

Abstract
Synaptic plasticity in the most general terms represents the flexibility of neurotransmission in response to neuronal activity. Synaptic plasticity is essential both for the moment-by-moment modulation of neural activity in response to dynamic environmental cues and for long-term learning and memory formation. These temporal characteristics are served by an array of pre- and post-synaptic mechanisms that are frequently modulated by ethanol exposure. This modulation likely makes significant contributions to both alcohol abuse and dependence. In this review, I discuss the modulation of both short-term and long-term synaptic plasticity in the context of specific ethanol-sensitive cellular substrates. A general discussion of the available preclinical, animal-model based neurophysiology literature provides a comparison between results from in vitro and in vivo studies. Finally, in the context of alcohol abuse and dependence, the review proposes potential behavioral contributions by ethanol modulation of plasticity.
Keywords: ethanol, withdrawal, synaptic plasticity, review
1. Introduction – What is synaptic plasticity?
The chemical synapse allows both the complex integration of information among neurons within a brain region as well as distant communication between distinct areas. Fine-tuning the strength of these synapses, referred to synaptic plasticity, provides flexibility and ultimately regulates behavioral outcomes associated with specific neural circuits. For example, synaptic plasticity in brain regions like the hippocampus or amygdala has been defined as making critical cellular contributions to learning and memory processes. It is important to recognize that plasticity is a relative description. Synaptic strength can either be facilitated or inhibited relative to some basal condition. This can create some caveats and miss-understandings if there is not reliable information about the basal state of a particular synapse. In addition to this directional characteristic, synaptic plasticity is also characterized by the time-course over which it is expression, typically defined as either short-term (spanning milliseconds-to-tens of minutes) or long-term (lasted hours-to-days or longer). These directional and temporal characteristics are of course artificial demarcations with some synapses expressing intermediate, mixed, or multiple phenotypes depending upon the extracellular and intracellular environments and the experimental preparation. However, important for the purposes of this review, distinct cellular mechanisms have been shown to regulate both the direction and time-course. More important still, drugs of abuse and particularly ethanol exposure can modulate these distinct mechanisms in dramatically important ways. The available evidence suggests that synaptic plasticity confers behavioral flexibility. Ethanol modulation of short- or long-term changes in synaptic strength may therefore play a substantial role in the transition from abuse to addiction.
The current review focuses on ethanol modulation of glutamatergic and GABAergic synaptic plasticity in the context of functional measures made with in vitro preparations from young adult/adult animals. The reader is directed to additional reviews or primary literature related to ethanol modulation of developmental plasticity (Berman and Hannigan, 2000; Klintsova et al., 2000; Medina and Krahe, 2008) and plasticity associated with other neurotransmitter systems (Gottesfeld et al., 1989).
2. Ethanol Modulation of Synaptic Plasticity In Vitro
2.1. Short-term Plasticity
Short-term synaptic plasticity has historically been thought of as a presynaptic phenomena related to calcium homeostasis in the synaptic terminal. At least on a time-frame relative to the frequency of action potentials invading a presynaptic terminal, calcium concentrations can raise substantially above resting levels due to incomplete clearance during closely-spaced or trains of presynaptic depolarizations (Zucker and Regehr, 2002). The resulting residual calcium can increase neurotransmitter release via a number of related mechanisms. For example, residual calcium can regulate presynaptic calcium channel function (Cuttle et al., 1998) such that subsequent depolarizations in the terminal result in greater or lesser presynaptic calcium entry. Alternatively, residual calcium can modulate vesicle release probability by directly regulating calcium-dependent components of the release machinery or modulating the size of the readily releasable pool of synaptic vesicles (reviewed in (Neher and Sakaba, 2008)). These mechanisms can alter synaptic responses across a wide range of time frames depending upon the complexity of the signaling pathways required for their initiation, expression, and down-regulation. This section focuses on the effects of acute ethanol administration to tissue slices in vitro and the resulting modulation of synaptic plasticity.
2.1.1. Ethanol modulation of Paired-pulse Plasticity
Experimentally, short-term plasticity has commonly been studied following delivery of two stimuli with inter-stimulus intervals ranging from tens to several hundred milliseconds. These paired pulses can lead to transient facilitation of the second synaptic response of the pair at synapses either characterized by a low intrinsic release probability or following treatments that decrease release probability. Conversely, paired-pulse depression, where the second synaptic response is smaller than the first, is typically found at synapses with a high intrinsic release probability or following treatments that enhance presynaptic neurotransmitter release. The paired pulse ratio calculated from these synaptic responses is therefore inversely related to release probability (Katz et al., 1993). This approach has been used extensively to study the presynaptic effects of ethanol. A wide variety effects are evident in the literature and we have summarized several of these below and in Table 1.
Table 1.
Ethanol Modulation of Paired-Pulse Synaptic Plasticity Measured with In Vitro Neurophysiological Approaches
| Neuro- transmitter | Region/Preparation | Measurea | [EtOH]b | Effect on Ratioc | Commentsd | Citation(s) |
|---|---|---|---|---|---|---|
| Glutamate | Lateral/Basolateral Amygdala | Kainate EPSC |
80mM | No Effect | 50msec interval | Lack et al., 2008 |
| Central Amygdala | AMPA EPSC |
44mM | ↑ | 40msec interval | Zhu et al., 2007 | |
| NMDA EPSC/EPSP |
44mM | No Effect and ↓ | 50-180msec interval, altered by chronic EtOH history | Roberto et al., 2006; Roberto et al., 2004b | ||
| CA1 Hippocampus | NMDA EPSC |
50mM, 120mM | ↑ or No Effect | 50msec interval, preparation- dependent? | Hendricson et al., 2004; Proctor et al., 2006 | |
| CA3 Hippocampus | AMPA EPSC |
50mM | ↑ | Age-dependent, 50msec interval | Mameli et al., 2005 | |
| Dorsal Striatum | EPSC | 100mM | No Effect | 50msec interval | Choi et al., 2006 | |
| Nucleus Accumbens (Shell) | NMDA EPSC |
75mM | ↑ | 50msec interval | Zhang et al., 2005 | |
| VTA Dopamine Neurons | EPSC | 40mM | ↓ | 50msec interval | Xiao et al., 2009 | |
| Crayfish Neuromuscular Junction | EPSP | 60–434mM | No Effect | 5–200msec interval | Blundon and Bittner, 1992 | |
| GABA | Lateral/Basolateral Amygdala | IPSC | 80mM | ↓ and No Effect | 50msec interval, input-dependent | Silberman et al., 2008 |
| Central Amygdala | IPSP/IPSC | 11–66mM | ↓ | 50–100msec interval, concentration- dependent | Kang-Park et al., 2009; Nie et al., 2004; Nie et al., 2009; Roberto et al., 2010; Roberto et al., 2003a; Roberto et al., 2004a; Roberto and Siggins, 2006 | |
| CA1 Hippocampus | IPSC | 80–120mM | ↓ and No Effect | 50–100msec interval, input- dependent | Proctor et al., 2006; Sanna et al., 2004; Wu et al., 2005 | |
| Cerebellar Purkinje Neurons | IPSC | 50mM | ↓ and No Effect | 50msec interval, input-dependent | Criswell et al., 2008; Kelm et al., 2008; Mameli et al., 2008 | |
| Lateral Septum | IPSC | 50mM | No Effect | 50msec interval | Criswell et al., 2008 | |
| Ventrobasal Thalamus | IPSC | 20–100mM | No Effect | 150msec interval | Jia et al., 2008 | |
| VTA Dopamine Neurons | IPSC | 40–50mM | ↓ or ↑ | 50–70msec interval | Guan and Ye, 2010; Theile et al., 2008; Xiao and Ye, 2008 |
In vitro measures of isolated synaptic responses to electrical stimuli recorded with intracellular or whole-cell electrodes. Pharmacologically identified responses are indicated by including the receptor subtype. EPSC = excitatory postsynaptic current (measured with whole-cell voltage-clamp). EPSP = excitatory postsynaptic potential (measured with current clamp). IPSP = inhibitory postsynaptic potential. IPSC = inhibitory postsynaptic current.
Indicates the concentration of ethanol used in the study or studies.
The ratio of amplitudes from fast synaptic responses following pairs of electrical stimuli (“response 2 amplitude/response 1 amplitude” or some variation thereof) is generally held as being inversely related to the probability of neurotransmitter release at that synapse (Zucker, 1989). Increased ratios would therefore reflect decreased release while decreased ratios would indicate increased neurotransmitter release.
“Interval” indicates the inter-stimulus interval between the paired electrical stimuli used in the study or studies.
At glutamatergic synapses for example, acute ethanol can either enhance or inhibit synaptic responses to paired stimuli depending upon the brain region and developmental age of the preparation. Ethanol enhances paired-pulse facilitation at neonatal rat CA3 hippocampal synapses suggesting that it inhibits glutamate release onto these neurons. This modulation is occluded by antagonists of presynaptic N-type voltage-gated calcium channels in a developmentally regulated fashion (Mameli et al., 2005). Ethanol similarly decreases presynaptic glutamate release in adult central amygdala synapses (Zhu et al., 2007) and CA1 hippocampal synapses (Hendricson et al., 2004). The Zhu et al. study and parallel studies by the Morrisett lab (Maldve et al., 2004) suggest that presynaptic calcium channels might be the target for the presynaptic effects of ethanol on glutamate terminals in both cases. Although the precise molecular character of presynaptic calcium channels that might confer acute ethanol sensitivity is not clear, ethanol-induced release of other modulatory neurotransmitters acting specifically on these channels should not be ruled out. For example, ethanol can enhance presynaptic GABA release (see below) which subsequently activates inhibitory presynaptic GABAB hetero-receptors located on glutamate terminals (Steffensen et al., 2000). Acute ethanol can also enhance glutamate release (decrease paired-pulse facilitation or increase paired-pulse depression) at some glutamate synapses. In postnatal day 3–4 CA1 hippocampus for example, ethanol enhances the production of pregnenolone-like neuroactive steroids (Caldeira et al., 2004) that facilitate the presynaptic, calcium-permeable, NMDA-type glutamate receptors expressed by neonatal rats (Mameli and Valenzuela, 2006). Acute ethanol similarly decreases paired-pulse facilitation of glutamate synapses in VTA dopamine neurons from P22-32 rats but via an entirely distinct signaling pathway. Here, ethanol stimulates local dopamine release which activate stimulatory D1 dopamine receptors that enhance glutamate release (Xiao et al., 2009). These D1-mediated effects are sensitive to tetrodotoxin (TTX) suggesting that voltage-dependent processes stimulate either presynaptic signaling pathways (Chu et al., 2010) or the release of postsynaptic retrograde messengers (Andre et al., 2010; Yang, 1999). Acute ethanol modulation of paired-pulse glutamatergic plasticity thus can involve a variety of pre- and post-synaptic elements. However, the precise intra-terminal signaling cascades remain to be fully characterized at most ethanol-sensitive glutamatergic presynapses.
