

Abstract
Acrylamide (ACR) intoxication is associated with selective nerve terminal damage in the central and peripheral nervous systems. As a soft electrophile, ACR could form adducts with nucleophilic sulfhydryl groups on cysteine residues of kelch-like erythroid cell-derived protein with CNS homology-associated protein 1 (Keap1) leading to dissociation of the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 activation of the antioxidant-responsive element (ARE) and subsequent upregulated gene expression of phase II detoxification enzymes and anitoxidant proteins might provide protection in neuronal regions with transcriptional capabilities (e.g., cell body). In contrast, non-transcriptional cell regions (axons, nerve terminals) might be vulnerable to electrophile-induced damage. To test this possibility, immunoblot analysis was used to measure protein products of Nrf2-activated ARE genes in nerve terminals and in cytosolic/nuclear factions of neuronal cell bodies isolated from rats intoxicated at two different ACR dose-rates; i.e., 50 mg/kg/d × 10 days, 21 mg/kg/d × 38 days. To detect possible differences in cell-specific induction, the cytoprotective response to ACR intoxication was determined in hepatic cells. Results show that control brain and hepatic cell fractions exhibited distinct subcellular distributions of Nrf2, Keap1 and several ARE protein products. ACR intoxication, however, did not alter the levels of these proteins in synaptosomal, brain cytoplasm or liver cell fractions. These data indicate that ACR was an insufficient electrophilic signal for ARE induction in all subcellular fractions tested. Because a cytoprotective response was not induced in any fraction, nerve terminal vulnerability to ACR cannot be ascribed to the absence of transcription-based defense mechanisms in this neuronal region.
Keywords: acrylamide, axonopathy, phase II enzymes, Keap1, Nrf2, antioxidant/electrophile response element
1. Introduction
Exposure of humans and laboratory animals to acrylamide (ACR) produces ataxia, skeletal muscle weakness and changes in cognition. In laboratory animal models, this neurotoxicity is associated with highly selective damage to distal axon and nerve terminal regions (reviewed in LoPachin et al., 2002a, 2003; LoPachin and Gavin, 2008). Although the exact mechanism is unknown, this selective damage might be due to the differential abilities of cell bodies and nerve terminals to mount inducible cytoprotective processes (LoPachin and Gavin, 2008). Specifically, ACR is an electrophile that forms Michael-type adducts with nucleophilic residues on proteins (Barber and LoPachin, 2004; Barber et al., 2007; LoPachin et al., 2004, 2006; 2007a,b; reviewed in LoPachin et al., 2008). Cells are protected from electrophilic attack by xenobiotic chemicals (e.g., acrolein) and reactive oxygen species (ROS) through induction of the antioxidant-responsive element (ARE) and other transcriptionally-based mechanisms. The ARE is a cis-acting regulatory element found in the promotor regions of genes encoding many phase II biotransformation enzymes (e.g., hemeoxygenase-1, γ-glutamyl cysteine ligase) and antioxidant proteins (e.g., glutathione reductase; Frilling et al., 1990; Lee and Johnson, 2004; Zhu et al., 2008). Binding of the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2), to the ARE activates gene transcription. During basal conditions, Nrf2 is associated with cytosolic kelch-like erythroid cell-derived protein with CNS homology-associated protein 1 (Keap1), which targets the transcription factor for ubiquitination and proteosomal degradation. Electrophile adduction of specific cysteine residues (Cys257, Cys273, Cys288 and Cys297) on Keap1 promotes dissociation of the Nrf2-Keap1 complex, which leads to nuclear translocation of Nrf2 and subsequent transcriptional activation of ARE-driven genes and their cytoprotective protein products (Dinkova-Kostova et al., 2002; Hong et al., 2005; Wakabayashi et al., 2004; Zhang and Hanninck, 2003; Zhang et al., 2004). In contrast to other cell types, nerve cells are unique since the cell body and nucleus are anatomically separated from their nerve terminals by an axon of variable length. Nerve terminals are anuclear regions that lack transcriptional machinery and, consequently, the distances separating these distal regions from the perikaryon could limit the availability of cell body-derived cytoprotective proteins. The absence of inducible defense mechanisms could render nerve terminals vulnerable to electrophilic attack.
The present study was designed to test the hypothesis that distal nerve regions, in contrast to the perikaryon, are selectively vulnerable to ACR attack due to their inability to mount a transcriptionally-based ARE response. ARE induction is a possible consequence of ACR intoxication since the αβ-unsaturated carbonyl structure of this neurotoxicant is common among electrophilic chemicals that induce the ARE (Talalay et al., 1988). Glycidamide, the epoxide metabolite of ACR, is also an electrophile and could, therefore, activate the ARE. Furthermore, ACR might indirectly stimulate the antioxidant response via glutathione depletion leading to oxidative stress and ROS production (Catalgol et al., 2009; Yousef and El-Demerdash, 2006). In the present study we measured the levels of selected ARE-derived cytoprotective proteins in nerve terminals and in cytosolic/nuclear factions of neuronal cell bodies isolated from rats intoxicated at two different ACR dose-rates. To detect possible differences in cell-specific induction, the ARE response to ACR intoxication was determined in hepatic cells. Results indicate that, despite the induction of significant neurotoxicity, ACR exposure did not alter the content of phase II enzymes/antioxidants proteins in any cell fraction examined. These data do not support our original hypothesis and, instead, suggest previously unrecognized limitations in the electrophile responsiveness of the Nrf2-ARE pathway.
