

Abstract
HIV protease inhibitors are the backbone of HIV therapy. In addition to blocking intracellular HIV protease and dramatically decreasing viral burden, the protease inhibitors also regulate apoptosis. A growing body of data has confirmed the immunomodulatory effects of HIV protease inhibitors which block CD4+ and CD8+ T cell death in models of HIV infection. The mechanism of this apoptosis inhibition is still under active investigation and supported by several proposed hypothesis for how they alter the fate of the cell. More recently, the anti-apoptotic effects of the HIV protease inhibitors has been extended to the non-HIV, non-immune cell, whereby protease inhibitors prevent apoptosis, and disease, in animal models of sepsis, hepatitis and stroke. Interestingly, when HIV protease inhibitors are used at supra-therapeutic concentrations, they exert pro-apoptotic effects. This has been demonstrated in a number of tumor models. Although it is unclear how HIV protease inhibitors can induce apoptosis at increased concentrations, future research will define the targets of the immunomodulation and reveal the full clinical potential of this intriguing class of drugs.
INTRODUCTION
HIV protease inhibitors are chemical inhibitors of the HIV protease enzyme which act intracellularly. These drugs were designed to mimic the chemical structure of the HIV viral peptides that are normally recognized and cleaved by the HIV protease. The compounds have a strong affinity for the active site of the HIV protease and irreversibly inhibit the catalytic activity of the enzyme. Currently, there are nine HIV protease inhibitors that are approved for clinical use in humans (Saquinavir, Indinavir, Nelfinavir, Lopinavir, Amprenavir, Atazanavir, Tipranavir, Ritonavir, and Fosamprenavir). Most of the current HIV protease inhibitors (PI) are pseudopeptide compounds that are classified as pepti-domimetic compounds. Without a functional HIV protease, viral particles are produced, but they are immature, do not package properly, and are not infectious [1, 2]. Although these compounds have limited bioavailability and stability, they are the basis of effective anti- HIV therapy, and in combination with other anti-retroviral agents, produce a sustained decrease in HIV viral load. The first HIV protease inhibitor was FDA approved for anti-HIV therapy in humans in 1995. With over a decade of experience in humans, there is accumulating evidence that HIV protease inhibitors have host cell effects beyond blocking HIV protease enzymatic activity. It appears that HIV protease inhibitors also modulate cellular apoptosis.
IN VIVO BENEFIT OF HIV PROTEASE INHIBITORS DURING HIV INFECTION
As HIV protease inhibitors have been used in HIV infected individuals, it was observed that patients frequently have an improved CD4+T cell counts that was not related to viral suppression. In one of the earliest clinical trials with the HIV protease inhibitor, saquinivir, the data suggested that the protease inhibitor had immunologic effects independent of the effect on HIV replication. In a double-blinded study, HIV-infected subjects were randomized to receive triple therapy (saquinivir plus zidovudine plus zalictabine) or dual therapy with either zidovudine plus saquinivir, or to zidovudine plus zalictabine. Two hundred and eighty subjects completed 24 weeks of therapy. Even though there was inferior virologic suppression in the saquinivir plus zidovudine group, compared to the zidovudine plus zalictabine group, the subjects who received the HIV PI, had improved CD4+T cell counts [3]. Subsequent clinical trials confirmed that HIV protease inhibitors improved CD4+T cell counts in HIV–infected individuals, independent of the viral suppression [4–7]. In a meta-analysis comparing anti-HIV drug regimens that maintained a PI to those that switched the PI component to a Non-nucleoside reverse transcriptase inhibitor (NNRTI), PI- based therapy resulted in superior CD4+ T cell counts than switching to a non-PI based regimen [8]. The analysis required that both PI and non- PI based regimens have complete HIV viral suppression. Therefore, the CD4+T cell benefit with HIV protease inhibitor therapy was unrelated to the effect of HIV viral suppression. In a trial comparing treatment with HIV protease containing highly active anti-retroviral therapy (HAART) to protease inhibitor therapy alone, there was worse virologic suppression of the subjects on PI mono-therapy. However, there was no decrease in CD4+T cell counts over one year, even in the group who received PI mono-therapy and virologically failed [9]. Additionally, in a comparison of PI-based anti-HIV therapy to NNRTI-based anti-HIV therapy, equal viral suppression was reported in both groups; however, PI-based regimens had a superior increase in CD4+T cell counts [10].