In addition to acute modulation of glutamatergic paired-pulse plasticity, ethanol also regulates short-term synaptic plasticity at GABAergic synapses. In an early study, ethanol was shown to increase the frequency, but not the amplitude, of TTX-insensitive spontaneous inhibitory postsynaptic currents (IPSCs) measured in spinal motor neurons (Ziskind-Conhaim et al., 2003). This suggested to the authors that acute ethanol might enhance presynaptic GABA release. There has subsequently been robust interest in the acute actions of ethanol at presynaptic GABA terminals that has generated several excellent reviews on the subject (Criswell and Breese, 2005; Kumar et al., 2009; Siggins et al., 2005; Weiner and Valenzuela, 2006). We briefly focus here only on short-term GABAergic synaptic plasticity as it is represented by responses to paired-electrical stimuli. As with glutamate, ethanol modulation of GABAergic short-term plasticity is robust in some brain regions and absent in others. In a survey across several brain regions, Criswell and Breese (Criswell et al., 2008) recently found that acute ethanol shifted basal paired-pulse facilitation to paired pulse inhibition in cerebellar Purkinje cells. This apparent increase in GABA release appears to be specific to stellate cell-Purkinje cell synapses as basket cell IPSCs are not similarly modulated (Mameli et al., 2008). The ethanol-dependent increase in GABA release onto Purkinje neurons is qualitatively similar to inhibitory synapses in several other brain regions including proximal inputs onto CA1 hippocampal neurons (Wu et al., 2005), local feed-back inhibitory inputs onto the lateral/basolateral amygdala projection neurons (Silberman et al., 2008), and GABAergic inputs onto substantia nigral neurons (Criswell et al., 2008), VTA dopamine neurons (Theile et al., 2008), and central amygdala neurons (Roberto et al., 2003a). The precise signaling cascades responsible for this ethanol modulation are not understood in every case. However, the mobilization of intra-terminal calcium stores (Kelm et al., 2007), perhaps via ethanol-dependent increases in G protein-coupled receptor signaling (Theile et al., 2009), the activation of phospholipase C, and ethanol-sensitive presynaptic protein kinases (Kelm et al., 2008; Kelm et al., 2010b) appear to all be good candidates. Activation of these presynaptic signaling cascades may be the result of specific interactions between ethanol and presynaptic proteins or may be related to ethanol-dependent release of additional neuromodulators. For example, ethanol-dependent decreases in paired-pulse facilitation at central amygdala GABAergic synapses is blocked by a corticotropin-releasing factor (CRF) type 1 receptor antagonist and is absent in CRFR1 knock-out mice (Nie et al., 2009). Central amygdala CRFR1 receptors are found on synaptic terminals (Jaferi and Pickel, 2009) and can stimulate intracellular calcium mobilization (Riegel and Williams, 2008) and cAMP accumulation (Blank et al., 2003), and can activate various protein kinases (Bajo et al., 2008; Ugolini et al., 2008; Wanat et al., 2008). These findings suggest that a diverse array of direct or indirect signaling pathways could underlie acute ethanol modulation of short-term synaptic plasticity at a given GABAergic terminal. George Breese and colleagues recently published a review of ethanol-enhanced GABA release that focuses on potential signaling and G protein-coupled receptor contributions (Kelm et al., 2010a). It seems reasonable to suggest that similar processes might be involved at ethanol-sensitive presynaptic compartments in both the GABAergic and glutamatergic systems.
Despite these examples, ethanol modulation of short term GABAergic plasticity is not apparent in every brain region or evident under every set of experimental conditions. For example, ethanol does not modulate paired-pulse plasticity in thalamic relay neurons (Jia et al., 2008), lateral septal neurons (Criswell et al., 2008), and the feed-forward inhibitory synapses onto lateral/basolateral amygdala projection neurons (Silberman et al., 2008). These instances appear to represent a genuine insensitivity to the acute ethanol modulation of short-term plasticity at these synapses. However, in some cases, the appearance of an ethanol-insensitive GABAergic presynaptic terminal may be related to the ethanol-dependent increases in GABA release and subsequent activation of presynaptic inhibitory GABAB autoreceptors. This has been shown to mask the apparent facilitation of presynaptic GABA release by ethanol (Silberman et al., 2009; Wu et al., 2005; Zhu and Lovinger, 2006). Additional research into the regional expression of cellular mechanisms distinguishing ethanol-sensitive from – insensitive short-term plasticity might provide important insight into the acute impairing effects associated with ethanol intoxication.
2.1.2. Acute Ethanol Modulation of Post-tetanic Plasticity
The time-course of paired-pulse plasticity (tens to hundreds of milliseconds) suggests it is a prime candidate for providing second-to-second flexibility in information processing. However, exposure of terminals to a more robust stimulation, typically consisting of trains of stimuli lasting several hundred to thousands of milliseconds (tetanus), can result in synaptic plasticity lasting tens of minutes (post-tetanic potentiation or PTP). This type of response provides a more obvious candidate for brief behavioral adaptations like short-term or working memory formation. Importantly, acute ethanol accelerates the decay of PTP, decreasing the time needed for synapses to return to baseline (Gage and Hubbard, 1966). Although the ethanol-specific mechanisms are unclear, the decay of post-tetanic potentiation appears to be directly related to the slow removal of intra-terminal calcium that raises above resting levels during tetanic stimulation (Habets and Borst, 2005). Any cellular mechanism regulating intra-terminal calcium clearance, including plasma membrane and intracellular calcium pumps/exchangers (Kelm et al., 2007), should be considered a potential ethanol-sensitive candidate in this process. Alternatively, post-tetanic facilitation also depends upon the mobilization of the reserve pool of synaptic vesicles to the readily releasable pool (Habets and Borst, 2007). This process requires a number of presynaptic signaling pathways, including cAMP, Ca2+, and PKC (Kuromi and Kidokoro, 2000; Smith, 1999), cytoskeletal rearrangements (Wang et al., 1996), and phosphorylation of a number of vesicle-associated proteins including synapsins (reviewed in (Turner et al., 1999)). Many of these pathways and proteins have already been identified as acute ethanol targets in other systems (Gordon and Diamond, 1993; Kelm et al., 2010b; Popp and Dertien, 2008). It is worth noting that PTP is frequently expressed as a robust synaptic facilitation occurring just prior to the onset of long-term potentiation (LTP) at many synapses. Since ethanol accelerates PTP decay but does not alter its magnitude, ethanol effects on this type of plasticity might not be obvious in most LTP studies. So, despite the obvious behavioral implications of post-tetanic plasticity, ethanol modulation of this form of short-term plasticity has not been explicitly or extensively studied.
2.2. Acute Ethanol Modulation of Long-term Plasticity
As the name implies, the most prominent difference between short-term and long-term plasticity is the enduring nature of the latter. Long-term synaptic plasticity, either facilitation or inhibition, lasts for many hours in vitro (essentially as long as one can measure it) and can last for days/weeks/months/years in vivo. It is this characteristic that has encouraged investigators to define long-term plasticity as the cellular/molecular mechanism responsible for learning and memory. Another characteristic of long-term plasticity that differentiates it from short-term plasticity is the pronounced contributions by postsynaptic signaling cascades.
Long-term facilitation of glutamatergic synapses onto CA1 hippocampal principal neurons requires robust synaptic glutamate release, postsynaptic depolarization, and activation of calcium-permeable NMDA-type glutamate receptors expressed by CA1 neurons themselves (Malenka, 1991). The resulting elevation of intracellular calcium in the neuronal soma activates a number of intracellular signaling pathways including calcium/calmodulin-dependent protein kinase and PKC (Malenka et al., 1989; Malinow et al., 1989). These kinases initiate a series of biochemical events that ultimately increases the trafficking of AMPA receptors from intracellular and extrasynaptic sites to the postsynaptic compartment (Malenka, 2003). While this trafficking process can potentially occur relatively rapidly, it is ultimately consolidated into long-term alterations by changes in gene transcription/translation (Nguyen et al., 1994; Stanton and Sarvey, 1984). Acute ethanol can modulate a number of these cellular/molecular steps. For example, NMDA receptors are inhibited by clinically relevant ethanol concentrations (Lovinger et al., 1989). And, this is consistent with ethanol inhibition of NMDA receptor-dependent LTP initiation at numerous synapses (see Table 2). As noted above, acute ethanol can also enhance GABAergic synaptic transmission which can suppress NMDA receptor activation by hyperpolarizing the postsynaptic membrane (or shunting depolarizing currents) and reinforcing magnesium-block of the NMDA receptor. Acute ethanol can also modulate PKC-dependent intracellular signaling pathways involved in LTP initiation (Bajo et al., 2008; Choi et al., 2008; Kelm et al., 2010b; Kumar et al., 2010; Wilkie et al., 2007; Yao et al., 2008) although this likely does not involve direct ethanol modulation of the kinase (Machu et al., 1991). Ethanol modulation of the cellular/molecular mechanisms directly involved with LTP expression or maintenance, rather than initiation, has not been as extensively studied.
Table 2.