2. Materials and methods
2.1 Chemicals and materials
All chemicals were purchased from Aldrich Chemical Company (Milwaukee, WI). EDTA-free Protease Inhibitor Cocktail was purchased from Roche Applied Sciences (Indianapolis, IN). Antibodies were purchased from following sources: Novus – Nrf2, the modifier subunit of glutamate-cysteine ligase (GCLM), heme oxygenase-1 (HO-1) and cystine/glutamic acid transporter (xCT); Santa Cruz – nuclear transcription factor Y subunit α (NF-Ya), NAD(P)H:quinone oxidoreductase 1 (NQO1) and glutathione reductase (GR); Abcam – glutathione transferase M1 (GST-M1); R&D System – Keap1; Sigma - glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Goat anti-mouse or anti-rabbit IgG conjugated to alkaline phosphatase and Western Blue® (alkaline phosphatase substrate) were purchased from Promega Life Sciences (Madison, WI). Pre-made gels were purchased from Invitrogen Co. (Carlsbad, CA). Pierce bicinchoninic acid (BCA) protein assay kits and radio-immunoprecipitation assay (RIPA) buffer were purchased from Thermo Fisher Scientific Inc. (Rockford, IL). Amicon Ultra-4 centrifuge filter units (3 kDa molecular weight cutoff) were purchased from Millipore Co. (Billerica, MA).
2.2 Animals and ACR intoxication
All aspects of this study were approved by the Montefiore Medical Center Animal Care Committee. Adult male Sprague-Dawley rats (300-325 g; Taconic Farms, Germantown, NY) were housed individually with drinking water and chow available ad libitum. Rats (n=4-6 per group) were exposed to ACR at dose-rates of either 50 mg/kg/d × 10 days (i.p.) or 21 mg/kg/d × 38 days (p.o.). Both dosing schedules produced moderate-severe levels of neurotoxicity as assessed via bi-weekly measurements of body weights and gait scores. Previous neurological studies have shown that body weight changes and gait scoring were sensitive indices of developing chemical-induced neurotoxicity (LoPachin et al., 2002b). To measure the development of gait abnormalities, rats were placed in a plexiglass box and were observed for 3 mins by a trained, blinded observer who was not involved in animal care or ACR exposure. Following observations, a gait score was assigned from 1 to 4 where: 1 = a normal gait; 2 = a slightly abnormal gait (slight ataxia, hopping gait and foot splay); 3 = moderately abnormal gait (obvious ataxia and foot splay with limb abduction during ambulation); 4 = severely abnormal gait (inability to support body weight and foot splay).
2.3 Differential centrifugation methods
The tissue fractionation procedures described in the following subsections are well-characterized and have been used extensively to determine the respective subcellular distribution of various soluble proteins and factors (e.g., see Gullo et al., 1987; Mishra et al., 2002). Furthermore, our general approach is based on previous studies (e.g., see Watai et al., 2007) using subcellular fractionation and immunoblot analysis to define the responses of the Nrf2-Keap1 system to oxidative/electrophilic stress. Brain and liver fractions were prepared 24 hrs after termination of ACR exposure.
2.3.1 Hepatic cell fractionation
Hepatic cell nuclear/endoplasmic reticulum and cytosolic fractions were prepared by the method of Dyer and Herzog (1995). Briefly, liver (1 gm) was minced and then homogenized in liver lysis buffer (pH 7.9) containing 10mM HEPES, 1.5mM MgCl2, 10mM KCl, 0.2% β-mercaptoethanol and 0.5mM phenylmethylsulfonyl fluoride (PMSF) using a Dounce Tissue Grinder. The homogenate was filtered through 70-μm nylon mesh and the filtrate was centrifuged at 11,000 × g for 20 min. The supernatant was retained as the liver cell cytosolic fraction. To prepare nuclei, the corresponding pellet was washed in lysis buffer and was resuspended with extraction buffer (0.66 volume) containing 20mM HEPES, 1.5mM MgCl2, 420mM NaCl, 0.2mM EDTA and 25% glycerol (pH 7.9). The pellet suspension was shaken gently for 30mins (4°C) and then centrifuged at 11,000 × g (20 mins). The supernatant was de-salted using Amicon Ultra-4 centrifuge filter units. The retained filtrate was resuspended in RIPA buffer and was designated the liver nuclear/ER (LNE) fraction.