A recent trial by Landay et al. compared patients with suppressed HIV viral replication on a PI based anti-HIV regimen or non-PI based drug regimen. One week after enrollment in the study, subjects in the PI containing arm had significantly less spontaneous T cell apoptosis that those in the non-PI containing arm [11]. Although there are likely multiple reasons for CD4+T cell decline during the course if HIV infection, these studies detail that patients on HIV protease inhibitors have less spontaneous CD4+T cell apoptosis and have improved CD4+ T cell counts, further demonstrating that the HIV protease inhibitors block CD4+T cell apoptosis, independent of their effect on HIV replication.
IN VITRO EFFECTS OF HIV PROTEASE INHIBITORS ON APOPTOSIS
In 1998, we proposed that highly active antiretroviral therapy (HAART) might decrease apoptosis due to findings that PI therapy reduced lymphocyte apoptosis in HIV infected patients [12]. We subsequently tested and confirmed this finding ex vivo [13] and in vitro [14]. One of the earlier reports of PI’s anti-apoptotic effects was investigating ritonavir use in cultures of bone marrow cells from HIV-infected patients or normal controls. Adding Ritonavir to hematopoietic colonies increased colony forming unit replication. In addition, when Ritonavir was added to HIV bone marrow cultures, there was 45% less apoptosis than untreated cultures. The authors also report a decrease in caspase 1 expression after Ritonavir treatment [15].
Additional reports investigated the effects of HIV PIs on T cell death in the setting of HIV infection. CD4+T cells isolated from healthy uninfected individuals had an increase in Fas expression and Fas and anti-CD3− induced apoptosis when incubated with HIV virions. The inducible Fas expression and apoptosis were blocked when the cells were pre-incubated with the HIV PI, saquinavir [16]. These findings were supported by subsequent investigations demonstrating that saquinavir and ritonavir inhibited TNF mediated apoptosis in U937 cells in a dose dependent fashion with 38–60% reduction in apoptosis in PI treated cells [17]. During HIV infection, the HIV envelope gp120 binds to the CD4+ T cell through the receptors CD4 and CXCR4, and signals the cell to undergo apoptosis. This bystander death is one of the ways that HIV depletes the immune system. Matarrese et al. pre-treated human CD4+ T cells with HIV gp120 which made the cell sensitive to Fas mediated apoptosis, and resulted in mitochondrial changes and apoptosis after Fas exposure. When the CD4+ T cells were pre-treated with a PI, mitochondrial hyperpolorization was blocked in a whole cell or cell free system, or isolated mitochondria [18]. Taken together, this data indicates that patient’s receiving HIV protease inhibitors had an improved CD4+ T cell counts unrelated to the state of HIV viral replication, in vitro work confirmed that HIV PIs can inhibit T cell apoptosis, specifically that induced by HIV.
MECHANISM OF HIV PROTEASE INHIBITOR MEDIATED APOPTOSIS INHIBITION
The mechanism of HIV Protease inhibitor mediated apoptosis inhibition is under active investigation (Table 1). Because HIV PIs inhibit the proteolytic activity of HIV viral protease, it has been postulated that they might have a similar effect on other cellular proteases. Together with the early observations that HIV PIs block Fas-mediated apoptosis, many investigators hypothesize that HIV PIs may inhibit the caspases family members and block apoptosis. Although this model is appealing, caspases are cysteine proteases and HIV protease is an aspartyle protease [19, 20]. Several reports have investigated the direct effect of HIV PIs on caspases activity. When recombinant caspases -1, -3, -6, -7, or -8 were incubated with a fluorogenic tetrapeptide substrate for each caspases in the presence of absence of nelfinavir, the HIV PI inhibited HIV protease cleavage of gag/pol, but did not inhibit the activity of any of the caspases [21]. These results were expanded in a study that demonstrated that caspases -3, -6, and -8 activity were not inhibited by indinavir in U937 cells at drug concentrations that effectively inhibited U937 apoptosis [22]. Nelfinavir did not inhibit activation of caspases -1, -3, -4, -5, -9, and -8 in lysates of Jurkat T cells undergoing Fas-mediated apoptosis [23].