Ethanol Modulation of Long-term Synaptic Plasticity Measured In Vitroa
| Region | EtOH Exposureb | BEC/[EtOH]c | EtOH Effect/Comments | Citation(s) |
|---|---|---|---|---|
| Long-term Potentiation (LTP) | ||||
| Lateral/Basolateral Amygdala | Acute (in vitro) | 80mM | ↓, kainate receptor- dependent | Lack et al., 2008 |
| 7–10 days, liquid diet, 13–17 g/kg/day | N.D. | ↓ | Stephens et al., 2005 | |
| 10 day, vapor inhalation, 12hr/day | 180–250 mg/dL | ↓, kainate receptor- dependent | Lack et al., 2009 | |
| Bed Nucleus Stria Terminalis | Acute (in vitro) | 100mM | ↓, Dorsolateral nucleus | Weitlauf et al., 2004 |
| 4 weeks, vapor inhalation, 14hr/day | 140–170 mg/dL | No Effect and ↓, withdrawal dependent, Juxtacapsular nucleus | Francesconi et al., 2009 | |
| Hippocampus (CA1) | Acute (in vitro) | 5–100 mM | ↓, age-dependent | Blitzer et al., 1990; Grover and Frye, 1996; Izumi et al., 2007; Izumi et al., 2005; Schummers and Browning, 2001; Sinclair and Lo, 1986; Sugiura et al., 1995; Swartzwelder et al., 1995; Tokuda et al., 2007 |
| 7–18 days, liquid diet, 13–17 g/kg/day | 95–180 mg/dL | ↓ | Johnsen-Soriano et al., 2007; Stephens et al., 2005 | |
| 12 weeks, liquid diet | ~60–70 mg/dL | ↑, acute EtOH tolerance | Fujii et al., 2008 | |
| 18 weeks, EtOH in drinking water, 10–14 g/kg/day | N.D. | ↓ | Ripley and Little, 1995 | |
| 7–9 months, liquid diet, ~10g/kg/day | N.D. | ↓, partial recovery after 2–5 month withdrawal | Durand and Carlen, 1984; Tremwel and Hunter, 1994 | |
| 12–14 day, vapor inhalation, 14hr/day | 150–200 mg/dL | ↑ and/or ↓, age-dependent | Roberto et al., 2003b; Sabeti and Gruol, 2008 | |
| Hippocampus (Dentate Gyrus) | Acute (in vitro) | 25–100 mM | ↓ | Morrisett and Swartzwelder, 1993 |
| Dorsal Striatum | Acute (in vitro) | 2–50 mM | ↓, high [EtOH] may convert to LTD | Xie et al., 2009; Yin et al., 2007 |
| 16 days, liquid diet | N.D. | ↑, intracellular EPSP | Yamamoto et al., 1999 | |
| Ventral Tegmental Area | Acute (in vitro) | 40mM | ↓ (GABA) | Guan and Ye, 2010 |
| 2 g/kg, I.P. | N.D. | ↓ (GABA & Glutamate), mouse line-dependent | Guan and Ye, 2010; Wanat et al., 2009 | |
| Long-term Depression (LTD) | ||||
| Bed Nucleus Stria Terminalis | 4 days, vapor inhalation, 16hr/day or continuous | 150–185 mg/dL | ↓, α1 adrenergic- dependent | McElligott et al., 2010 |
| Cerebellum | Acute (in vitro) | 50mM | ↓, climbing & parallel fiber-Purkinje cell synapses | Belmeguenai et al., 2008; Carta et al., 2006; Su et al., 2010 |
| Hippocampus (CA1) | Acute (in vitro) | 60–75 mM | ↑ or ↓ | Hendricson et al., 2002; Izumi et al., 2005 |
| 9–11 months, liquid diet, 15–16 g/kg/day | 150–175 mg/dL | ↓ | Thinschmidt et al., 2003 | |
| Nucleus Accumbens | 4 days, vapor inhalation, 16hr/day | 150–200 mg/dL | ↓, shift to LTP in the shell | Jeanes et al., 2010 |
| Dorsal Striatum | 10–30 days, EtOH in water (6%, forced) | 85 mg/dL | ↓ | Xia et al., 2006 |
| 16 days, liquid diet | N.D. | ↓, shift to LTP | Yamamoto et al., 1999 | |
Refers to the form of synaptic plasticity, either long-term potentiation (LTP) or long-term depression (LTD) measured with in vitro electrophysiologic approaches.
Duration, format, and level of exposure are indicated. “Acute” refers to exposures made during in vitro studies on acute tissue preparations. These studies have been included here for the sake of comparisons with the in vivo exposures discussed in the text.
Blood-ethanol concentrations (BEC) are expressed as mg/dL. In vitro concentrations are reported here as mmol/L (mM). 80mg/dL is approximately equivalent to 17mM.
In addition to long-term facilitation, acute ethanol also modulates long-term depression in a number of brain regions (Table 2). Long-term depression of parallel fiber glutamatergic inputs onto cerebellar Purkinje neurons requires coincidental activation of parallel and climbing fibers. This activates postsynaptic Group I metabotropic glutamate receptors (mGluR1/5) which mobilize intracellular calcium stores and ultimately lead to the phosphorylation and internalization of postsynaptic AMPA receptors (reviewed in (Luscher and Huber, 2010)). Acute ethanol selectively inhibits parallel fiber LTD (but not LTP) in vitro by suppressing mGluR1 function (Carta et al., 2006) and intracellular signaling (Belmeguenai et al., 2008). A similar outcome is evident for mGluR1-mediated, NMDA receptor-dependent LTD at juvenile CA1 hippocampal neurons (Overstreet et al., 1997). However, some forms of LTD are dependent solely upon NMDA receptor activation (reviewed in (MacDonald et al., 2006)) and would presumably be sensitive to ethanol inhibition as well; but, this needs more extensive investigation. Finally, acute ethanol modulation of long-term plasticity can be complex and representative of the flexibility at individual synapses. For example, ethanol inhibits NMDA receptor-dependent LTP at glutamate synapses onto medium spiney dorsal striatal neurons revealing instead long-term depression (Yin et al., 2007). This ethanol-induced LTD is blocked by a cannabinoid receptor type 1 (CB1) antagonist suggesting that the production of retrograde endocannabinoids known to suppress glutamatergic synapses (Gerdeman et al., 2002), is stimulated by acute ethanol. Acute ethanol modulation of long-term synaptic plasticity therefore appears to involve a wide variety of alcohol-sensitive targets spanning from receptors in the plasma membrane to intracellular signaling pathways to retrograde messengers.
3. Ethanol Exposure In Vivo and the Plasticity of Plasticity
The acute ethanol sensitivity of neurotransmitter receptors and synaptic plasticity itself quickly led to investigations of the effects of in vivo ethanol exposure and withdrawal. With a few notable, more recent exceptions (see below), much this literature has focused on chronic ethanol exposure paradigms. And, much of the early work in this area focused on neurotransmitter receptors and system that were known to be sensitive to acute ethanol in vitro. For example, chronic exposure of mice to an ethanol-containing liquid diet causes increased NMDA receptor binding density in hippocampus, cortex, thalamus, and striatum (Gulya et al., 1991), all regions with NMDA receptor-dependent synaptic plasticity. These initial binding studies were complimented by functional approaches showing that chronic ethanol increases NMDA receptor function at both synaptic and extrasynaptic compartments across a wide variety of brain regions and preparations (reviewed in (Nagy, 2008)). Chronic ethanol exposure also increases expression of Group I mGluRs in several brain regions (Obara et al., 2009). And, withdrawal from chronic ethanol increases Group I mGluR-mediated signaling in cultured cerebellar Purkinje neurons (Netzeband et al., 2002). In general, chronic ethanol appears to up-regulate many of the receptors involved with the initiation of long-term synaptic plasticity as well as several relevant intracellular signaling cascades (reviewed in (Pandey, 1998)). Finally, the effects of chronic ethanol exposure on GABAergic neurotransmission have been the subject of several excellent reviews (Crews et al., 1996; Kumar et al., 2009). Again, the exposure-related changes in GABAergic neurotransmission appear to support the notion that chronic ethanol/withdrawal alter the general balance between excitatory and inhibitory systems in favor of the former. These findings together suggest that chronic ethanol might set up favorable cellular environments for the initiation or expression of long-term synaptic plasticity. Importantly, the physiological outcomes associated with this ethanol-induced plasticity-of-plasticity (“metaplasticity”, (Abraham and Bear, 1996; Deisseroth et al., 1995)) have been investigated in a number of different brain regions (Table 2). Many of these are summarized in the following section.
3.1. Chronic Ethanol/Withdrawal and Hippocampal Synaptic Plasticity
Similar to the effects of acute ethanol, early studies suggested that chronic exposure to an ethanol-containing liquid attenuated LTP induction at Schaffer collateral-CA1 synapses (Durand and Carlen, 1984). In this study, LTP inhibition was not associated with any differences in the overall responsiveness of these synapses to electrical stimuli. However, this seems inconsistent with the withdrawal-associated hyperactivity of Schaffer collateral-CA1 synapses measured in latter studies (Hendricson et al., 2007; Thomas et al., 1998b). And more recent findings suggest that chronic exposure to a liquid diet or ethanol vapor may actually enhance LTP at CA1 glutamate synapses (Fujii et al., 2008; Sabeti and Gruol, 2008). In fact, the Sabeti and Gruol study found that the effects of chronic ethanol on LTP in this brain region are dependent upon the age of the animal during exposure, with increased LTP expression occurring in younger adolescents (28–36 days old) and decreased LTP expression found in older adolescent animals (45–50 days old). This suggests age as a possible variable regulating LTP modulation by chronic ethanol. Regardless, the mechanism governing chronic ethanol-related LTP modulation is not entirely clear. Chronic ethanol exposure up-regulates hippocampal NMDA receptor expression/function (Follesa and Ticku, 1995; Hendricson et al., 2007; Morrow et al., 1994; Nelson et al., 2005; Snell et al., 1996; Thomas et al., 1998a). Ethanol exposure also increases the trafficking of NMDA receptors to synaptic compartments in cultured hippocampal neurons (Carpenter-Hyland et al., 2004). In those cases where chronic ethanol enhances Schaffer collateral-CA1 LTP, it seems reasonable to suggest that this NMDA receptor up-regulation may simply improve the likelihood that a given stimulus would initiate long-term plasticity at those synapses. In those contrasting cases where ethanol exposure attenuates LTP initiation/expression, the exposure may 1) un-couple CA1 NMDA receptors from important intracellular signaling pathways or 2) undermine the mechanisms responsible for LTP expression at these synapses. Specific outcomes associated with chronic ethanol and LTP in CA1 hippocampus may be sensitive to the duration of ethanol exposure or intervening periods of self-imposed abstinence (depending on the form of chronic exposure).
3.2. Chronic Ethanol/Withdrawal Modulation of Amygdala Long-term Synaptic Plasticity
3.2.1. Lateral/Basolateral Amygdala
Classic fear conditioning, where animals learn to associate environmental or interoceptive cues with aversive emotionally-relevant stimuli, is dependent upon the activation of NMDA receptors and increased AMPA receptor-mediated synaptic function in the lateral/basolateral amygdala (BLA; reviewed in (Maren, 2005; Sigurdsson et al., 2007)). These subdivisions represent important input regions that receive processed information from cognitive, sensory, and memory systems. BLA projection neurons are glutamatergic and send axons throughout the extended amygdala (including the central amygdala and bed nucleus of the stria terminalis; (Davis et al., 2010)), to portions of the reward circuit like the nucleus accumbens (Kita and Kitai, 1990), and to many different prefrontal cortical areas (Ghashghaei and Barbas, 2002). So, in addition to learning/memory associated with fearful stimuli, long-term synaptic plasticity in the BLA is also thought to contribute to cue-relapse of drug seeking (Feltenstein and See, 2008) and psychiatric diseases like post-traumatic stress disorder (Adamec, 1997). Thus, ethanol modulation of long-term changes in synaptic efficacy in the BLA would potentially impact a great many cognitive and emotional systems.
Early work in the amygdala suggested that chronic ethanol exposure and withdrawal generally suppressed long-term plasticity or plasticity-dependent behaviors. For example, repeated long-term exposure of rats to an ethanol-containing liquid diet produced deficits in amygdala-mediated fear conditioning (Stephens et al., 2001). This observation was later paralleled by conditioning studies in human binge drinkers (Stephens et al., 2005). In this same study (Figure 1), chronic ethanol/withdrawal diminished electrically-evoked LTP in both the lateral amygdala and CA1 hippocampus. At the time, it was not clear if these adaptations might represent a simple suppression of the neurobiological systems responsible for the initiation or expression of long-term synaptic plasticity. However, there were several observations inconsistent with this. First, chronic ethanol exposure enhanced the expression of anxiety-like behavior during withdrawal and increased c-fos immediate-early gene expression levels throughout the amygdala including the BLA (Borlikova et al., 2006; Knapp et al., 1998). Second, the withdrawal-related increase in anxiety-like behavior is sensitive to pharmacological inhibition of basolateral amygdala glutamatergic neurotransmission (Lack et al., 2007 ). These data suggest that BLA principal neurons are activated by chronic ethanol exposure and withdrawal in a manner that is similar to fear conditioning.
