Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in NSCLC Cell Lines through De-Repression of FGFR2 and FGFR3 Expression. PLoS One. 2010;5:e14117. doi: 10.1371/journal.pone.0014117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8:1439–52. doi: 10.1158/1541-7786.MCR-10-0168. [DOI] [PubMed] [Google Scholar]
- 21.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 22.Greulich H, Pollock PM. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol Med. 2011;17:283–92. doi: 10.1016/j.molmed.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Stockton DW, Ittmann M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin Cancer Res. 2004;10:6169–78. doi: 10.1158/1078-0432.CCR-04-0408. [DOI] [PubMed] [Google Scholar]
- 24.Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–24. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- 25.Ezzat S, Huang P, Dackiw A, Asa SL. Dual inhibition of RET and FGFR4 restrains medullary thyroid cancer cell growth. Clin Cancer Res. 2005;11:1336–41. [PubMed] [Google Scholar]
- 26.St Bernard R, Zheng L, Liu W, Winer D, Asa SL, Ezzat S. Fibroblast growth factor receptors as molecular targets in thyroid carcinoma. Endocrinology. 2005;146:1145–53. doi: 10.1210/en.2004-1134. [DOI] [PubMed] [Google Scholar]
- 27.Ozen M, Medrano EE, Ittmann M. Inhibition of proliferation and survival of melanoma cells by adenoviral-mediated expression of dominant negative fibroblast growth factor receptor. Melanoma Res. 2004;14:13–21. doi: 10.1097/00008390-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Logie A, Dunois-Larde C, Rosty C, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet. 2005;14:1153–60. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 29.Kono SA, Marshall ME, Ware KE, Heasley LE. The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors in non-small cell lung cancer. Drug Resist Updat. 2009;12:95–102. doi: 10.1016/j.drup.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakkar AA, Wallerand H, Radvanyi F, et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. 2003;63:8108–12. [PubMed] [Google Scholar]
- 31.Bernard-Pierrot I, Brams A, Dunois-Larde C, et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis. 2006;27:740–7. doi: 10.1093/carcin/bgi290. [DOI] [PubMed] [Google Scholar]
- 32.Streit S, Bange J, Fichtner A, Ihrler S, Issing W, Ullrich A. Involvement of the FGFR4 Arg388 allele in head and neck squamous cell carcinoma. Int J Cancer. 2004;111:213–7. doi: 10.1002/ijc.20204. [DOI] [PubMed] [Google Scholar]
- 33.Theodorou V, Kimm MA, Boer M, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nature Genetics. 2007;39:759–69. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 34.Weiss J, Sos ML, Seidel D, et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–91. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 36.McDermott LA, Simcox M, Higgins B, et al. RO4383596, an orally active KDR, FGFR, and PDGFR inhibitor: synthesis and biological evaluation. Bioorg Med Chem. 2005;13:4835–41. doi: 10.1016/j.bmc.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010;70:3857–60. doi: 10.1158/0008-5472.CAN-10-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakulich C, Jackson-Boeters L, Daley TD, Wysocki GP. Immunohistochemical localization of growth factors fibroblast growth factor-1 and fibroblast growth factor-2 and receptors fibroblast growth factor receptor-2 and fibroblast growth factor receptor-3 in normal oral epithelium, epithelial dysplasias, and squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:573–9. doi: 10.1067/moe.2002.124461. [DOI] [PubMed] [Google Scholar]
- 39.Shemirani B, Crowe DL. Head and neck squamous cell carcinoma lines produce biologically active angiogenic factors. Oral Oncol. 2000;36:61–6. doi: 10.1016/s1368-8375(99)00052-4. [DOI] [PubMed] [Google Scholar]
- 40.Leunig A, Tauber S, Spaett R, Grevers G, Leunig M. Basic fibroblast growth factor in serum and urine of patients with head and neck cancer. Oncol Rep. 1998;5:955–8. doi: 10.3892/or.5.4.955. [DOI] [PubMed] [Google Scholar]
- 41.Freier K, Schwaenen C, Sticht C, et al. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC) Oral Oncol. 2007;43:60–6. doi: 10.1016/j.oraloncology.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Hiraishi Y, Wang H, et al. Constitutive activating mutation of the FGFR3b in oral squamous cell carcinomas. Int J Cancer. 2005;117:166–8. doi: 10.1002/ijc.21145. [DOI] [PubMed] [Google Scholar]
- 43.Aubertin J, Tourpin S, Janot F, Ahomadegbe JC, Radvanyi F. Analysis of fibroblast growth factor receptor 3 G697C mutation in oral squamous cell carcinomas. Int J Cancer. 2007;120:2058–9. doi: 10.1002/ijc.22285. author reply 60. [DOI] [PubMed] [Google Scholar]
- 44.Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–44. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 45.de Giorgi V, Sestini S, Massi D, Ghersetich I, Lotti T. Keratinocyte growth factor receptors. Dermatol Clin. 2007;25:477–85. vii. doi: 10.1016/j.det.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 46.Nagy N, Bata-Csorgo Z, Kopasz N, et al. The expression of keratinocyte growth factor receptor (FGFR2-IIIb) correlates with the high proliferative rate of HaCaT keratinocytes. Exp Dermatol. 2006;15:596–605. doi: 10.1111/j.1600-0625.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- 47.Petiot A, Conti FJ, Grose R, Revest JM, Hodivala-Dilke KM, Dickson C. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development. 2003;130:5493–501. doi: 10.1242/dev.00788. [DOI] [PubMed] [Google Scholar]
- 48.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–34. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.