Table 1.
Possible Mechanisms for HIV Protease Inhibitor Mediating Apoptosis
| Theories | Evidence |
|---|---|
| Alter caspase activity | HIV PIs block Fas and TNF mediated apoptosis HIV PIs not block caspase -1, -3, -4, -5, -6, -7, -8, or -9 activity in cell models |
| Inhibit Calpain activity | HIV PIs inhibit apoptosis in cell systems where calpains are activated HIV PIs blocked m-calpain activation in U937 cells HIV PIs inhibited activity of both m- and -μ calpain isoforms in PC12 cells HIV PI did not inhibit m- or μ-calpain hydrolysis or activation at lower, physiologic, concentrations |
| Alter levels of apoptosis regulatory proteins | No change in Fas protein levels after HIV PI treatment No change in RNA levels of Fas, Fas L, and TNF after HIV PI treatment No change in Bcl-2, Bax, and Bcl-XL after HIV PI treatment |
| Maintain Mitochondrial Transmembrane Potential | HIV PIs maintain mitochondrial membrane integrity after apoptosis stimuli HIV PIs prevent cytochrome c release from mitochondria after apoptosis stimuli ANT (adenine nucleotide translocator) necessary for HIV PI to block mitochondria mediated apoptosis |
In addition to caspases, other proteases such as calpains are implicated in apoptosis and have been considered as a possible site for HIV PIs to mediate apoptosis. Calpains are a set of Ca++-dependent cysteine proteases that are involved in several models of apoptosis, including U937 cells, but are not absolutely required for apoptosis [24]. Because HIV PIs are designed to inhibit the HIV aspartyle protease, they may modulate apoptosis by blocking calpain activation and function. Ghibelli et al. demonstrate in a U937 model of apoptosis that indinavir and ritonavir directly inhibit apoptosis in cell systems where calpains are activated and block m-calpain activation [22]. Other investigators have demonstrated that in PC12 cells, ritonavir competitively inhibited activity of both m- and -μ calpain isoforms [25]. However, results from a different group that specifically investigated calpain activity and hydrolysis did not show that HIV PI could inhibit m- or μ-calpain activation. These authors postulate that previous reports used concentrations of ritonavir close to the maximum solubility of the drug which may have artificially altered the results [26]. A recent report demonstrated that HIV related cellular apoptosis is associated with increased calpain activity and was reversed after HIV PI including highly active anti-retroviral therapy. This was confirmed using calpain inhibitors in vitro [27]. An inhibitor of calpain activity would have significant therapeutic implications beyond HIV apoptosis, including neurodegenerative diseases; however, the evidence remains unclear whether HIV PIs have a significant effect on calpain activity.
An alternative model of how HIV PIs inhibit apoptosis suggests that the drugs might alter the expression of apoptotic regulatory proteins. Although early reports demonstrated a change in Fas expression after PI treatment [16], subsequent work did not show a change in Fas levels with HIV PI therapy [28–30] In the bone marrow of HIV infected individuals incubated with ritonavir and indinavir there was no change in RNA levels of Fas, Fas L or mFas. Only one report has specifically investigated the levels of intracellular apoptotic regulatory proteins with and without HIV PI treatment. Protein levels of Bcl-2, Bax, and Bcl-XL were evaluated by flow cytometry. The levels were unchanged after PI administration [31]. Therefore, it is clear that HIV PIs can block Fas and TNF mediated apoptosis, [16–18], however, this effect does not appear to be due to changes in intra- or extracellular apoptotic regulatory protein levels.