Figure 1. Chronic ethanol exposure occludes distinct forms of long-term synaptic plasticity in the lateral/basolateral amygdala.

(A) The panels are reprinted from Biological Psychiatry (58), Stephens et al., “Repeated ethanol exposure and withdrawal impairs human fear conditioning and depresses long-term potentiation in rat amygdala and hippocampus”, 392–400, Copyright (2005), with permission from Elsevier. (Left) Representation of the horizontal slice preparation used in this study illustrates the relationships between the recording electrode (R), the stimulator (S), the lateral amygdala (LA), and anatomical boundaries like the external capsule. (Right) Theta burst stimulation (TBS) of the external capsule caused robust long-term potentiation of synaptic responses in slices prepared from control animals (M) while repeated withdrawal from a chronic exposure to an ethanol-containing liquid diet (F) reduced the magnitude of this form of plasticity. (B) Panels reprinted from Alcohol (43), Läck et al., “Chronic ethanol and withdrawal effects on kainate receptor-mediated excitatory neurotransmission in the rat basolateral amygdala”, 25–33, Copyright (2009), with permission from Elsevier. (Left) Sample fEPSP responses recorded in basolateral amygdala in control brain slices (CON) or 24hr after a chronic intermittent exposure to ethanol vapor (24WD). Kainate receptor-dependent long-term synaptic potentiation was initiated by a 15min exposure to the agonist ATPA ((RS)-2-Amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid; 5 μM). (Right) The magnitude of ATPA-induced LTP was significantly depressed by chronic ethanol exposure (not shown) and withdrawal.
What about other forms of BLA plasticity? Kainate-type ionotropic glutamate receptors (KA receptors) are structurally and biophysically related to the better studied AMPA receptors (reviewed in (Pinheiro and Mulle, 2006)). In the BLA, KA receptors are expressed on both glutamatergic projection neurons and GABAergic interneurons (reviewed in (Aroniadou-Anderjaska et al., 2007; Braga et al., 2004)). Kainate receptor synaptic currents can be measured from BLA principal neurons using standard electrophysiological approaches (Li and Rogawski, 1998). KA receptors are calcium-permeable; and, activation of BLA KA receptors initiates long-term increases in glutamatergic synaptic transmission in this brain region (Li et al., 2001). Both KA receptors (Crowder et al., 2002; Lack et al., 2008; Valenzuela et al., 1998) and KA receptor-mediated synaptic plasticity in the BLA (Lack et al., 2008) can be inhibited by acute ethanol exposure. This naturally led to studies with chronic ethanol which found that, like BLA NMDA receptors, kainate receptor synaptic function is up-regulated during ethanol exposure. However, unlike NMDA receptors, this increase is transient and returns to control levels during a 24hr withdrawal period (Lack et al., 2009); and, importantly, KAR-dependent plasticity was also disrupted during chronic ethanol and withdrawal (Lack et al., 2009). So, despite elevated synaptic function of kainate receptors, long-term synaptic plasticity mediated by this type of glutamate receptor is impaired (Figure 1).
How can one reconcile the up-regulation of BLA NMDA and KA receptor synaptic function and the apparent attenuation of long-term synaptic plasticity mediated by these receptors? We recently examined AMPA receptors and AMPA receptor-mediated neuronal responses to provide a better understanding of these relationships. Chronic ethanol and especially withdrawal dramatically increased synaptic function of AMPA-type glutamate receptors measured in BLA neurons. The amplitude of both spontaneous and tetrodotoxin-resistant miniature EPSCs were increased during chronic ethanol exposure and particularly during withdrawal (Lack et al., 2007). The postsynaptic nature of this change was confirmed by the treatment-dependent alterations in the kinetics of the miniature EPSCs. Thus, in addition to the neurotransmitter receptors responsible for the initiation of long-term plasticity at glutamate synapses (NMDA & KA receptors), the receptors responsible for the expression of this plasticity (AMPA receptors) were likewise up-regulated (Figure 2). This increased AMPA receptor synaptic function was associated with increased field excitatory postsynaptic potentials (fEPSPs) generated by groups of BLA neurons in response to electrical stimulation (Lack et al., 2009). These alterations together are remarkably similar to those that characterize fear-potentiated startle (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). This suggests that chronic ethanol exposure chemically conditions the BLA such that the mechanisms responsible for the initiation and expression of long-term synaptic plasticity are usurped and up-regulated by the treatment itself. This BLA “Ethanol-LTP” would help explain both the altered fear/aversive-memory formation (Bertotto et al., 2006; Quadros et al., 2003; Ripley et al., 2003) and the increased anxiety following chronic alcohol exposure.
Figure 2. Chronic intermittent ethanol exposure and withdrawal upregulate the synaptic function of AMPA-type glutamate receptors in the lateral/basolateral amygdala.
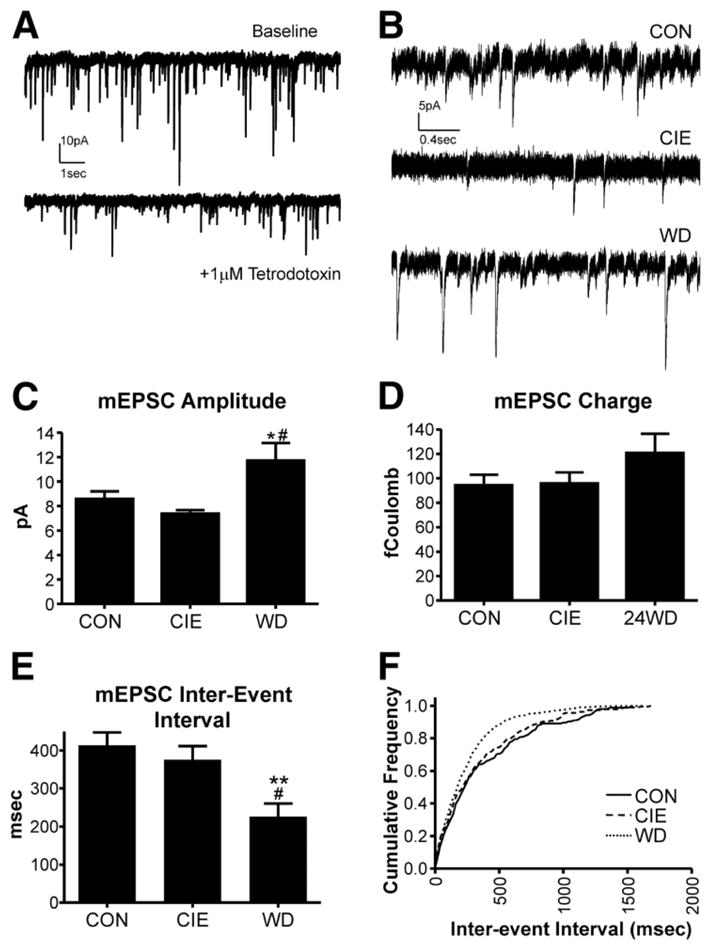
The figure is republished with permission of the American Physiological Society from “Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala”, Läck et al., Journal of Neurophysiology 98(6), 3185–96, 2007; permission conveyed through Copyright Clearance Center, Inc. (A) Exemplar traces of spontaneous glutamatergic synaptic activity recorded from lateral/basolateral amygdala (BLA) principal neurons using whole-cell patch-clamp electrophysiology. In the presence of the voltage-gated sodium channel blocker tetrodotoxin, the resulting miniature EPSCs (mEPSCs) are smaller in amplitude and occur less frequently. (B) Exemplar mEPSCs recorded from control (CON) BLA slices, immediately following a chronic intermittent ethanol exposure (CIE), or 24hr after withdrawal from CIE (WD). (C) Summary of mEPSC amplitude data from the three treatment groups. mEPSC amplitude was significantly greater in WD neurons compared to both CON and CIE. (D) The charge carried by mEPSCs was not significantly different between the treatment groups, largely due to an increase in the decay rate of individual mEPSCs recorded from WD BLA neurons (not shown). (E&F) The interval between mEPSCs was significantly shorter in recordings from WD BLA neurons compared to the other treatment groups.
3.2.2. Central Amygdala
The central amygdala (CeA) is found medial to the BLA and receives heavy glutamatergic input from this and other amygdala subdivisions. This subdivision projects extensively to brain regions that regulate many of the physical and psychological manifestations of fear/anxiety and is critical for the expression of learned-fear behaviors (reviewed in (Davis et al., 2010; LeDoux, 1993)). CeA projection neurons are GABAergic and morphologically resemble medium spiney neurons found in the dorsal striatum and nucleus accumbens (McDonald, 1982; Sun and Cassell, 1993). In addition to excitatory glutamatergic inputs, the activity of CeA projection neurons is regulated by both feed-forward (Royer et al., 1999) and intrinsic (Nose et al., 1991) GABAergic inhibitory circuits. CeA neurons express a variety of neuropeptides including CRF which has been extensively demonstrated to regulate dependence-associated alcohol consumption (Funk et al., 2006).
Like the BLA, chronic ethanol appears to regulate numerous CeA-dependent, withdrawal-related behaviors including enhanced anxiety and ethanol-seeking (reviewed in (Koob, 2009)). Chronic intermittent exposure to ethanol inhalation diminished paired-pulse facilitation of GABAergic responses suggesting increased GABA release in this brain region (Roberto et al., 2004a). It is not presently clear whether this represents altered short-term plasticity of intrinsic GABAergic circuits or feed-forward inhibitory inputs. Regardless, a similar chronic ethanol exposure reduced AMPA-mediated glutamate synaptic function and altered the acute sensitivity of short-term glutamatergic plasticity to ethanol (Roberto et al., 2004b). The parallel increases in both glutamatergic and GABAergic synaptic transmission make it difficult to predict the effects of chronic ethanol on CeA long-term plasticity; and this has not yet been directly assessed.