2.3.2 Brain cell fractionation
Brain cell nuclear/ER and cytosolic fractions were prepared by a modification of Giufrida et al. (1975) method. Briefly, whole brains were minced and then homogenized in a brain lysis buffer (pH 6.4) containing 0.32M sucrose, 1.0 mM KH2PO4, 3.0 mM MgCl2, 1.0% Brij-35, 0.2% β-mercaptoethanol and 0.5 mM PMSF using a Dounce Tissue Grinder. The homogenate was then centrifuged at 11,000 × g for 20 min and the supernatant was retained as the brain cell (neuronal plus glial) cytosolic fraction. To prepare nuclei, the pellet was washed in brain lysis buffer (excluding Brij-35) and resuspended in extraction buffer (see subsection 2.3.1). The pellet suspension was shaken gently for 30mins (4°C) and then centrifuged at 11,000 × g (20 mins). The supernatant was de-salted using Amicon Ultra-4 centrifuge filter units. The retained filtrate was resuspended in RIPA buffer and was designated the brain nuclear/ER (BNE) fraction.
2.3.3 Preparation of synaptosomes
Synaptosomes were prepared by the Percoll gradient method of Dunkley et al. as modified by LoPachin et al. (2004). In brief, whole brains were rapidly removed and minced in cold (4°C, pH 7.4) gradient buffer containing 0.32 M sucrose, 1 mM EDTA and 0.25 mM dithiothreitol (SED). Tissue was gently homogenized in SED buffer (10 passes in a Teflon-glass homogenizer; 700 RPM) and the resulting homogenate was centrifuged at 1,000 g (10 min, 4°C). The pellet (P1) was washed once and supernatants (S1 and S2) were combined. Protein content of the pooled supernatant was determined by the Pierce BCA protein assay and was adjusted with SED to 5 mg/ml and then layered on top of a freshly prepared 4-step discontinuous Percoll gradient (3%, 10%, 15% and 23% Percoll in SED, pH 7.4). Gradients were centrifuged at 32,000 g for 6 mins and synaptosomes were collected at the last interface (15%/23%) and homogenized in RIPA buffer.
2.4 Gel Electrophoresis and Semi-Quantitative Immunoblot Analysis
Tris-glycine continuous gradient gels (8-16%) were used to separate proteins. Protein contents were measured by the Pierce BCA assay and proteins were loaded on gels at 25 μg per lane. Following electrophoretic separation, proteins were transferred to polyvinyldene fluoride (PVDF) membranes overnight (20mA current). After transfer, membranes were blocked with 5.0% dried milk in TBS/0.1% TWEEN 20 for 1 hr and then rinsed. Membranes prepared from fractions were incubated for 1hr with a selected antibody diluted in 5% dried milk/TBS. Following primary antibody incubation, membranes were washed in TBS and incubated (1hr) with alkaline phosphatase (AP)-conjugated secondary antibody (goat anti-mouse or anti-rabbit IgG). Membranes were washed again and immunoreactive bands were visualized with alkaline phosphatase substrate (Western Blue®). Immunoreactive protein bands were scanned with a densitometer, digitized and analyzed as pixels per total area of the immunoreactive band using the freehand selection tool of the NIH Imaging Program (LoPachin et al., 2004). Densitometric data from the different subcellular fractions were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). To determine statistical differences in the fractional distribution of ARE and associated proteins, normalized data (mean ± SEM; n ≥ 3) from control liver and brain cell fractions were compared using Student’s t test (p < 0.05). ANOVA followed by Dunnett’s post hoc test (p < 0.05) was used for multiple range comparisons among treated and control group mean data.
2.4 Calculation of HSAB Parameters
The Lowest Unoccupied Molecular Orbital (LUMO) energy (ELUMO) and Highest Occupied Molecular Orbital (HOMO) energy (EHOMO), were calculated using Spartan08 (version 1.1.1) software (Wavefunction Inc., Irvine CA). For each chemical, ground state equilibrium geometries were calculated with Density Functional BSLYP 6-31G* in water starting from 6-31G* geometries. Global (whole molecule) hardness (η) was calculated as η = (ELUMO-EHOMO)/2 and softness (σ) was calculated as the inverse of hardness or σ= 1/η The electrophilicity index (ω) was calculated as ω = μ2/2η, where μ is chemical potential of the electrophile and was calculated as μ = (ELUMO+EHOMO)/2. The nucleophilicity index (ω−) was calculated as ω− = ηA (μA − μB)2/2(ηA − ηB)2, where η = (ELUMO−EHOMO)/2, A = reacting nucleophile (sulfhydryl thiolate state) and B = reacting electrophile (see LoPachin et al., 2008 for details).