A final possible mechanism by which HIV PIs may modulate cellular apoptosis includes stabilizing the mitochondria. The mitochondrial transmembrane potential that occurs from asymmetric distribution of ions on both sides of the inner mitochondrial membrane is maintained by the mitochrondrial permeability transition pore complex (PTPC). After an apoptotic signal, the PTPC opens, disrupts the membrane potential, and releases apoptogenic factors, including cytochrome c, and pro-caspase-9. As such, the mitochondria serves as a regulatory checkpoint of apoptotic signaling, with many regulatory proteins including Bcl-2 family members and IAPs interacting at the level of the mitochondria to alter apoptosis. Multiple groups have questioned whether HIV PIs alter the transmitochondrial membrane potential and block apoptosis at the level of the mitochondria. In one of the first reports, Jurkat cell Fas-mediated apoptosis was inhibited with 10 μM of nelfinavir, a dose that would simulate physiologic levels if taken clinically. Nelfinavir treated cells maintained intact mitochondrial trans-membrane potential, as determined by DioC6 staining, which is a lipophilic dye that stains the mitochondria [21]. The authors also report that 10 μM of nelfinavir inhibited cytochrome c release during Fas apoptosis. The HIV accessory molecule Vpr causes PTPC opening and loss of transmitochondrial potential when added to mitochondria. Nelfinavir pre-treatment of Jurkat cells prevented Vpr induced DioC6 release from the mitochondria and cell death. HIV envelope mediated HIV apoptosis is reported to correlate with HIV gp41-induced hemifusion. In CD4+T cells this cellular apoptosis was inhibited by Nelfinavir but not other HIV protease inhibitors over a narrow range of concentrations [32]. Numerous other groups have confirmed that HIV protease inhibitors block mitochondrial transmembrane potential loss in multiple models of apoptosis [18, 21, 31, 32–35]. Recently, Weaver et al. further investigated how HIV PIs influence mitochondrial integrity by investigating yeast mitochondrial apoptotic models. Wild type or yeast deficient in voltage-dependent anion channel (VDAC) or adenine nucleotide translocator (ANT) isoforms, the components of the PTPC, were treated with Vpr or H2O2 which induce mitochondrial apoptosis. Apoptosis only occurred after Vpr or H2O2 treatment when ANT was present. When Jurkat cells were pre-treated with nelfinavir and then an agonist for VDAC, there was no inhibition of mitochondrial potential loss or apoptosis. However, 10 μM of nelfinavir blocked ANT agonist induced mitochondrial transmembrane potential loss and apoptosis. (Fig. 1) [36]. Furthermore, proteoliposomes reconstituted with ANT which release a fluorescent dye after pore opening did not demonstrate pore opening when pre-treated with nelfinavir before ANT agonist treatment. In summary, there are several theories for how HIV PIs inhibit cellular apoptosis. The greatest and most consistent data suggests that HIV PIs maintain mitochondrial integrity to block apoptosis. This is likely due to PIs preventing pore function of the adenine nucleotide translocator subunit of the mitochondrial permeability transition pore complex.
Fig. 1.
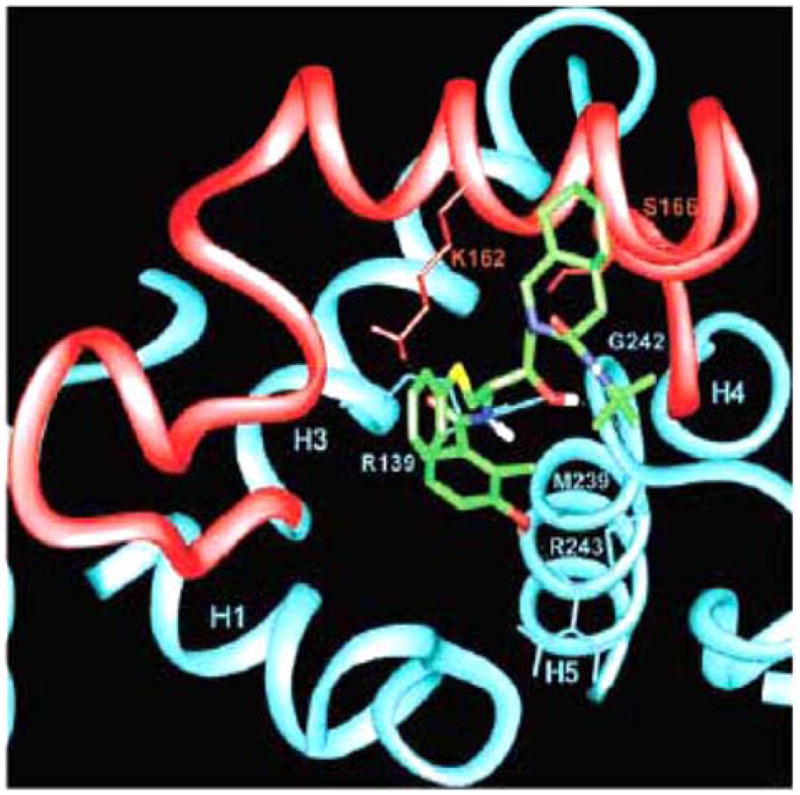
Computer-simulated model of NFV (green) interaction with ANT (blue and red). Close-up of the proposed binding mode of NFV to the matrix side of ANT. Parts of helices 1, 3, 4, and 5 (H1, H3, H4, and H5) are shown. Loop M2, connecting helices 3 and 4, is shown in red. Hydrogen bonds are displayed as dotted lines.