3.3. Chronic Ethanol Modulation of Synaptic Plasticity in the Dorsal and Ventral Striatum
The effects of in vivo chronic ethanol exposure on synaptic plasticity in the dorsal striatum paint a somewhat more complex picture than it does in either the hippocampus or amygdala. Forced, chronic ethanol consumption suppresses LTD initiation/expression in the dorsal striatum (Xia et al., 2006). This LTD requires postsynaptic co-activation of Group I mGluRs and dopamine D2 receptors and is dependent upon the release endocannabinoids which act as retrograde messengers to suppress presynaptic glutamate release (reviewed in (Lovinger, 2010)). The ethanol-related suppression of LTD might suggest that chronic exposure uncouples mGluR/D2-mediated signaling, endocannabinoid production, or both. Alternatively, striatal LTP is produced in vitro under different experimental conditions that accentuate NMDA receptor function (Lovinger, 2010). Since chronic ethanol increases NMDA receptor function (Gulya et al., 1991; Navamani et al., 1997; Wang et al., 2010) and expression (Raeder et al., 2008) in the striatum, it is also possible that chronic ethanol might alter the relative balance between striatal LTD and LTP. This interpretation is consistent with the observation that withdrawal increases the percentage of striatal slices where LTP (compared to LTD) can be observed experimentally (Xia et al., 2006). It is noteworthy that acute ethanol promotes LTD initiation/expression over LTP (Yin et al., 2007) presumably by inhibiting NMDA receptors. However, acute ethanol does not appear to inhibit striatal LTD (Clarke and Adermark, 2010). Thus, the adaptations of dorsal striatal glutamatergic plasticity to chronic ethanol seem to represent alterations predicted by the acute sensitivity of the individual signaling components responsible for these forms of plasticity.
Recent data in the shell of the nucleus accumbens suggests that similar alterations in long term plasticity might also occur in the ventral striatum following chronic ethanol exposure (Jeanes et al., 2010). Here, acute ethanol inhibits a NMDA receptor-dependent, post-synaptic form of long-term depression at glutamatergic synapses via enhanced D1 dopamine receptor signaling. Within 24hr following chronic ethanol exposure, the same stimulation paradigm produces NMDA receptor-dependent long-term potentiation, instead of depression, at these synapses. Notably, both forms of plasticity were absent 72hr post-withdrawal. These effects very similar to abstinence-related suppression of plasticity in the nucleus accumbens core following prolonged abstinence from cocaine self-administration (Martin et al., 2006). This suggests that chronic exposure to different drugs of abuse with distinct behavioral profiles (stimulant versus sedative) produce similar changes in accumbens synaptic plasticity. However, additional studies are needed to identify the precise neuroadaptations governing these changes in accumbal long-term plasticity.
3.4. Chronic Ethanol Modulation of Synaptic Plasticity in the Bed Nucleus of the Stria Terminalis
In the bed nucleus of the stria terminalis (BNST), chronic ethanol exposure reduces long-term depression of glutamatergic synaptic responses following exposure to the α1 adrenergic receptor agonist methoxamine (McElligott et al., 2010). Methoxamine stimulates clathrin-dependent endocytosis of AMPA-type glutamate receptors by BNST neurons; and, notably, other forms of stress likewise disrupt this adrenergic receptor-dependent process. These findings might suggest that the sensitivity of BNST LTD to chronic ethanol may be specifically related to altered adrenergic receptor function or signaling. Although the effects of chronic ethanol have not been examined in this same brain region, acute ethanol suppresses the early-phase of long-term facilitation of glutamatergic field EPSPs (Weitlauf et al., 2004). This acute modulation (Table 2) appears dependent on ethanol inhibition of NMDA receptors. These findings together suggest that chronic ethanol could potentially disrupt neurotransmitters other than noradrenaline, but this remains to be established.
3.5. Ethanol and Synaptic Plasticity in the Ventral Tegmental Area
Finally, much of the in vivo ethanol-plasticity literature has focused on robust chronic exposures. This raises the question: how long or robust of an exposure is required to cause these changes related synaptic plasticity? Studies with VTA dopamine neurons shed some light on this and suggest that ethanol-induced changes in synaptic plasticity may take place quite rapidly. Here, a single ethanol exposure converts GABAergic paired-pulse facilitation to a GABAB/PKA-dependent depression measured just a day after the exposure (Melis et al., 2002; Wanat et al., 2009). This suggests that long-term modulation of GABAergic plasticity may only require a single ethanol exposure; and, recent evidence suggests this is precisely the case. First, long-term depression of GABAergic inputs onto VTA dopamine neurons following high-frequency electrical stimuli is totally abolished by a single intraperitoneal (IP) dose of ethanol. This is dependent on mu-opioid receptors (Guan and Ye, 2010). Second, a single IP ethanol exposure also reduces postsynaptic synaptic contributions by both AMPA and NMDA receptors and reduces the initiation/expression of long-term potentiation at glutamatergic synapses onto VTA dopamine neurons (Wanat et al., 2009). These single-dose effects are reminiscent of a chemically-induced long-term depression that might represent an adaptive response to acute ethanol-induced facilitation of glutamate release in this brain region (Xiao et al., 2009). Despite these single-dose effects, repeated ethanol exposure via prolonged self-administration enhances post-synaptic contributions by AMPA receptors on VTA dopamine neurons (Stuber et al., 2008) in a manner that is quite similar to long-term potentiation in this brain region (Bonci and Malenka, 1999). It remains to be established whether ethanol attenuation of VTA GABAergic and glutamatergic plasticity reflects an inhibition of the initiation/expression mechanisms or, similar to the amygdala, represents an occlusion resulting from ethanol-induced plasticity.
4. Conclusions & Perspectives
The in vitro modulation of synaptic plasticity can be characterized by a wide array of cellular mechanisms associated with ethanol intoxication. The facilitation of short-term GABAergic plasticity and inhibition of glutamatergic short-term plasticity for example support a growing literature suggesting that acute ethanol tips the balance between GABA and glutamate neurotransmission towards the inhibitory systems. On another level, short-term synaptic plasticity serves as a “frequency filter” (see Fortune & Rose 2002) that can diminish or accentuate closely-spaced synaptic events in order to ultimately alter behaviors directed at or dictated by repeated environmental and intrinsic stimuli. Since acute ethanol exposure modulates goal-oriented attentional processes under some conditions (Olmstead et al., 2006; Zeichner et al., 1993), it seems reasonable to suggest then that modulation of short-term synaptic plasticity might contribute to this aspect of alcohol intoxication. Similarly, ethanol modulation of post-tetanic synaptic plasticity, although not well characterized, potentially modulates behavior over the span of several minutes. This has implications for behaviors related to the intoxication process itself including loss of social inhibitions and loss of control -type behaviors (Loeber and Duka, 2009) associated with binge drinking. It is somewhat easier to imagine the how acute ethanol modulation of long-term plasticity would make substantial behavioral contributions since this form of plasticity underlies memory formation and learning. Inhibition of long-term plasticity by acute ethanol is consistent with the alcohol-induced decrements in working memory (Grattan-Miscio and Vogel-Sprott, 2005), short-term memory (Goodwin and Hill, 1973), and long-term or conditioned memory formation (Land and Spear, 2004; Matthews and Silvers, 2004). It is worth noting that we have artificially segregated short- and long-term plasticity here to emphasize their distinct cellular mechanisms and potential behavioral contributions. However, these forms of plasticity actually represent a continuum of synaptic processes that ultimately regulate behavior on a moment-by-moment time scale.
The effects of in vivo ethanol exposure on synaptic plasticity also seem to vary considerably from brain region to brain region. In the hippocampus, chronic ethanol exposure has complex effects, but most appear consistent with the inhibition of experimentally-induced long-term plasticity. If this inhibition is representative of the behavioral outcomes directed by the hippocampus, these adaptations may attenuate the ability of an alcohol-dependent individual to learn new goal-directed behaviors (Kennedy and Shapiro, 2009). In the amygdala, parallels between learning/memory-related changes on glutamate signaling and the effects of chronic ethanol/withdrawal suggest these alterations may provide the cellular machinery to help negatively reinforce the withdrawal state (Koob, 2009). In the dorsal striatum, the behavioral ramifications of the ethanol-dependent shift from long-term depression to long-term potentiation at glutamate inputs is more difficult to conceptualize until we know more about anatomical and cellular specificity. Regardless, it is tempting to speculate that these adaptations would participate in the shift from more goal-directed to habitual behaviors during the addiction process (Balleine and O’Doherty, 2010; Gerdeman et al., 2003; Goldstein et al., 2009; Palmiter, 2008). Finally, in the VTA, exposure to a single ethanol dose appears to be sufficient to produce a chemical depression of VTA dopamine neurons. This is remarkably contrasted with the robust excitatory effects of acute ethanol on VTA dopamine neurons (Morikawa and Morrisett, 2010) and dopamine release in VTA neuron terminal fields (Robinson et al., 2009). These plasticity-related high and low dopamine states may parallel intoxication/withdrawal cycles in alcoholics that could well contribute to continued ethanol seeking in dependent individuals. It is important to note that in vivo exposure studies provide only a snap-shot of the underlying neuropathology. And, this is contrasted with the longitudinal nature of the addiction process. Ultimately, chronic ethanol/withdrawal may down-regulate plastic processes required for the inhibition of dependence-related behaviors and up-regulate plasticity that is important for either reinforcing drug-reward relationships or negatively reinforcing abstinence itself. Significant additional work is needed to provide a more mechanistic understanding of the contributions made by ethanol modulation of long-term synaptic plasticity.