3. Results
3.1 Protein distribution in control brain and liver cell fractions
Preliminary studies were conducted to determine the validity of our subcellular fractionation procedures by determining the distribution of selected marker proteins. Thus, synaptotagmin, a nerve terminal marker (Calakos and Scheller, 1996), was highly enriched in the synaptosomal fraction and was not detected in either the liver cytoplasmic or nuclear/ER fractions (Fig. 1A; see also LoPachin et al., 2004). As expected, the liver-/brain-enriched transcription factor, NF-Yα was present in the nuclear/ER fractions of liver and brain (Fig. 1A; Schmidt and Schibler, 1995).
Figure 1.

Nrf2 and Keap1 have unique distributions in subcellular fractions of brain and liver. (A). Representative immunoblots are presented for Nrf2, the accessory protein, Keap1, the liver/brain-enriched transcription factor, NF-Yα, the synaptosomal marker protein, synaptotagmin and the loading control, GAPDH, in brain and hepatic cell fractions from control animals. Proteins were loaded on gels at 25 μg per lane. S = synaptosomes; BC = brain cytosol; LC = liver cytosol; BNE = brain nuclear/ER fraction and LNE = liver nuclear/ER fraction. (B). Relative immunoblot intensities for individual subcellular fractions of brain and liver are presented. * p<0.05, n = 4-6/group.
Immunoblot analyses of control brain and liver cell fractions (Fig. 1) revealed distinct subcellular distributions for Nrf2, Keap1 and selected ARE-derived gene products that are involved in different cytoprotective processes. Thus, Nrf2 and Keap1 were not detectable in control synaptosomes and were present only in relatively low abundance in the brain cytosol and nuclear/ER fractions (Fig. 1B). In contrast, these proteins were significantly more prevalent in liver cell fractions (Fig. 1B). The phase II enzymes and other cytoprotective proteins exhibited distinct patterns of distribution in the different neuronal and hepatic cell fractions (Fig. 2A). Specifically, proteins involved in xenobiotic detoxification, NAD(P)H:quinone oxidoreductase 1 (NQO1) and glutathione transferase M1 (GST-M1) were found in the brain (BC) and liver (LC) cytosolic fractions (Edlund et al., 1982). To a lesser extent, GST-M1 was also present in the synaptosomal fraction and the nuclear/ER fractions of liver and brain (Fig. 2B). Heme oxygenase-1 (HO-1), which catalyzes the rate-limiting step in pro-oxidant heme/Fe metabolism, was detected in all subcellular fractions, except synaptosomes (Johnson et al., 1993; Lin et al., 2007; Rogers et al., 2002). It is noteworthy that HO-1 was highly enriched in the liver cell compartments (Fig. 2B). Our distribution data indicated that the modifier subunit of glutamate-cysteine ligase (GCLM), the rate-limiting enzyme in glutathione (GSH) synthesis, was prevalent in the cytosolic fractions of the brain and liver preparations (Franklin et al., 2009). Glutathione reductase (GR), also involved in GSH synthesis, was detected in the hepatic compartments, whereas the cystine/glutamate transporter (xCT) was relatively abundant in the synaptosomal preparation and the nuclear/ER fractions of brain and liver (Fig. 2B; Flynn and McBean, 2000; LaBella et al., 2007). In general, the phase II detoxifying enzymes and antioxidant proteins were more prevalent in liver fractions when compared to those of the brain (Fig. 2B).
Figure 2.

The Nrf-targeted ARE products have unique distributions in subcellular fractions of brain and liver. (A). Representative immunoblots are presented for the cytoprotective proteins and the loading control, GAPDH, in brain and hepatic cell fractions from control animals. Proteins were loaded on gels at 25 μg per lane. S = synaptosomes; BC = brain cytosol; LC = liver cytosol; BNE = brain nuclear/ER fraction and LNE = liver nuclear/ER fraction. (B). Relative immunoblot intensities for individual subcellular fractions of brain and liver are presented. * p<0.05, n = 4-6/group.
3.2 Effects of ACR on protein distribution in brain and liver cell fractions
Regardless of daily dose-rate, ACR did not induce an ARE response in brain or liver cells of moderate-to-severely intoxicated rats. ACR intoxication did not cause changes in the respective immunoblot densities for Nrf2 or Keap1 or the cytoprotective proteins in brain synaptosomes or the nuclear/ER fractions of brain or liver (Fig. 3). Nor were significant changes in immunopositive band densities for Nrf2, Keap1 or ARE-derived protein products evident in the cytoplasmic fractions of brain or liver from ACR-intoxicated rats (data not shown).
Figure 3.

ACR intoxication does not upregulate phase II detoxifying enzymes or antioxidant proteins in subcellular fractions of brain or liver. (A). Representative immunoblots are presented for Nrf2, Keap1, the cytoprotective proteins in brain and subcellular fractions from control and ACR-intoxicated animals. Also shown is the loading control, GAPDH. Proteins were loaded on gels at 25 μg per lane. CON = control; IP = intraperitoneal injection, 50 mg/kg/d × 10 days; ORAL = oral intoxication; 21 mg/kg/d × 38 days. (B). Relative immunoblot intensities for individual subcellular fractions of brain and liver are presented. Immunoblots for brain and liver cytosolic compartments (BC, LC respectively) are not shown, but were not altered by ACR intoxication.
