

Abstract
Epidermal growth factor receptor (EGFR) is overexpressed in up to 90% of head and neck cancer (HNC), where increased expression levels of EGFR correlate with poor prognosis. To date, EGFR expression levels have not predicted the clinical response to the EGFR-targeting therapies. Elucidation of the molecular mechanisms underlying anti-EGFR-induced antitumor effects may shed some light on the mechanisms of HNC resistance to EGFR-targeting therapeutics and provide novel targets for improving the treatment of HNC. Here, we conducted a quantitative proteomics analysis to determine the molecular networks regulated by EGFR levels in HNC by specifically knocking-down EGFR and employing stable isotope labeling with amino acids in cell culture (SILAC). Following data normalization to minimize systematic errors and Western blotting validation, 12 proteins (e.g., p21, stratifin, and maspin) and 24 proteins (e.g., cdc2 and MTA2) were found to be significantly upregulated or downregulated by EGFR knockdown, respectively. Bioinformatic analysis revealed that these proteins were mainly involved in long-chain fatty acid biosynthesis and β-oxidation, cholesterol biosynthesis, cell proliferation, DNA replication, and apoptosis. Cell cycle analysis confirmed that G2/M phase progression was significantly inhibited by EGFR knockdown, a hypothesis generated from network modeling. Further investigation of these molecular networks may not only enhance our understanding of the antitumor mechanisms of EGFR targeting but also improve patient selection and provide novel targets for better therapeutics.
Keywords: cholesterol, epidermal growth factor receptor, fatty acids, head and neck cancer, mass spectrometry, molecular network, P53, quantitative proteomics, small interfering RNA, stable isotope labeling with amino acids in cell culture
Introduction
Head and neck cancer (HNC), a group of biologically similar cancers originating from the upper aerodigestive tract, is one of the most common malignancies worldwide.1 Despite improvements in the diagnosis and standard treatment of HNC, the overall survival rate for advanced HNC patients has not been significantly improved over the last three decades.2 In addition, standard therapies including surgery, chemotherapy, and radiotherapy often lead to functional deficits and disfigurement. Therefore, novel approaches (e.g., targeted therapeutics) that are more effective and have fewer associated toxicities than standard therapies are currently under active investigation.
Epidermal growth factor receptor (EGFR), also known as ErbB1 and HER1, plays a pivotal role in a wide range of biological processes such as cell proliferation, survival, differentiation, and migration.3 The EGFR was anticipated to be an excellent drug target for HNC treatment because it is overexpressed in up to 90% of HNC4 and its higher levels in HNC positively correlate with decreased patient survival.5 Two main EGFR-targeting strategies, that is, monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKIs), have been demonstrated to be highly effective in HNC preclinical models and promising in HNC clinical trials.6 Cetuximab, one of the anti-EGFR mAbs, became the first targeted therapy approved by the U.S. Food and Drug Administration for use in advanced HNC in 2006. In addition to the mAb and TKI approaches, we and other groups have reported that EGFR knockdown using nucleotide-based approaches, such as antisense nucleotides and small interfering RNA (siRNA), effectively inhibits the proliferation of EGFR-overexpressing HNC cells but not normal mucosal epithelial cells.7 We recently completed a phase I clinical trial of EGFR antisense DNA in advanced HNC where 5 out of 17 of HNC patients achieved a clinical response without evidence of toxicity.8
Although EGFR-targeted therapies have been demonstrated to prolong survival when anti-EGFR agents are administered in conjunction with conventional therapies,9 the responses are generally modest (~10%) when EGFR antagonists are delivered as monotherapies for the treatment of HNC.6 Currently, relatively little is known about the molecular mechanisms underlying the intrinsic and acquired HNC resistance as well as the HNC response to EGFR inhibition.10 Studies to date have focused on testing the role of known downstream signaling effectors to promote cell proliferation and/or metastasis in the HNC resistance.11,12 However, applying an unbiased approach (e.g., quantitative proteomics) to identify proteins that are altered in the setting of EGFR downregulation may elucidate unexpected targets that can be exploited for therapeutic benefit. Among all quantitative proteomics approaches, stable isotope labeling with amino acids in cell culture (SILAC)13 is probably the method with lowest technical variations, since minimal manipulations are required before the differentially labeled samples are mixed.14 In fact, to take full advantage of the high accuracy of SILAC, many researchers use a fold change of 1.3–1.5 as a cutoff to filter out the significantly changed proteins.15 Since our group and co-workers published the first application of SILAC in cancer,16 additional groups have subsequently applied this technique to various cancer models (e.g., prostate cancer,17 breast cancer,18–20 melanoma,21 and hepatocellular carcinoma22,23). In the present study, we applied SILAC-based quantitative proteomics to discover the molecular networks regulated by EGFR protein expression level in HNC cells. Further investigation of certain molecules within the networks may reveal biomarkers to predict HNC response to EGFR targeting as well as novel drug targets to improve the treatment of HNC.