Acknowledgments
This work was supported by grants from the National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism (R01 AA014445 and P01 AA017056). I am grateful to Dr. Marvin Diaz and Mr. Dan Christian for their constructive comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Adamec R. Transmitter systems involved in neural plasticity underlying increased anxiety and defense--implications for understanding anxiety following traumatic stress. Neurosci Biobehav Rev. 1997;21:755–765. doi: 10.1016/s0149-7634(96)00055-3. [DOI] [PubMed] [Google Scholar]
- Andre VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010;31:14–28. doi: 10.1111/j.1460-9568.2009.07047.x. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Qashu F, Braga MF. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: implications for epilepsy and anxiety disorders. Amino Acids. 2007;32:305–315. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M. Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci U S A. 2008;105:8410–8415. doi: 10.1073/pnas.0802302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69. doi: 10.1038/npp.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmeguenai A, Botta P, Weber JT, Carta M, De Ruiter M, De Zeeuw CI, Valenzuela CF, Hansel C. Alcohol impairs long-term depression at the cerebellar parallel fiber-Purkinje cell synapse. J Neurophysiol. 2008;100:3167–3174. doi: 10.1152/jn.90384.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RF, Hannigan JH. Effects of prenatal alcohol exposure on the hippocampus: spatial behavior, electrophysiology, and neuroanatomy. Hippocampus. 2000;10:94–110. doi: 10.1002/(SICI)1098-1063(2000)10:1<94::AID-HIPO11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Bertotto ME, Bustos SG, Molina VA, Martijena ID. Influence of ethanol withdrawal on fear memory: Effect of D-cycloserine. Neuroscience. 2006;142:979–990. doi: 10.1016/j.neuroscience.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J. Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci. 2003;23:700–707. doi: 10.1523/JNEUROSCI.23-02-00700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–208. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- Blundon JA, Bittner GD. Effects of ethanol and other drugs on excitatory and inhibitory neurotransmission in the crayfish. J Neurophysiol. 1992;67:576–587. doi: 10.1152/jn.1992.67.3.576. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlikova GG, Le Merrer J, Stephens DN. Previous experience of ethanol withdrawal increases withdrawal-induced c-fos expression in limbic areas, but not withdrawal-induced anxiety and prevents withdrawal-induced elevations in plasma corticosterone. Psychopharmacology (Berl) 2006;185:188–200. doi: 10.1007/s00213-005-0301-3. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Li H. The physiological role of kainate receptors in the amygdala. Mol Neurobiol. 2004;30:127–141. doi: 10.1385/MN:30:2:127. [DOI] [PubMed] [Google Scholar]
- Caldeira JC, Wu Y, Mameli M, Purdy RH, Li PK, Akwa Y, Savage DD, Engen JR, Valenzuela CF. Fetal alcohol exposure alters neurosteroid levels in the developing rat brain. J Neurochem. 2004;90:1530–1539. doi: 10.1111/j.1471-4159.2004.02686.x. [DOI] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci. 2004;24:7859–7868. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol potently modulates climbing fiber-->Purkinje neuron synapses: role of metabotropic glutamate receptors. J Neurosci. 2006;26:1906–1912. doi: 10.1523/JNEUROSCI.4430-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DS, Wei W, Deitchman JK, Kharazia VN, Lesscher HM, McMahon T, Wang D, Qi ZH, Sieghart W, Zhang C, Shokat KM, Mody I, Messing RO. Protein kinase Cdelta regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J Neurosci. 2008;28:11890–11899. doi: 10.1523/JNEUROSCI.3156-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SJ, Kim KJ, Cho HS, Kim SY, Cho YJ, Hahn SJ, Sung KW. Acute inhibition of corticostriatal synaptic transmission in the rat dorsal striatum by ethanol. Alcohol. 2006;40:95–101. doi: 10.1016/j.alcohol.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Chu HY, Yang Z, Zhao B, Jin GZ, Hu GY, Zhen X. Activation of phosphatidylinositol-linked D1-like receptors increases spontaneous glutamate release in rat somatosensory cortical neurons in vitro. Brain Res. 2010;1343:20–27. doi: 10.1016/j.brainres.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Clarke RB, Adermark L. Acute ethanol treatment prevents endocannabinoid-mediated long-lasting disinhibition of striatal output. Neuropharmacology. 2010;58:799–805. doi: 10.1016/j.neuropharm.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Crews FT, Morrow AL, Criswell H, Breese G. Effects of ethanol on ion channels. Int Rev Neurobiol. 1996;39:283–367. doi: 10.1016/s0074-7742(08)60670-4. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Breese GR. A conceptualization of integrated actions of ethanol contributing to its GABAmimetic profile: a commentary. Neuropsychopharmacology. 2005;30:1407–1425. doi: 10.1038/sj.npp.1300750. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Kelm MK, Breese GR. Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J Pharmacol Exp Ther. 2008;326:596–603. doi: 10.1124/jpet.107.135418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder TL, Ariwodola OJ, Weiner JL. Ethanol antagonizes kainate receptor-mediated inhibition of evoked GABA(A) inhibitory postsynaptic currents in the rat hippocampal CA1 region. J Pharmacol Exp Ther. 2002;303:937–944. doi: 10.1124/jpet.102.038471. [DOI] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512 ( Pt 3):723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology. 2010;35:105–135. doi: 10.1038/npp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Schulman H, Tsien RW. Synaptic plasticity: A molecular mechanism for metaplasticity. Curr Biol. 1995;5:1334–1338. doi: 10.1016/s0960-9822(95)00262-4. [DOI] [PubMed] [Google Scholar]
- Durand D, Carlen PL. Impairment of long-term potentiation in rat hippocampus following chronic ethanol treatment. Brain Res. 1984;308:325–332. doi: 10.1016/0006-8993(84)91072-2. [DOI] [PubMed] [Google Scholar]
- Feltenstein MW, See RE. The neurocircuitry of addiction: an overview. Br J Pharmacol. 2008;154:261–274. doi: 10.1038/bjp.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follesa P, Ticku MK. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Brain Res Mol Brain Res. 1995;29:99–106. doi: 10.1016/0169-328x(94)00235-7. [DOI] [PubMed] [Google Scholar]
- Francesconi W, Berton F, Repunte-Canonigo V, Hagihara K, Thurbon D, Lekic D, Specio SE, Greenwell TN, Chen SA, Rice KC, Richardson HN, O’Dell LE, Zorrilla EP, Morales M, Koob GF, Sanna PP. Protracted withdrawal from alcohol and drugs of abuse impairs long-term potentiation of intrinsic excitability in the juxtacapsular bed nucleus of the stria terminalis. J Neurosci. 2009;29:5389–5401. doi: 10.1523/JNEUROSCI.5129-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Yamazaki Y, Sugihara T, Wakabayashi I. Acute and chronic ethanol exposure differentially affect induction of hippocampal LTP. Brain Res. 2008;1211:13–21. doi: 10.1016/j.brainres.2008.02.052. [DOI] [PubMed] [Google Scholar]
- Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PW, Hubbard JI. An investigation of the post-tetanic potentiation of end-plate potentials at a mammalian neuromuscular junction. J Physiol. 1966;184:353–375. doi: 10.1113/jphysiol.1966.sp007919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115:1261–1279. doi: 10.1016/s0306-4522(02)00446-3. [DOI] [PubMed] [Google Scholar]
- Goldstein RZ, Craig AD, Bechara A, Garavan H, Childress AR, Paulus MP, Volkow ND. The neurocircuitry of impaired insight in drug addiction. Trends Cogn Sci. 2009;13:372–380. doi: 10.1016/j.tics.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin DW, Hill SY. Short-term memory and the alcoholic blackout. Ann N Y Acad Sci. 1973;215:195–199. doi: 10.1111/j.1749-6632.1973.tb28268.x. [DOI] [PubMed] [Google Scholar]
- Gordon AS, Diamond I. Adenosine mediates the effects of ethanol on the cAMP signal transduction system. Alcohol Alcohol Suppl. 1993;2:437–441. [PubMed] [Google Scholar]
- Gottesfeld Z, Garcia CJ, Lingham RB, Chronister RB. Prenatal ethanol exposure impairs lesion-induced plasticity in a dopaminergic synapse after maturity. Neuroscience. 1989;29:715–723. doi: 10.1016/0306-4522(89)90143-7. [DOI] [PubMed] [Google Scholar]
- Grattan-Miscio KE, Vogel-Sprott M. Effects of alcohol and performance incentives on immediate working memory. Psychopharmacology (Berl) 2005;181:188–196. doi: 10.1007/s00213-005-2226-2. [DOI] [PubMed] [Google Scholar]
- Grover CA, Frye GD. Ethanol effects on synaptic neurotransmission and tetanus-induced synaptic plasticity in hippocampal slices of chronic in vivo lead-exposed adult rats. Brain Res. 1996;734:61–71. [PubMed] [Google Scholar]
- Guan YZ, Ye JH. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving mu-opioid receptors. Neuropsychopharmacology. 2010;35:1841–1849. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulya K, Grant KA, Valverius P, Hoffman PL, Tabakoff B. Brain regional specificity and time-course of changes in the NMDA receptor-ionophore complex during ethanol withdrawal. Brain Res. 1991;547:129–134. [PubMed] [Google Scholar]
- Habets RL, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Dynamics of the readily releasable pool during post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2007;581:467–478. doi: 10.1113/jphysiol.2006.127365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricson AW, Maldve RE, Salinas AG, Theile JW, Zhang TA, Diaz LM, Morrisett RA. Aberrant synaptic activation of N-methyl-D-aspartate receptors underlies ethanol withdrawal hyperexcitability. J Pharmacol Exp Ther. 2007;321:60–72. doi: 10.1124/jpet.106.111419. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA. Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther. 2002;301:938–944. doi: 10.1124/jpet.301.3.938. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Sibbald JR, Morrisett RA. Ethanol alters the frequency, amplitude, and decay kinetics of Sr2+-supported, asynchronous NMDAR mEPSCs in rat hippocampal slices. J Neurophysiol. 2004;91:2568–2577. doi: 10.1152/jn.00997.2003. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Murayama K, Tokuda K, Krishnan K, Covey DF, Zorumski CF. GABAergic neurosteroids mediate the effects of ethanol on long-term potentiation in rat hippocampal slices. Eur J Neurosci. 2007;26:1881–1888. doi: 10.1111/j.1460-9568.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Nagashima K, Murayama K, Zorumski CF. Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience. 2005;136:509–517. doi: 10.1016/j.neuroscience.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Pickel VM. Mu-opioid and corticotropin-releasing-factor receptors show largely postsynaptic co-expression, and separate presynaptic distributions, in the mouse central amygdala and bed nucleus of the stria terminalis. Neuroscience. 2009;159:526–539. doi: 10.1016/j.neuroscience.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanes ZM, Buske TR, Morrisett RA. In vivo chronic intermittent ethanol exposure reverses the polarity of synaptic plasticity in the nucleus accumbens shell. J Pharmacol Exp Ther. 2010 doi: 10.1124/jpet.110.171009. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Chandra D, Homanics GE, Harrison NL. Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. J Pharmacol Exp Ther. 2008;326:475–482. doi: 10.1124/jpet.108.139303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen-Soriano S, Bosch-Morell F, Miranda M, Asensio S, Barcia JM, Roma J, Monfort P, Felipo V, Romero FJ. Ebselen prevents chronic alcohol-induced rat hippocampal stress and functional impairment. Alcohol Clin Exp Res. 2007;31:486–492. doi: 10.1111/j.1530-0277.2006.00329.x. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Roberto M, Madamba SG, Siggins GR, Moore SD. Mu-opioid receptors selectively regulate basal inhibitory transmission in the central amygdala: lack of ethanol interactions. J Pharmacol Exp Ther. 2009;328:284–293. doi: 10.1124/jpet.108.140749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz PS, Kirk MD, Govind CK. Facilitation and depression at different branches of the same motor axon: evidence for presynaptic differences in release. J Neurosci. 1993;13:3075–3089. doi: 10.1523/JNEUROSCI.13-07-03075.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR. Calcium release from presynaptic internal stores is required for ethanol to increase spontaneous gamma-aminobutyric acid release onto cerebellum Purkinje neurons. J Pharmacol Exp Ther. 2007;323:356–364. doi: 10.1124/jpet.107.126144. [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR. The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J Neurophysiol. 2008;100:3417–3428. doi: 10.1152/jn.90970.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR. Ethanol-enhanced GABA release: A focus on G protein-coupled receptors. Brain Res Rev. 2010a doi: 10.1016/j.brainresrev.2010.09.003. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Weinberg RJ, Criswell HE, Breese GR. The PLC/IP 3 R/PKC pathway is required for ethanol-enhanced GABA release. Neuropharmacology. 2010b;58:1179–1186. doi: 10.1016/j.neuropharm.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PJ, Shapiro ML. Motivational states activate distinct hippocampal representations to guide goal-directed behaviors. Proc Natl Acad Sci U S A. 2009;106:10805–10810. doi: 10.1073/pnas.0903259106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Amygdaloid projections to the frontal cortex and the striatum in the rat. J Comp Neurol. 1990;298:40–49. doi: 10.1002/cne.902980104. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Goodlett CR, Greenough WT. Therapeutic motor training ameliorates cerebellar effects of postnatal binge alcohol. Neurotoxicol Teratol. 2000;22:125–132. doi: 10.1016/s0892-0362(99)00052-5. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Duncan GE, Crews FT, Breese GR. Induction of Fos-like proteins and ultrasonic vocalizations during ethanol withdrawal: further evidence for withdrawal-induced anxiety. Alcohol Clin Exp Res. 1998;22:481–493. [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56:18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS, Morrow AL. The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (Berl) 2009;205:529–564. doi: 10.1007/s00213-009-1562-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Suryanarayanan A, Boyd KN, Comerford CE, Lai MA, Ren Q, Morrow AL. Ethanol reduces GABAA alpha1 subunit receptor surface expression by a protein kinase Cgamma-dependent mechanism in cultured cerebral cortical neurons. Mol Pharmacol. 2010;77:793–803. doi: 10.1124/mol.109.063016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/s0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Lack AK, Ariwodola OJ, Chappell AM, Weiner JL, McCool BA. Ethanol inhibition of kainate receptor-mediated excitatory neurotransmission in the rat basolateral nucleus of the amygdala. Neuropharmacology. 2008;55:661–668. doi: 10.1016/j.neuropharm.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Christian DT, Diaz MR, McCool BA. Chronic ethanol and withdrawal effects on kainate receptor-mediated excitatory neurotransmission in the rat basolateral amygdala. Alcohol. 2009;43:25–33. doi: 10.1016/j.alcohol.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol. 2007;98:3185–3196. doi: 10.1152/jn.00189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land C, Spear NE. Fear conditioning is impaired in adult rats by ethanol doses that do not affect periadolescents. Int J Dev Neurosci. 2004;22:355–362. doi: 10.1016/j.ijdevneu.2004.04.008. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotional memory: in search of systems and synapses. Ann N Y Acad Sci. 1993;702:149–157. doi: 10.1111/j.1749-6632.1993.tb17246.x. [DOI] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology. 1998;37:1279–1286. doi: 10.1016/s0028-3908(98)00109-9. [DOI] [PubMed] [Google Scholar]
- Loeber S, Duka T. Acute alcohol impairs conditioning of a behavioural reward-seeking response and inhibitory control processes--implications for addictive disorders. Addiction. 2009;104:2013–2022. doi: 10.1111/j.1360-0443.2009.02718.x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Luscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- Machu TK, Olsen RW, Browning MD. Ethanol has no effect on cAMP-dependent protein kinase-, protein kinase C-, or Ca(2+)-calmodulin-dependent protein kinase II-stimulated phosphorylation of highly purified substrates in vitro. Alcohol Clin Exp Res. 1991;15:1040–1044. doi: 10.1111/j.1530-0277.1991.tb05208.x. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Chen X, Zhang TA, Morrisett RA. Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: real-time visualization in intact hippocampal slices. Alcohol Clin Exp Res. 2004;28:143–152. doi: 10.1097/01.ALC.0000106304.39174.AD. [DOI] [PubMed] [Google Scholar]
- Malenka RC. The role of postsynaptic calcium in the induction of long-term potentiation. Mol Neurobiol. 1991;5:289–295. doi: 10.1007/BF02935552. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Mameli M, Botta P, Zamudio PA, Zucca S, Valenzuela CF. Ethanol decreases Purkinje neuron excitability by increasing GABA release in rat cerebellar slices. J Pharmacol Exp Ther. 2008;327:910–917. doi: 10.1124/jpet.108.144865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Valenzuela CF. Alcohol increases efficacy of immature synapses in a neurosteroid-dependent manner. Eur J Neurosci. 2006;23:835–839. doi: 10.1111/j.1460-9568.2006.04597.x. [DOI] [PubMed] [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci. 2005;25:8027–8036. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Matthews DB, Silvers JR. The use of acute ethanol administration as a tool to investigate multiple memory systems. Neurobiol Learn Mem. 2004;82:299–308. doi: 10.1016/j.nlm.2004.06.007. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cytoarchitecture of the central amygdaloid nucleus of the rat. J Comp Neurol. 1982;208:401–418. doi: 10.1002/cne.902080409. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL, Winder DG. Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci U S A. 2010;107:2271–2276. doi: 10.1073/pnas.0905568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Medina AE, Krahe TE. Neocortical plasticity deficits in fetal alcohol spectrum disorders: lessons from barrel and visual cortex. J Neurosci Res. 2008;86:256–263. doi: 10.1002/jnr.21447. [DOI] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Morrisett RA. Ethanol action on dopaminergic neurons in the ventral tegmental area: interaction with intrinsic ion channels and neurotransmitter inputs. Int Rev Neurobiol. 2010;91:235–288. doi: 10.1016/S0074-7742(10)91008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett RA, Swartzwelder HS. Attenuation of hippocampal long-term potentiation by ethanol: a patch- clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci. 1993;13:2264–2272. doi: 10.1523/JNEUROSCI.13-05-02264.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow AL, Devaud LL, Bucci D, Smith FD. GABAA and NMDA receptor subunit mRNA expression in ethanol dependent rats. Alcohol Alcohol Suppl. 1994;2:89–95. [PubMed] [Google Scholar]
- Nagy J. Alcohol related changes in regulation of NMDA receptor functions. Curr Neuropharmacol. 2008;6:39–54. doi: 10.2174/157015908783769662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navamani M, Morgan M, Williams RJ. Ethanol modulates N-methyl-D-aspartate-evoked arachidonic acid release from neurones. Eur J Pharmacol. 1997;340:27–34. doi: 10.1016/s0014-2999(97)01396-4. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL. Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res. 2005;1048:69–79. doi: 10.1016/j.brainres.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Netzeband JG, Schneeloch JR, Trotter C, Caguioa-Aquino JN, Gruol DL. Chronic ethanol treatment and withdrawal alter ACPD-evoked calcium signals in developing Purkinje neurons. Alcohol Clin Exp Res. 2002;26:386–393. [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science. 2004;303:1512–1514. doi: 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Nie Z, Zorrilla EP, Madamba SG, Rice KC, Roberto M, Siggins GR. Presynaptic CRF1 receptors mediate the ethanol enhancement of GABAergic transmission in the mouse central amygdala. ScientificWorldJournal. 2009;9:68–85. doi: 10.1100/tsw.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose I, Higashi H, Inokuchi H, Nishi S. Synaptic responses of guinea pig and rat central amygdala neurons in vitro. J Neurophysiol. 1991;65:1227–1241. doi: 10.1152/jn.1991.65.5.1227. [DOI] [PubMed] [Google Scholar]
- Obara I, Bell RL, Goulding SP, Reyes CM, Larson LA, Ary AW, Truitt WA, Szumlinski KK. Differential effects of chronic ethanol consumption and withdrawal on homer/glutamate receptor expression in subregions of the accumbens and amygdala of P rats. Alcohol Clin Exp Res. 2009;33:1924–1934. doi: 10.1111/j.1530-0277.2009.01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead MC, Hellemans KG, Paine TA. Alcohol-induced impulsivity in rats: an effect of cue salience? Psychopharmacology (Berl) 2006;184:221–228. doi: 10.1007/s00213-005-0215-0. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Pasternak JF, Colley PA, Slater NT, Trommer BL. Metabotropic glutamate receptor mediated long-term depression in developing hippocampus. Neuropharmacology. 1997;36:831–844. doi: 10.1016/s0028-3908(97)00031-2. [DOI] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC. Neuronal signaling systems and ethanol dependence. Mol Neurobiol. 1998;17:1–15. doi: 10.1007/BF02802021. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Popp RL, Dertien JS. Actin depolymerization contributes to ethanol inhibition of NMDA receptors in primary cultured cerebellar granule cells. Alcohol. 2008;42:525–539. doi: 10.1016/j.alcohol.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor WR, Diao L, Freund RK, Browning MD, Wu PH. Synaptic GABAergic and glutamatergic mechanisms underlying alcohol sensitivity in mouse hippocampal neurons. J Physiol. 2006;575:145–159. doi: 10.1113/jphysiol.2006.112730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadros IM, Souza-Formigoni ML, Fornari RV, Nobrega JN, Oliveira MG. Is behavioral sensitization to ethanol associated with contextual conditioning in mice? Behav Pharmacol. 2003;14:129–136. doi: 10.1097/00008877-200303000-00004. [DOI] [PubMed] [Google Scholar]
- Raeder H, Holter SM, Hartmann AM, Spanagel R, Moller HJ, Rujescu D. Expression of N-methyl-d-aspartate (NMDA) receptor subunits and splice variants in an animal model of long-term voluntary alcohol self-administration. Drug Alcohol Depend. 2008;96:16–21. doi: 10.1016/j.drugalcdep.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Williams JT. CRF facilitates calcium release from intracellular stores in midbrain dopamine neurons. Neuron. 2008;57:559–570. doi: 10.1016/j.neuron.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley TL, Brown G, Dunworth SJ, Stephens DN. Aversive conditioning following repeated withdrawal from ethanol and epileptic kindling. Eur J Neurosci. 2003;17:1664–1670. doi: 10.1046/j.1460-9568.2003.02604.x. [DOI] [PubMed] [Google Scholar]