3.3 HSAB parameters for unsaturated carbonyl compounds and their potential cysteine sulfhydryl targets
The covalent interactions of ARE inducers such as acrolein or 3-methylene-2-norbornanone (Prestera et al., 1993; Talalay et al., 1988) with cysteine residues is governed by the energies of the respective frontier molecular orbitals (FMO). Since the relevant FMO energies can be accurately calculated, these adduct reactions can be described quantitatively through calculations of Hard-Soft Acid-Base (HSAB) parameters such as softness (σ), electrophilicity (ω) and nucleophilicity (ω−; reviewed in LoPachin et al., 2008a, 2009). Thus, the softness of an electrophile is considered the ease with which electron redistribution occurs during adduct (covalent bond) formation. Consequently, the softer the electrophile (larger σ value), the faster it will accept electrons from a given nucleophile; e.g. cysteine sulfhydryl group, to form a bond. The electrophilic index (ω) is a higher order parameter that combines softness (σ) with chemical potential (μ; ability of electrophile to accept electron density) and is a general measure of electrophilic reactivity. As illustrated in Table 1, the respective σ and ω values indicate that acrolein and 3-methylene-2-norbornanone are relatively soft, reactive electrophiles, whereas methylvinyl ketone, 1-cyclohexen-2-one and 1-cyclopenten-2-one are moderately soft, reactive unsaturated ketone derivatives (Table 1). Previous in vitro studies showed that these electrophiles were relatively potent ARE inducers (Prestera et al., 1993; Talalay et al., 1988). In contrast, ACR was a substantially harder, less reactive electrophile than the other unsaturated ketones (Table 1) and did not cause ARE induction (Talalay et al., 1988).
Table 1.
Calculated HSAB parameters for Unsaturated Carbonyl Compounds and Their Possible Reactions with Nucleophilic Targets
| Compound | Evokes ARE Response? |
σ (x 10−3 ev1) |
ω (ev) |
Thiolate ω− (x 10−3 ev) |
Thiol ω− (x 10−3 ev) |
|---|---|---|---|---|---|
|
yes | 371 | 3.81 | 258 | 66 |
|
yes | 363 | 3.38 | 207 | 43 |
|
yes | 383 | 3.32 | 188 | 32 |

|
yes | 370 | 3.13 | 173 | 27 |
|
yes | 359 | 2.90 | 152 | 21 |
|
no | 315 | 2.61 | 140 | 21 |
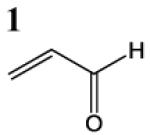
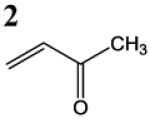
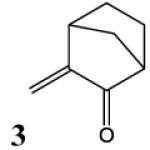
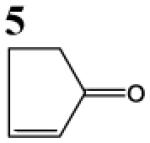
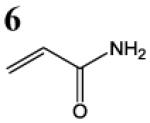
HSAB parameters were calculated as described in Materials and Methods section. Based on HSAB calculations, the unsaturated carbonyl compounds that activate the Nrf2-ARE pathway are relatively soft (σ) reactive electrophiles (ω). These compound preferentially form Michael-type adducts with the thiolate-state of cysteine sulfhydryl groups. Chemical identification: 1 = acrolein; 2 = methylvinyl ketone; 3 = 3-methylene-2-norbornanone; 4 = 1-cyclohexen-2-one; 5 =1-cyclopenten-2-one and 6 = acrylamide (see Talalay et al., 1988; Prestera et a.., 1993).
According to the selectivity principle of the HSAB theory (LoPachin et al., 2009), soft electrophiles preferentially react with soft nucleophiles. The softness of a nucleophile reflects the relative ability to rapidly transfer electron density to the vacant FMO of the electrophile. Calculations of nucleophilic softness (data not shown) indicate that sulfhydryl groups in the anionic thiolate-state (−1) are substantially softer than corresponding thiol groups (0; LoPachin et al., 2007a,b). This suggests that thiolate groups are the preferential targets of soft electrophiles such as acrolein and 3-methylene-2-norbornanone. The probability that a given nucleophile will form an adduct with an unsaturated carbonyl electrophile can be determined by calculating the nucleophilicity index (ω−). This higher order HSAB parameter considers the hardness (η) and chemical potential (μ) of both the electrophile and the nucleophilic thiolate reactants (LoPachin et al., 2008a, 2009). Based on the significant differences (4-7 fold) in the respective ω− values (Table 1), the cysteine sulfhydryl thiol (0) state is substantially less reactive with the electrophiles than the highly nucleophilic thiolate. These data suggest that electrophilic ARE inducers will preferentially form adducts with sulfhydryl thiolates of cysteine residues on Keap1. The rank order of the ω− values indicate that, of the electrophiles evaluated, ACR is least likely to form adducts with cysteine sulfhydryl thiolates (Table 1).