PARADOXICAL PRO-APOPTOTIC EFFECTS OF HIV PROTEASE INHIBITORS
In addition to the considerable evidence that HIV PIs have anti-apoptotic properties, there is a growing body of literature that suggests these drugs may be pro-apoptotic as well. Because other protease inhibitors can block apoptosis in tumor cell lines that are otherwise resistant to programmed cell death, ritonavir was investigated as an anti-tumor agent. High concentrations of ritonavir (10–50 μM) inhibit the proliferation of murine and human tumor cell lines. DNA laddering demonstrated that 15–30 μM of ritonavir induced apoptosis in the same cell lines [37]. Of note, the concentrations of ritonavir used were over 15 times what most adults achieve at FDA approved doses and at which the drug blocks apoptosis. This led to further work developing the pro-apoptotic effects of HIV PIs. Adult T-cell leukemia (ATL) is an aggressive malignancy associated with human T-cell leukemia virus (HTLV) and is very resistant to conventional chemotherapy. When ATL cells were incubated with 20–40 μM of ritonavir, there was a five fold increase in spontaneous apoptosis, resulting in a similar decrease in tumor cell survival [38]. In ATL cell lines and primary ATL cells 40 μM of ritonavir inhibited transcriptional activation of NF-κB. In addition, HIV PIs inhibited the expression of the targets of NF-κB, Bcl-XL, survivin, c-Myc, and cyclin D2 [38]. Thus, confirming that high concentrations of HIV PIs have pro-apoptotic effects in tumors of T-cell origin.
There is also evidence that HIV protease inhibitors are pro-apoptotic in models of solid tumors. Freshly isolated multiple myeloma cells from patients under went apoptosis when incubated with 40–50 μM of ritonavir, saquinavir, and nelfinavir. This was associated with a decrease in the anti-apoptotic protein Mcl-1, and blocked IL-6 phosphorylation of ERK 1/2 and STAT 3 [39]. Different tumor types may have different mechanisms by which HIV PIs inhibit tumor growth and promote apoptosis. Ritonavir used at 20 μM in solid and hematologic tumor models caused an increase in the cellular concentrations of the anti-proliferative and pro-apoptotic proteasome substrate cdk inhibitor, p21. This was associated with a block in proteolytic degradation [37]. Accumulation of intracellular p21 resulted in cell cycle arrest in G1 phase and subsequent apoptosis in ritonavir treated tumor cells. Furthermore, different HIV PIs have differential effects on tumors. In a prostate cancer model HIV PIs Nelfinavir, Ritonavir, and Saquinivir induced cellular apoptosis, disrupted STAT signaling and blocked Akt activation. Indinavir, however, failed to show similar results [40].