Materials and Methods
Materials
Anti-TK1 (thymidine kinase), anti-β-actin, and anti-β-tubulin were purchased from Abcam (Cambridge, MA). Anti-EGFR antibody and propidium iodide were from BD Biosciences (San Jose, CA). Bradford protein assay kit, Coomassie Blue R-250 staining solution, and secondary antibodies including antirabbit and antimouse IgG-horseradish peroxidase conjugates were from Bio-Rad (Hercules, CA). 13C615N2-lysine (Lys8) was from Cambridge Isotope Laboratory (Andover, MA). The human EGFR-specific siRNA (siEGFR) with the sequence 5′-CUCUGGAGGAAAAGAAAGU-3′ was manufactured by Dharmacon. Dialyzed fetal bovine serum (FBS) and Lipofectamine 2000 were from Invitrogen (Carlsbad, CA). Anti-ALDH1A3 (aldehyde dehydrogenase family 1 member A3), anti-LGALS7 (galectin 7), and anti-MAT2A (methionine adenosyltransferase IIα) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). 12C614N2-lysine (Lys0) was from Sigma-Aldrich (St. Louis, MO). Lysine-depleted Dulbecco’s Modified Eagle Medium was from Specialty Media/Millipore (Billerica, MA).
Cell Culture, SILAC Labeling, and EGFR siRNA Transfection
HNC cell line PCI-15B was established at the University of Pittsburgh School of Medicine.24 For SILAC labeling, PCI-15B cells were grown at 37 °C in lysine-depleted DMEM supplemented with 10% (v/v) FBS and either Lys0 or Lys8. Both “light” and “heavy” culture media were replaced every two to three days. On day 5, PCI-15B cells grown in serum-containing “light” medium were cultured in serum-free medium containing Lys0 for 3 days and the medium were changed daily. PCI-15B cells cultured in serum-containing “heavy” medium were divided into two groups: one was transfected with 100 nM human siEGFR using Lipofectamine 2000 according to the manufacturer’s instructions, and the other was untransfected and used as a control to minimize the systematic errors introduced by differential SILAC labeling. These two groups of cells maintained in serum-containing “heavy” medium were then cultured in serum-free medium containing Lys8 for 3 days and the medium was changed daily. On day 8, all groups of cells were harvested for whole cell protein lysate isolation.
Protein Preparation, Separation, and Tryptic Digestion
Whole cell protein lysates were prepared as described previously.25 Protein concentration of whole cell lysates was measured using a Bradford protein assay kit according to the manufacturer’s protocol. Proteins extracted from Lys0-labeled control cells were mixed at equal amounts with proteins extracted from Lys8-labeled control cells or Lys8-labeled cells transfected with EGFR siRNA to generate two samples for quantitative proteomics analysis. Forty micrograms proteins from each sample were resolved on a 12.5% SDS-gel and visualized with Coomassie Blue R-250 staining solution. Each gel lane was excised into 10 slices of similar size, which were further cut into ~1 mm3 particles. Subsequently, gel particles were subjected to in-gel reduction, alkylation, and tryptic digestion essentially as previously described.26,27 Tryptic peptides were extracted and all samples were dried down in vacuo using a SpeedVac concentrator (Thermo Savant, Waltham, MA).