4. Discussion
In this study we tested the ability of ACR intoxication to upregulate the levels of phase II detoxifying enzymes and antioxidant proteins in liver and brain cell fractions. The α,β-unsaturated carbonyl structure of ACR is a soft electrophile (LoPachin et al., 2008; LoPachin and Gavin, 2008) and, therefore, this neurotoxicant could induce Nrf2-driven ARE gene expression by forming adducts with soft nucleophilic cysteine residues (e.g., Cys257, Cys273, Cys288, Cys297) that act as electrophile sensors on Keap1 (Dinkova-Kostova et al., 2001; Hong et al., 2005; Itoh et al., 2004; Wakabayashi et al., 2004). Theoretically, this defense mechanism could provide cell body protection against electrophile attack, whereas anuclear distal axon regions that lack the required transcriptional machinery might be relatively vulnerable (LoPachin and Gavin, 2008). However, immunoblot detection indicated that ACR did not alter the levels of cytoprotective proteins (e.g., GST, NQO1 and GCLM) in any cell fraction examined suggesting a lack of ARE induction. This is consistent with early in vitro research, which showed that ACR was similarly inactive, whereas Michael acceptors such acrolein, methylvinyl ketone and other α,β–unsaturated aldehydes induced phase II enzymes in exposed hepatoma cells (Talalay et al., 1988). Also consistent with the observed lack of induction, previous mass spectrometric analyses of brain fractions from ACR-intoxicated rats (Barber et al., 2007; see also Barber and LoPachin, 2004) failed to detect ACR adducts of Keap1. Furthermore, although the ARE might be activated indirectly through ROS generation (Introduction), previous research suggests that ACR does not cause oxidative stress in CNS of intoxicated animals (Johnson and Murphy, 1977; LoPachin et al., 2006).
Together, these data suggest that ACR intoxication is an insufficient stimulus for ARE activation in rat brain or liver. These results, therefore, do not support our proposal that nerve terminal vulnerability to ACR is due to the differential abilities of neuronal perikaryal and distal axon regions to mount cytoprotective transcriptionally-based mechanisms. As an alternative mechanism, the inherently slow turnover rate of proteins in distal axons and nerve terminals might render these regions selectively vulnerable to cumulative neurotoxicity induced by weak electrophiles such as ACR (LoPachin et al., 2008). Thus, in the absence of transcriptional capacity, the distal axon proteome must be maintained through cell body synthesis and subsequent anterograde transport. To conserve resources, protein turnover rates in distal axon regions are exceptionally slow relative to that in neuronal cell bodies or other cell-types, (Calakos and Scheller, 1996; Katyare and Shallom, 1988). Indeed, our previous research indicates that, as a result of their slow turnover, ACR-adducted, dysfunctional proteins are replaced slowly. This leads to their progressive accumulation (Barber and LoPachin, 2004; Barber et al., 2007) and the parallel development of cumulative neurotoxicity (LoPachin et al., 2002a).
The inability of ACR to activate the ARE is likely a function of the low electrophilicity of this neurotoxicant and the possibly lower than expected nucleophilicity of “reactive” Keap1 cysteine residues. Specifically, HSAB calculations of softness (σ) and the electrophilic index (ω) show that ACR is a harder and relatively weak electrophile, when compared to other Michael acceptors that act as ARE inducers; e.g., acrolein or 3-methylene-2-norbornanone (Table 1; Prestera et al., 1993; Talalay et al., 1988). As a weak electrophile, ACR at low in vivo target-site concentrations will preferentially form adducts with highly reactive cysteine residues; i.e., those exhibiting greater nucleophilicity (reviewed in LoPachin and Barber, 2006; LoPachin et al., 2008). Thus, calculations of nucleophilic indices (ω−) demonstrate that the reaction of Michael acceptor electrophiles with sulfhydryl thiol groups on cysteine residues is kinetically unfavored; i.e., the respective ω− values are relatively low (Table 1). In contrast, these calculations indicate that the Michael electrophiles, including ACR, react preferentially with highly nucleophilic cysteine thiolates, which can be found in protein microenvironments (e.g., catalytic triads) that lower the pKa of sidechain sulfhydryl groups (Barber et al., 2007; LoPachin et al., 2006, 2007a,b; reviewed in LoPachin et al., 2008). However, these calculations also indicate that ACR is not likely to form adducts with sulfhydryl thiolate sites (Table 1). Although the Keap1 sensory cysteines are considered to be reactive, it is not known whether these residues exist in low pKa microenvironments, nor have the respective nucleophilicities been determined experimentally (Liebler, 2008). Therefore, the nucleophilic reactivities of these cysteines might be too low to form adducts with weak electrophiles such as ACR. Finally, it is also unlikely that the ACR metabolite, glycidamide (GLY), can activate ARE (Introduction). This epoxide metabolite is a relatively hard electrophile with a low ω value (GLY ω = 1.72 ev vs. ACR ω = 2.61 ev). Therefore, the adduct reaction of this hard electrophile with the soft nucleophilic cysteines of Keap 1 is kinetically unfavored (see discussion in LoPachin and Gavin, 2008).