IN VIVO EFFECTS OF HIV PROTEASE INHIBITORS IN NON-HIV RELATED DISEASE
The novel anti-apoptotic properties of HIV PIs are now being used in early trials as therapy for disease states associated with increased levels of apoptosis. The earliest description of in vivo use of HIV PIs for non-HIV related disease was in a mouse sepsis model. Sepsis is the leading cause of death in critically ill patients. Despite many medical and technical advances in supportive care, mortality from sepsis remains high. Animal studies demonstrate that sepsis results in extensive lymphocyte apoptosis, as well as intestinal epithelial cell apoptosis [41, 42]. These findings have been confirmed during autopsy studies in humans who died of sepsis [43, 44]. In a mouse model of sepsis, created by cecal ligation and perforation, mice pretreated with HIV PIs had improved survival and reduced lymphocyte apoptosis [45]. The HIV PI treated mice had an increase in the Th1 cytokine TNFα and a reduction in the TH2 cytokines IL-6 and IL-10. It appears the beneficial effect of PI treatment was due to reduced lymphocyte apoptosis, because lymphocyte deficient Rag 1 −/− mice had no benefit from HIV PI treatment. Follow-up work by Weaver et. al. investigated the effect of HIV PIs during Staphylococcal enterotoxin B-induced shock. There was a 60% improvement in 24 hour survival in mice pre-treated with HIV PIs than those treated with vehicle control. In addition, the authors also demonstrated that HIV PI pre-treatment reduced mouse death from Fas-induced fatal hepatitis and middle cerebral artery occlusion-induced stroke. Improved survival was associated with an improvement in T cell apoptosis during the sepsis model, and reduced transaminasemia in the hepatitis model. Mice pre-treated with HIV PIs before stroke had reduced cerebral infarct size, decreased cerebral apoptosis, and performed better in behavioral recovery indices [36]. In addition, the HIV protease inhibitor Nelfinavir is being used in early clinical trials to treat recurrent or refractory solid tumors and Liposarcomas.
HIV protease inhibitors have cellular effects beyond their anti-HIV, anti-viral properties. At concentrations that match what is achieved during therapy with the FDA approved doses, the protease inhibitors clearly have anti-apoptotic effects on multiple cell types. The mechanism of this effect is under active investigation but centers around mitochondrial stabilization. Interestingly, when used at higher concentrations, the HIV PIs are pro-apoptotic in cells that were otherwise resistant to apoptosis, such as tumor cell lines. These serendipitous properties of HIV PIs are now being used in early animal studies as therapy for diseases associated with increased apoptosis for normal concentrations of PIs and tumors states at high concentrations of PIs, respectively. Future studies are necessary to determine the many possible benefits of HIV protease inhibitors for human disease.
Acknowledgments
Stacey Rizza is supported by a grant from the Mayo Program in Translational Immunovirology and Biodefense and a Kogod Award in Aging from the Mayo Foundation. Andrew Badley is supported by NIH RO1 AI62261, RO1 AI40384 and a Burroughs Wellcome award ID# 1005160.
References
- 1.Chou KC. J Biol Chem. 1993;268:16938. [PubMed] [Google Scholar]
- 2.Chou KC. Anal Biochem. 1996;233:1. doi: 10.1006/abio.1996.0001. [DOI] [PubMed] [Google Scholar]