Mass Spectrometric Analysis
Peptide pellets were redissolved with 10 μL 1.5% acetic acid and 7.5% acetonitrile solution. Five μL samples were analyzed by online C18 nanoflow reversed-phase HPLC (Eksigent nanoLC · 2D) connected to an LTQ Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) as described previously.28,29 Briefly, samples were loaded onto an in-house packed 100 μm inner-diameter × 15 cm C18 column (Magic C18, 5 μm, 200 Å, Michrom Bioresource) and separated at about 200 nL/min with 80 min linear gradients from 5 to 35% acetonitrile in 0.4% formic acid. Survey spectra were acquired in the Orbitrap with the resolution set to a value of 30 000. Up to 5 of the most intense ions per cycle were fragmented and analyzed in the linear trap.
Database Searching
Peak lists of the 200 most abundant fragment ions from each product ion spectrum were extracted out of Thermo.raw files and converted into.mgf files using in-house written software30 as previously described.28 No further data processing such as smoothing, deisotoping, and filtering were performed. All mass spectrometry (MS) data sets were searched against the target and decoy (reversed) International Protein Index human protein database (v3.36; 69 012 sequences) using the MASCOT search engine (Matrix Science, v2.104). Protein modifications were set as Carbamidomethyl (C) (fixed) and Lysine (K-full), Oxidation (M), and N-Acetyl (Protein) (variable). Up to one missed tryptic cleavage was allowed. The MS tolerance was set as ±20 ppm and MS/MS ± 0.6 Da. All peptides were identified with ion scores no less than 33 (p < 0.05); all proteins were identified with scores no less than 40 (p < 0.01). The proteins identified with at least two unique peptides were classified as a high-confidence protein data set, for which the false discovery rate was assessed using a target-decoy search strategy. For all quantitated proteins identified with only one peptide, the MS/MS spectra were manually checked to ensure the accuracy of protein identification. Raw MS data files are freely available at Tranche (https://proteomecommons.org/dataset.jsp?i=74438).
Protein Quantification
Identified proteins were quantitated using an open-source software program MSQuant (v1.4.3a74, msquant.sourceforge.net),31,32 which automatically computed peptide and protein ratios by calculating the “heavy”/“light” ratios of areas under the curves of extracting ion chromatograms. All SILAC pairs were manually inspected to minimize potentially incorrect quantifications. Each quantitative data set was then normalized using a multiple-point normalization strategy to minimize the systematic errors introduced by the Bradford assay and sample mixing. Briefly, the distributions of protein ratios were plotted using the Statistical Package for the Social Sciences (SPSS, v16.0.2), followed by the calculation of 5% trimmed mean values. All protein ratios were then normalized against the 5% trimmed mean values so that most protein ratios were distributed in the 1.00 ± 0.10 zone. The siEGFR/CON (H/L) ratios were normalized against corresponding CON/CON (H/L) ratios to minimize the systematic errors introduced by differential SILAC labeling. Proteins with more than 1.4-fold changes (i.e., >1.400 or <0.714) in both biological replicates were accepted as significantly regulated by EGFR knockdown.
Western Blotting Validation
PCI-15B cells were cultured in regular medium and transfected with or without siEGFR as described above. Western blotting analyses were performed essentially as described.33 The protein levels of ALDH1A3, EGFR, MAT2A, and TK1 before and after EGFR knockdown were compared. To exclude the possibility that siRNA transfection per se may regulate the significantly changed proteins, PCI-15B cells were cultured in regular medium and transfected with siEGFR or nontargeting control siRNA (siCON) as described above. The protein levels of LGALS7 and TK1 in PCI-15B cells transfected with siEGFR or siCON were compared.
Bioinformatic Analysis
Genecards (www.genecards.org) and PubMed (www.pubmed.org) were explored to obtain information about the functions of the significantly regulated proteins and the biological processes that they mediate. The molecular networks containing the significantly changed proteins were constructed and depicted using Microsoft Office PowerPoint 2003.
Cell Cycle Analysis
PCI-15B cells were cultured in regular medium and treated with siEGFR or siCON as described above. After 48 h transfection, cells were trypsinized, resuspended in PBS containing 2% FBS, and fixed in absolute ethanol at 4 °C for 1 h. Cells were resuspended in 50 μg/mL propidium iodide staining solution and sorted within 48 h on a FACSCalibur flow cytometer (BD Biosciences). The percentage of cells in different phases of cell cycle was calculated by using CellQuest Pro (BD Biosciences). Statistical significance was evaluated by student’s t-test.