- Ripley TL, Little HJ. Nitrendipine prevents the decrease caused by chronic ethanol intake in the maintenance of tetanic long-term potentiation. Exp Brain Res. 1995;103:1–8. doi: 10.1007/BF00241959. [DOI] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, Schweitzer P. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology. 2010;35:1962–1972. doi: 10.1038/npp.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A. 2003a;100:2053–2058. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004a;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Nelson TE, Ur CL, Brunelli M, Sanna PP, Gruol DL. The transient depression of hippocampal CA1 LTP induced by chronic intermittent ethanol exposure is associated with an inhibition of the MAP kinase pathway. Eur J Neurosci. 2003b;17:1646–1654. doi: 10.1046/j.1460-9568.2003.02614.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004b;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Siggins GR. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci U S A. 2006;103:9715–9720. doi: 10.1073/pnas.0601899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DL, Howard EC, McConnell S, Gonzales RA, Wightman RM. Disparity between tonic and phasic ethanol-induced dopamine increases in the nucleus accumbens of rats. Alcohol Clin Exp Res. 2009;33:1187–1196. doi: 10.1111/j.1530-0277.2009.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Pare D. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci. 1999;19:10575–10583. doi: 10.1523/JNEUROSCI.19-23-10575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti J, Gruol DL. Emergence of NMDAR-independent long-term potentiation at hippocampal CA1 synapses following early adolescent exposure to chronic intermittent ethanol: role for sigma-receptors. Hippocampus. 2008;18:148–168. doi: 10.1002/hipo.20379. [DOI] [PubMed] [Google Scholar]
- Sanna E, Talani G, Busonero F, Pisu MG, Purdy RH, Serra M, Biggio G. Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci. 2004;24:6521–6530. doi: 10.1523/JNEUROSCI.0075-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Browning MD. Evidence for a role for GABA(A) and NMDA receptors in ethanol inhibition of long-term potentiation. Brain Res Mol Brain Res. 2001;94:9–14. doi: 10.1016/s0169-328x(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Doyere V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Silberman Y, Ariwodola OJ, Weiner JL. Differential effects of GABAB autoreceptor activation on ethanol potentiation of local and lateral paracapsular GABAergic synapses in the rat basolateral amygdala. Neuropharmacology. 2009;56:886–895. doi: 10.1016/j.neuropharm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Shi L, Brunso-Bechtold JK, Weiner JL. Distinct mechanisms of ethanol potentiation of local and paracapsular GABAergic synapses in the rat basolateral amygdala. J Pharmacol Exp Ther. 2008;324:251–260. doi: 10.1124/jpet.107.128728. [DOI] [PubMed] [Google Scholar]
- Sinclair JG, Lo GF. Ethanol blocks tetanic and calcium-induced long-term potentiation in the hippocampal slice. Gen Pharmacol. 1986;17:231–233. doi: 10.1016/0306-3623(86)90144-8. [DOI] [PubMed] [Google Scholar]
- Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19:589–598. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL. Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Brain Res Mol Brain Res. 1996;40:71–78. doi: 10.1016/0169-328x(96)00038-1. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Nie Z, Criado JR, Siggins GR. Ethanol inhibition of N-methyl-D-aspartate responses involves presynaptic gamma-aminobutyric acid(B) receptors. J Pharmacol Exp Ther. 2000;294:637–647. [PubMed] [Google Scholar]
- Stephens DN, Brown G, Duka T, Ripley TL. Impaired fear conditioning but enhanced seizure sensitivity in rats given repeated experience of withdrawal from alcohol. Eur J Neurosci. 2001;14:2023–2031. doi: 10.1046/j.0953-816x.2001.01824.x. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Ripley TL, Borlikova G, Schubert M, Albrecht D, Hogarth L, Duka T. Repeated ethanol exposure and withdrawal impairs human fear conditioning and depresses long-term potentiation in rat amygdala and hippocampus. Biol Psychiatry. 2005;58:392–400. doi: 10.1016/j.biopsych.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hopf FW, Hahn J, Cho SL, Guillory A, Bonci A. Voluntary ethanol intake enhances excitatory synaptic strength in the ventral tegmental area. Alcohol Clin Exp Res. 2008;32:1714–1720. doi: 10.1111/j.1530-0277.2008.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LD, Sun CL, Shen Y. Ethanol Acutely Modulates mGluR1-Dependent Long-Term Depression in Cerebellum. Alcohol Clin Exp Res. 2010;34:1140–1145. doi: 10.1111/j.1530-0277.2010.01190.x. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Shoyama Y, Saito H, Abe K. The effects of ethanol and crocin on the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Jpn J Pharmacol. 1995;67:395–397. doi: 10.1254/jjp.67.395. [DOI] [PubMed] [Google Scholar]
- Sun N, Cassell MD. Intrinsic GABAergic neurons in the rat central extended amygdala. J Comp Neurol. 1993;330:381–404. doi: 10.1002/cne.903300308. [DOI] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Age-dependent inhibition of long-term potentiation by ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995;19:1480–1485. doi: 10.1111/j.1530-0277.1995.tb01011.x. [DOI] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA. Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcohol Clin Exp Res. 2008;32:1040–1048. doi: 10.1111/j.1530-0277.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA. Role of 5-hydroxytryptamine2C receptors in Ca2+-dependent ethanol potentiation of GABA release onto ventral tegmental area dopamine neurons. J Pharmacol Exp Ther. 2009;329:625–633. doi: 10.1124/jpet.108.147793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinschmidt JS, Walker DW, King MA. Chronic ethanol treatment reduces the magnitude of hippocampal LTD in the adult rat. Synapse. 2003;48:189–197. doi: 10.1002/syn.10203. [DOI] [PubMed] [Google Scholar]
- Thomas MP, Davis MI, Monaghan DT, Morrisett RA. Organotypic brain slice cultures for functional analysis of alcohol- related disorders: novel versus conventional preparations. Alcohol Clin Exp Res. 1998a;22:51–59. [PubMed] [Google Scholar]
- Thomas MP, Monaghan DT, Morrisett RA. Evidence for a causative role of N-methyl-D-aspartate receptors in an in vitro model of alcohol withdrawal hyperexcitability. J Pharmacol Exp Ther. 1998b;287:87–97. [PubMed] [Google Scholar]
- Tokuda K, Zorumski CF, Izumi Y. Modulation of hippocampal long-term potentiation by slow increases in ethanol concentration. Neuroscience. 2007;146:340–349. doi: 10.1016/j.neuroscience.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremwel MF, Hunter BE. Effects of chronic ethanol ingestion on long-term potentiation remain even after a prolonged recovery from ethanol exposure. Synapse. 1994;17:141–148. doi: 10.1002/syn.890170210. [DOI] [PubMed] [Google Scholar]
- Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- Ugolini A, Sokal DM, Arban R, Large CH. CRF1 Receptor Activation Increases the Response of Neurons in the Basolateral Nucleus of the Amygdala to Afferent Stimulation. Front Behav Neurosci. 2008;2:2. doi: 10.3389/neuro.08.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela CF, Bhave S, Hoffman P, Harris RA. Acute effects of ethanol on pharmacologically isolated kainate receptors in cerebellar granule neurons: comparison with NMDA and AMPA receptors. J Neurochem. 1998;71:1777–1780. doi: 10.1046/j.1471-4159.1998.71041777.x. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Sparta DR, Hopf FW, Bowers MS, Melis M, Bonci A. Strain specific synaptic modifications on ventral tegmental area dopamine neurons after ethanol exposure. Biol Psychiatry. 2009;65:646–653. doi: 10.1016/j.biopsych.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Zheng JQ, Poo MM. Effects of cytochalasin treatment on short-term synaptic plasticity at developing neuromuscular junctions in frogs. J Physiol. 1996;491 ( Pt 1):187–195. doi: 10.1113/jphysiol.1996.sp021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Egli RE, Grueter BA, Winder DG. High-frequency stimulation induces ethanol-sensitive long-term potentiation at glutamatergic synapses in the dorsolateral bed nucleus of the stria terminalis. J Neurosci. 2004;24:5741–5747. doi: 10.1523/JNEUROSCI.1181-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie MB, Besheer J, Kelley SP, Kumar S, O’Buckley TK, Morrow AL, Hodge CW. Acute ethanol administration rapidly increases phosphorylation of conventional protein kinase C in specific mammalian brain regions in vivo. Alcohol Clin Exp Res. 2007;31:1259–1267. doi: 10.1111/j.1530-0277.2007.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PH, Poelchen W, Proctor WR. Differential GABAB Receptor Modulation of Ethanol Effects on GABA(A) synaptic activity in hippocampal CA1 neurons. J Pharmacol Exp Ther. 2005;312:1082–1089. doi: 10.1124/jpet.104.075663. [DOI] [PubMed] [Google Scholar]
- Xia JX, Li J, Zhou R, Zhang XH, Ge YB, Ru Yuan X. Alterations of rat corticostriatal synaptic plasticity after chronic ethanol exposure and withdrawal. Alcohol Clin Exp Res. 2006;30:819–824. doi: 10.1111/j.1530-0277.2006.00095.x. [DOI] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC, 3rd, Li KY, Krnjevic K, Zhou C, Ye JH. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology. 2009;34:307–318. doi: 10.1038/npp.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Ye JH. Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu-opioid receptors. Neuroscience. 2008;153:240–248. doi: 10.1016/j.neuroscience.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie GQ, Wang SJ, Li J, Cui SZ, Zhou R, Chen L, Yuan XR. Ethanol attenuates the HFS-induced, ERK-mediated LTP in a dose-dependent manner in rat striatum. Alcohol Clin Exp Res. 2009;33:121–128. doi: 10.1111/j.1530-0277.2008.00818.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Nakanishi H, Takai N, Shimazoe T, Watanabe S, Kita H. Expression of N-methyl-D-aspartate receptor-dependent long-term potentiation in the neostriatal neurons in an in vitro slice after ethanol withdrawal of the rat. Neuroscience. 1999;91:59–68. doi: 10.1016/s0306-4522(98)00611-3. [DOI] [PubMed] [Google Scholar]
- Yang SN. Presynaptic involvement of nitric oxide in dopamine D1/D5 receptor-induced sustained enhancement of synaptic currents mediated by ionotropic glutamate receptors in the rat hippocampus. Neurosci Lett. 1999;270:87–90. doi: 10.1016/s0304-3940(99)00474-7. [DOI] [PubMed] [Google Scholar]
- Yao L, Fan P, Jiang Z, Gordon A, Mochly-Rosen D, Diamond I. Dopamine and ethanol cause translocation of epsilonPKC associated with epsilonRACK: cross-talk between cAMP-dependent protein kinase A and protein kinase C signaling pathways. Mol Pharmacol. 2008;73:1105–1112. doi: 10.1124/mol.107.042580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Park BS, Adermark L, Lovinger DM. Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci. 2007;25:3226–3232. doi: 10.1111/j.1460-9568.2007.05606.x. [DOI] [PubMed] [Google Scholar]
- Zeichner A, Allen JD, Petrie CD, Rasmussen PR, Giancola P. Attention allocation: effects of alcohol and information salience on attentional processes in male social drinkers. Alcohol Clin Exp Res. 1993;17:727–732. doi: 10.1111/j.1530-0277.1993.tb00830.x. [DOI] [PubMed] [Google Scholar]
- Zhang TA, Hendricson AW, Morrisett RA. Dual synaptic sites of D(1)-dopaminergic regulation of ethanol sensitivity of NMDA receptors in nucleus accumbens. Synapse. 2005;58:30–44. doi: 10.1002/syn.20181. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurophysiol. 2006;96:433–441. doi: 10.1152/jn.01380.2005. [DOI] [PubMed] [Google Scholar]
- Zhu W, Bie B, Pan ZZ. Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci. 2007;27:289–298. doi: 10.1523/JNEUROSCI.3912-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Gao BX, Hinckley C. Ethanol dual modulatory actions on spontaneous postsynaptic currents in spinal motoneurons. J Neurophysiol. 2003;89:806–813. doi: 10.1152/jn.00614.2002. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]