- 3.Collier AC, Coombs RW, Schoenfeld DA, Bassett RL, Timpone J, Baruch A, Jones M, Facey K, Whitacre C, McAuliffe VJ, Friedman HM, Merigan TC, Reichman RC, Hooper C, Corey L. N Engl J Med. 1996;334:1011. doi: 10.1056/NEJM199604183341602. [DOI] [PubMed] [Google Scholar]
- 4.Kravcik S, Magill A, Sanghvi B, Ogden R, Cameron WD, Lewis R, Yu G, Badley AD. HIV Clin Trials. 2001;2:160. doi: 10.1310/F45L-FDKK-Y48N-N2BT. [DOI] [PubMed] [Google Scholar]
- 5.Deeks SG, Grant RM. Antivir Ther. 1999;4(Suppl 3):7. [PubMed] [Google Scholar]
- 6.Albrecht MA, Bosch RJ, Hammer SM, Liou SH, Kessler H, Para MF, Eron J, Valdez H, Dehlinger M, Katzenstein DA. N Engl J Med. 2001;345:398. doi: 10.1056/NEJM200108093450602. [DOI] [PubMed] [Google Scholar]
- 7.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. N Engl J Med. 1999;341(25):1865–73. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 8.Owen C, Kazim F, Badley AD. Aids. 2004;18:693. doi: 10.1097/00002030-200403050-00016. [DOI] [PubMed] [Google Scholar]
- 9.Arribas JR, Pulido F, Delgado R, Lorenzo A, Miralles P, Arranz A, Gonzalez-Garcia JJ, Cepeda C, Hervas R, Pano JR, Gaya F, Carcas A, Montes ML, Costa JR, Pena JM. J Acquir Immune Defic Syndr. 2005;40:280. doi: 10.1097/01.qai.0000180077.59159.f4. [DOI] [PubMed] [Google Scholar]
- 10.Fethi T, Asma J, Amine SM, Amel EB, Taoufik BC, Mohamed C, Amel LO, Mounira G. Curr HIV Res. 2005;3:271. doi: 10.2174/1570162054368066. [DOI] [PubMed] [Google Scholar]
- 11.Landay AL, Spritzler J, Kessler H, Mildvan D, Pu M, Fox L, O’Neil D, Schock B, Kuritzkes D, Lederman MM. J Infect Dis. 2003;188:1444. doi: 10.1086/379041. [DOI] [PubMed] [Google Scholar]
- 12.Badley AD, Dockrell DH, Algeciras A, Ziesmer S, Landay A, Lederman MM, Connick E, Kessler H, Kuritzkes D, Lynch DH, Roche P, Yagita H, Paya CV. J Clin Invest. 1998;102:79. doi: 10.1172/JCI2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Badley AD, Parato K, Cameron DW, Kravcik S, Phenix BN, Ashby D, Kumar A, Lynch DH, Tschopp J, Angel JB. Cell Death Differ. 1999;6:420. doi: 10.1038/sj.cdd.4400509. [DOI] [PubMed] [Google Scholar]
- 14.Phenix BN, Angel JB, Mandy F, Kravcik S, Parato K, Chambers KA, Gallicano K, Hawley-Foss N, Cassol S, Cameron DW, Badley AD. AIDS Res Hum Retroviruses. 2000;16:559. doi: 10.1089/088922200308972. [DOI] [PubMed] [Google Scholar]
- 15.Sloand EM, Maciejewski J, Kumar P, Kim S, Chaudhuri A, Young N. Blood. 2000;96:2735. [PubMed] [Google Scholar]
- 16.Estaquier J, Lelievre JD, Petit F, Brunner T, Moutouh-De Parseval L, Richman DD, Ameisen JC, Corbeil J. J Virol. 2002;76:5966. doi: 10.1128/JVI.76.12.5966-5973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf T, Findhammer S, Nolte B, Helm EB, Brodt HR. Eur J Med Res. 2003;8:17. [PubMed] [Google Scholar]
- 18.Matarrese P, Tinari A, Gambardella L, Mormone E, Narilli P, Pierdominici M, Cauda R, Malorni W. Antivir Ther. 2005;10(Suppl 2):M29. [PubMed] [Google Scholar]
- 19.Chou KC, Tomasselli AG, Heinrikson RL. FEBS Lett. 2000;470:249. doi: 10.1016/s0014-5793(00)01333-8. [DOI] [PubMed] [Google Scholar]
- 20.Chou KC, Jones D, Heinrikson RL. FEBS Lett. 1997;419:49. doi: 10.1016/s0014-5793(97)01246-5. [DOI] [PubMed] [Google Scholar]
- 21.Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. Blood. 2001;98:1078. doi: 10.1182/blood.v98.4.1078. [DOI] [PubMed] [Google Scholar]
- 22.Ghibelli L, Mengoni F, Lichtner M, Coppola S, De Nicola M, Bergamaschi A, Mastroianni C, Vullo V. Biochem Pharmacol. 2003;66:1505. doi: 10.1016/s0006-2952(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 23.Chavan S, Kodoth S, Pahwa R, Pahwa S. Blood. 2001;98:383. doi: 10.1182/blood.v98.2.383. [DOI] [PubMed] [Google Scholar]