Results
Protein Separation and Identification
SILAC labeling and EGFR siRNA transfection were repeated to generate two biological replicates. Proteins extracted from Lys0-labeled PCI-15B control cells were mixed at equal amounts with proteins isolated from Lys8-labeled PCI-15B control cells or PCI-15B cells transfected with EGFR siRNA to generate two samples (Figure 1A). The samples were separated by 12.5% SDS-PAGE and a representative gel image is shown in Figure 1B. No significant differences were observed between the two samples, suggesting that at least abundant cellular proteins were not significantly changed by siRNA transfection.
Figure 1.

Separation, identification, and quantification of SILAC-labeled proteins. (A) Schematic flowchart for the discovery of molecular networks regulated by EGFR level in HNC using SILAC and LC–MS/MS. (B) SDS-PAGE separation of SILAC samples. S1 indicates the mixture of proteins from Lys0-labeled control cells and Lys8-labeled EGFR knockdown cells. S2 indicates the mixture of proteins from Lys0-labeled control cells and Lys8-labeled control cells. (C) Representative tandem mass spectrum for protein identification. (D) Representative SILAC pairs showing the regulation of proteins by EGFR knockdown.
Each gel lane was cut into 10 slices (Figure 1B). After in-gel digestion, tryptic peptides were separated by nanoflow reverse phase-high performance liquid chromatography and analyzed on an LTQ-Orbitrap mass spectrometer. Figure 1C shows a representative tandem mass spectrum, which was derived from the analysis of a doubly charged peptide (m/z 773.86) from stratifin (SFN; 14–3–3σ). A total of 13 025 unique peptides were identified with ion scores no less than 33 (p < 0.05) and 2784 different proteins were identified with MOWSE scores no less than 40 (p < 0.01) (Table S1, Supporting Information). Among the 2784 proteins, 1862 were identified with at least two unique peptides and thus classified as a high-confidence protein data set, for which the false discovery rate was assessed using a target-decoy search strategy. Using the same database searching parameters and highly stringent criteria, that is, proteins identified with at least two unique peptides and each peptide with an ion score no less than 33, less than 0.2% false positives were discovered by searching against a reversed IPI_Human database. To our knowledge, the present study provides so far the largest proteomic data set with high-confidence identification for HNC.
Protein Quantification
The metabolic conversion of arginine to proline is, at least in some cell lines (e.g., HeLa), a serious issue for SILAC-based quantitative proteomics, which may substantially reduce the accuracy of quantitation.34 Although several methods have been developed to address this problem, each of them has some limitations.34 Therefore, in the present study, we only used lysine, rather than both lysine and arginine, to label cellular proteins. As a result, only 6,714 unique peptides containing lysine residues were quantitated, accounting for 51.5% of total identified unique peptides. Figure 1D shows two representative SILAC pairs, which were from upregulated fatty acid-binding protein 5 (FABP5) and downregulated cytosolic hydroxymethylglutaryl-Coenzyme A (CoA) synthase (HMGCS1), respectively. The ratios of all quantifiable peptides and proteins were automatically calculated by MSQuant.31,32 For those quantitated proteins identified with only one peptide hit, the MS/MS spectra were manually checked to ensure the accuracy of protein identification (Figure S1, Supporting Information). Due to the high complexity of proteins in each gel slice, errors in quantitation may be introduced if either Lys8-labeled or Lys0-labeled isotope cluster overlaps any of the isotopic peaks of an unrelated but (partially) coeluated peptide. In addition, poor MS spectral quality may lead to incorrect quantification. Hence, all quantitated peptides and proteins were carefully manually inspected to minimize the potentially erroneous quantifications.
Histogram plotting of all protein ratios revealed that the 5% trimmed means of CON/CON (H/L) ratios in the two biological replicates were 0.794 (Figure S2A, Supporting Information) and 0.869 (Figure S2B, Supporting Information) but not the theoretical value 1.00. Analysis of all peptide ratios showed very similar results (data not shown). LC-MS/MS analysis of proteins isolated from a small aliquot of PCI-15B cells cultured in “heavy” medium excluded the possibility that the relatively low ratios were caused by insufficient SILAC labeling (Figure S3, Supporting Information). Therefore, we attribute the deviation to the relatively low accuracy of Bradford assay and the common problem of inaccuracies during sample mixing. A multipoint normalization strategy was applied to normalize the data sets against the 5% trimmed mean values, which were adopted to minimize the effects of extreme outliers. Consequently, the protein ratios were centered around 1.00 (Figure S2C and S2D, Supporting Information).