Although negative, our findings suggest that despite being a Michael acceptor ACR cannot form adducts with sensory cysteine residues on Keap1. This implies a detectability threshold for these residues and, therefore, it cannot be assumed that the Nrf2-ARE pathway and possibly other inducible cytoprotective systems will be generically responsive to electrophilic environmental toxicants. Thus, weak electrophilic α,β-unsaturated carbonyl derivatives such as ACR and methyl acrylate that cause cumulative neurotoxicity (LoPachin et al., 2008; LoPachin and Gavin, 2008) might nonetheless escape Keap1 detection. Our findings suggest that future studies should determine the nucleophilicity and microenvironment of the sensory cysteine residues on Keap1. In addition, calculations of the electrophilic index (ω) and other HSAB parameters for a series of Michael acceptors (Prestera et al., 1993) could be used to define quantitatively the threshold electrophilicity for ARE induction.
Acknowledgments
This research was supported by a grant from the National Institute of Environmental Health to R.M.L. [R01 ES03830-24]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest None.
References
- Barber DS, LoPachin RM. Proteomic analysis of acrylamide-protein adduct formation in rat brain synaptosomes. Toxicol. Appl. Pharmacol. 2004;201:120–136. doi: 10.1016/j.taap.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Barber DS, Stevens S, LoPachin RM. Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose-rate. Toxicol. Sci. 2008;100:156–167. doi: 10.1093/toxsci/kfm210. [DOI] [PubMed] [Google Scholar]
- Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking and fusion: a molecular description. Physiol. Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- Catalgol B, Ozhan G, Alpertunga B. Acrylamide-induced oxidative stress in human erythrocytes. Human. Exp. Toxicol. 2009;28:611–617. doi: 10.1177/0960327109350664. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer RB, Herzog NK. Isolation of intact nuclei for nuclear extract preparation from fragile B-lymphocyte cell lines. Biotechniques. 1995;19:192–195. [PubMed] [Google Scholar]
- Edlund C, Elhammer A, Dallner G. Distribution of newly synthesized DT-diaphorase in rat liver. Biosci. Rep. 1982;2:861–865. doi: 10.1007/BF01114891. [DOI] [PubMed] [Google Scholar]
- Flynn J, McBean GJ. Kinetic and pharmacological analysis of L-[35S]cystine transport into rat brain synaptosomes. Neurochem. Int. 2000;36:513–521. doi: 10.1016/s0197-0186(99)00151-5. [DOI] [PubMed] [Google Scholar]
- Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function and post-translational regulation of the catalytic and modifier subunits of gluatmate cysteine ligase. Mole. Asp. Med. 2009;30:86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friling R, Bensimon A, Tichauer Y, Daniel V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. 1990;87:6258–6262. doi: 10.1073/pnas.87.16.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giufrida AM, Cox D, Mathias AP. RNA polymerase activity in various classes of nuclei from different regions of rat brain during postnatal development. J. Neurochem. 1975;24:749–755. [PubMed] [Google Scholar]
- Gullo D, Sinha AK, Woods R, Pervin K, Ekins RP. Triiodothyronine binding in adult rat brain: compartmentation of receptor populations in purified neuronal and glial nuclei. Endocrinology. 1987;120:325–331. doi: 10.1210/endo-120-1-325. [DOI] [PubMed] [Google Scholar]
- Hong F, Freeman ML, Leibler DC. Identification of sensor cysteines in human Keap1 modified by the cacer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005;18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- Itoh K, Tong KT, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway of adaptive respoinse to electrophiles. Free Rad. Biol. Med. 2004;10:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Murphy SD. Effects of acrylamide intoxication on pyridine nucleotide concentrations and functions in rat cerebral cortex. Biochem. Pharmacol. 1977;26:2151–2155. doi: 10.1016/0006-2952(77)90267-2. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Barbary AE, Dornguth SE, Brugge JF, Siegel FL. Glutathione S-transferase isoenzymes in rat brain neurons and glia. J. Neurosci. 1993;13:2013–2023. doi: 10.1523/JNEUROSCI.13-05-02013.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyare SS, Shallom JM. Altered cerebral protein turnover in rats following prolonged in vivo treatment with nicotine. J. Neurochem. 1988;50:1356–1363. doi: 10.1111/j.1471-4159.1988.tb03016.x. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBella V, Valentino F, Piccoli T, Piccoli F. Expression and developmental regulation of the cystine/glutamate exchanger (Xc) in the rat. Neurochem. Res. 2007;32:1081–1090. doi: 10.1007/s11064-006-9277-6. [DOI] [PubMed] [Google Scholar]
- Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mole. Biol. 2004;37:139–143. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- Leibler DC. Protein damage by reactive electrophiles: targets and consequences. Chem. Res. Toxicol. 2008;21:117–128. doi: 10.1021/tx700235t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Weis S, Yang G, Weng Y, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007;282:20621–20633. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Ross JF, Lehning EJ. Nerve terminals as the primary site of acrylamide action: A hypothesis. NeuroToxicology. 2002a;23:43–60. doi: 10.1016/s0161-813x(01)00074-2. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Ross JF, Reid ML, Das S, Mansukhami S, Lehning EJ. Neurological evaluation of toxic axonopathies in rats: acrylamide and 2,5-hexanedione. NeuroToxicology. 2002b;23:95–110. doi: 10.1016/s0161-813x(02)00003-7. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Balaban CD, Ross JF. Acrylamide axonopathy revisited. Toxicol. Appl. Pharmacol. 2003;188:135–153. doi: 10.1016/s0041-008x(02)00072-8. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Schwarcz AI, Gaughan CL, Mansukhani S, Das S. In vivo and in vitro effects of acrylamide on synaptosomal neurotransmitter uptake and release. NeuroToxicology. 2004;25:349–363. doi: 10.1016/S0161-813X(03)00149-9. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Barber DS, He D, Das S. Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Toxicol. Sci. 2006;89:224–234. doi: 10.1093/toxsci/kfj005. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Barber DS. Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Toxicol. Sci. 2006;94:240–255. doi: 10.1093/toxsci/kfl066. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Barber DS, Geohagen BC, Gavin T, He D, Das S. Structure-toxicity analysis of Type-2 alkenes: in vitro neurotoxicity. Toxicol. Sci. 2007a;95:136–146. doi: 10.1093/toxsci/kfl127. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Geohagen BC, Das S. Neurotoxic mechanisms of electrophilic type-2 alkenes: soft-soft interactions described by quantum mechanical parameters. Toxicol. Sci. 2007b;98:561–570. doi: 10.1093/toxsci/kfm127. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Barber DS, Gavin T. Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Toxicol. Sci. 2008;104:235–249. doi: 10.1093/toxsci/kfm301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T. Acrylamide-induced nerve terminal damage: relevance to neurotoxic and neurodegenerative mechanisms. J. Agri. Food Chem. 2008;56:5994–6003. doi: 10.1021/jf703745t. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra OP, Ashraf QM, Delivoria-Papadopoulos M. Phosphorylation of cAMP response element binding (CREB) protein during hypoxia in cerebral cortex of newborn piglets and the effect of nitric oxide synthase inhibition. Neuroscience. 2002;115:985–991. doi: 10.1016/s0306-4522(02)00275-0. [DOI] [PubMed] [Google Scholar]
- Prestera T, Holzclaw WD, Zhang Y, Talalay P. Chemcial and molecular regulation of enzymes that detoxify carcinogens. Proc. Natl. Acad. Sci. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers LK, Gupta S, Welty SE, Hansen TN, Smith CV. Nuclear and nucleolar glutathione reductase, peroxidase, and transferase activities in livers of male and female Fischer-344 rats. Toxicol. Sci. 2002;69:279–285. doi: 10.1093/toxsci/69.1.279. [DOI] [PubMed] [Google Scholar]
- Satoh T, Okamoto S, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic phase II inducers. Proc. Natl. Acad. Sci. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EE, Schibler U. Cell size regulation, a mechanism that controls cellular RNA accumulation: consequences on regulation of the ubiquitous transcription factors Oct1 and NF-Y, and the liver-enriched transcription factor DBP. J. Cell. Biol. 1995;128:467–483. doi: 10.1083/jcb.128.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talalay P, DeLong MJ, Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Pro. Natl. Acad. Sci. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volti GL, Ientile R, Abraham NG, Vanella A, Cannoavo G, Mazza F, Curro M, Raciti G, Avola R, Campisi A. Immuncytochemical localization and expression of heme oxygenase-1 in primary astroglial cell cultures during differentiation: effect of glutamate. Biochem. Biophys. Res. Comm. 2004;315:517–524. doi: 10.1016/j.bbrc.2004.01.090. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kosotova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, Itoh K, Yamamoto M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells. 2007;12:1163–1178. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- Yousef MI, El-Demerdash FM. Acrylamide-induced oxidative stress and biochemical perturbations in rats. Toxicology. 2006;219:133–141. doi: 10.1016/j.tox.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cellular Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adapter protein for a Cul3-dependent ubiquitin ligase complex. Mole. Cell. Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Jia Z, Zhang L, Yamamoto M, Misra HP, Trush MA, Li Y. Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. 2008;233:463–474. doi: 10.3181/0711-RM-304. [DOI] [PubMed] [Google Scholar]