- 24.Spinedi A, Oliverio S, Di Sano F, Piacentini M. Biochem Pharmacol. 1998;56:1489. doi: 10.1016/s0006-2952(98)00169-5. [DOI] [PubMed] [Google Scholar]
- 25.Wan W, DePetrillo PB. Biochem Pharmacol. 2002;63:1481. doi: 10.1016/s0006-2952(02)00907-3. [DOI] [PubMed] [Google Scholar]
- 26.Cuerrier D, Nie Z, Badley AD, Davies PL. Biochem Biophys Res Commun. 2005;327:208. doi: 10.1016/j.bbrc.2004.11.161. [DOI] [PubMed] [Google Scholar]
- 27.Lichtner M, Mengoni F, Mastroianni CM, Sauzullo I, Rossi R, De Nicola M, Vullo V, Ghibelli L. Apoptosis. 2006;11:781. doi: 10.1007/s10495-006-5699-5. [DOI] [PubMed] [Google Scholar]
- 28.Sloand EM, Kumar PN, Kim S, Chaudhuri A, Weichold FF, Young NS. Blood. 1999;94:1021. [PubMed] [Google Scholar]
- 29.Lu W, Andrieu JM. Blood. 2000;96:250. [PubMed] [Google Scholar]
- 30.Isgro A, Aiuti A, Mezzaroma I, Ruco L, Pinti M, Cossarizza A, Aiuti F. AIDS Res Hum Retroviruses. 2005;21:51. doi: 10.1089/aid.2005.21.51. [DOI] [PubMed] [Google Scholar]
- 31.Matarrese P, Gambardella L, Cassone A, Vella S, Cauda R, Malorni W. J Immunol. 2003;170:6006. doi: 10.4049/jimmunol.170.12.6006. [DOI] [PubMed] [Google Scholar]
- 32.Garg H, Blumenthal R. J Leukoc Biol. 2006;79:351. doi: 10.1189/jlb.0805430. [DOI] [PubMed] [Google Scholar]
- 33.Weichold FF, Bryant JL, Pati S, Barabitskaya O, Gallo RC, Reitz MS., Jr J Hum Virol. 1999;2:261. [PubMed] [Google Scholar]
- 34.Badley AD, Roumier T, Lum JJ, Kroemer G. Trends Pharmacol Sci. 2003;24:298. doi: 10.1016/S0165-6147(03)00125-1. [DOI] [PubMed] [Google Scholar]
- 35.Miro O, Villarroya J, Garrabou G, Lopez S, Rodriguez de la Concepcion M, Pedrol E, Martinez E, Giralt M, Gatell JM, Cardellach F, Casademont J, Villarroya F. Antivir Ther. 2005;10:945. [PubMed] [Google Scholar]
- 36.Weaver JG, Tarze A, Moffat TC, Lebras M, Deniaud A, Brenner C, Bren GD, Morin MY, Phenix BN, Dong L, Jiang SX, Sim VL, Zurakowski B, Lallier J, Hardin H, Wettstein P, van Heeswijk RP, Douen A, Kroemer RT, Hou ST, Bennett SA, Lynch DH, Kroemer G, Badley AD. J Clin Invest. 2005;115:1828. doi: 10.1172/JCI22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M, Galanos C, Niedermann G. Cancer Res. 2002;62:6901. [PubMed] [Google Scholar]
- 38.Dewan MZ, Uchihara JN, Terashima K, Honda M, Sata T, Ito M, Fujii N, Uozumi K, Tsukasaki K, Tomonaga M, Kubuki Y, Okayama A, Toi M, Mori N, Yamamoto N. Blood. 2006;107:716. doi: 10.1182/blood-2005-02-0735. [DOI] [PubMed] [Google Scholar]
- 39.Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H. Mol Cancer Ther. 2004;3:473. [PubMed] [Google Scholar]
- 40.Yang Y, Ikezoe T, Takeuchi T, Adachi Y, Ohtsuki Y, Takeuchi S, Koeffler HP, Taguchi H. Cancer Sci. 2005;96:425. doi: 10.1111/j.1349-7006.2005.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Blood. 1996;87:4261. [PubMed] [Google Scholar]
- 42.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Crit Care Med. 1997;25:1298. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 43.Husain KD, Coopersmith CM. Curr Opin Crit Care. 2003;9:159. doi: 10.1097/00075198-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 44.Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS. Cri Care Med. 2002;30:195. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 45.Weaver JG, Rouse MS, Steckelberg JM, Badley AD. FASEB J. 2004;18:1185. doi: 10.1096/fj.03-1230com. [DOI] [PubMed] [Google Scholar]