In this study, no significant morphological changes were observed for PCI-15B cells transfected with or without EGFR siRNA (data not shown) and no significant differences were detected for the electrophoretic patterns of mixed proteins (Figure 1B). Therefore, the vast majority of PCI-15B cellular proteins was assumed to be at the same or very similar levels before and after EGFR siRNA transfection. The aforementioned multipoint normalization strategy was again used to shift the 5% trimmed means of the siEGFR/CON (H/L) ratios to 1.00 (Figure S4, Supporting Information). After normalization, the mean SILAC ratios of two housekeeping protein β-actin (ACTB) and β-tubulin (TUBB) were all distributed in the 1.00 ± 0.05 region (see Table S2, Supporting Information), suggesting that the multipoint normalization strategy worked as envisioned.
After normalization, most siEGFR/CON (H/L) ratios were distributed in the 1.2-fold change zone (Figure S5A, Supporting Information). About 20 proteins were found to be regulated 2.0-fold or more (Figure S5A, Supporting Information). However, most of the changes are caused, at least in part, by differential SILAC labeling, as shown in Figure S5B (Supporting Information). Systematic errors introduced by differential SILAC labeling was also observed by Matthias Mann’s group.35 They found that about 1% of all quantitated proteins may be regulated 2-fold or more by differential SILAC labeling.35 To minimize the false discoveries introduced by differential SILAC labeling, the siEGFR/CON (H/L) data set were normalized against the CON/CON (H/L) data set and the normalized ratio distributions were shown in Figure S5C (Supporting Information). All protein ratios before and after normalization were summarized in Table S2 (Supporting Information). Given that the standard deviation (SD) for each siEGFR/CON (H/H) data set is about 0.16 (Figure S6, Supporting Information), we set 1.4-fold (>1.0 ± 2 SD) as the cutoff to guarantee a confident distance to experimental variations. Only the proteins with more than 1.4-fold changes in both biological replicates were accepted as significantly changed. According to these criteria, 12 and 23 proteins were found to be significantly upregulated and downregulated, respectively (Figure 2 and Table 1).
Figure 2.

SILAC ratio distributions of significantly changed proteins. The siEGFR/CON (H/L) ratios in two biological replicates are indicated with horizontal and diagonal gold crosses while the siEGFR/CON (H/H) ratios, which were calculated to minimize false discoveries introduced by differential SILAC labeling, are indicated with horizontal and diagonal green crosses. Notably, most of the changes are modest (1.4–2.0-fold).
Table 1.
Proteins Significantly Regulated by EGFR Knockdown
| gene symbol | description | pep IDa | pep quantb | regc | siEGFR/CONd |
blot? | |||
|---|---|---|---|---|---|---|---|---|---|
| H/Le |
H/Hf |
||||||||
| R1g | R2h | R1 | R2 | ||||||
| AARS | Alanyl-tRNA synthetase, cytoplasmic | 14 | 6 | ↓ | 1.53 | 1.55 | 1.47 | 1.44 | |
| ACACA | Acetyl-CoA carboxylase 1 | 7 | 2 | ↓ | 1.29 | 1.50 | 1.44 | 1.42 | |
| ACADVL | Very long-chain specific acyl-CoA dehydrogenase | 18 | 4 | ↑ | 1.62 | 1.64 | 1.46 | 1.49 | |
| ACAT2 | Acetyl-CoA acetyltransferase, cytosolic | 10 | 5 | ↓ | 1.45 | 1.47 | 1.40 | 1.43 | |
| ACLY | ATP-citrate synthase | 27 | 14 | ↓ | 1.39 | 1.42 | 1.43 | 1.41 | |
| ALDH1A3 | Aldehyde dehydrogenase family 1 member A3 | 22 | 8 | ↓ | 1.88 | 1.84 | 1.46 | 1.58 | Yes |
| ANK3 | Ankyrin-3 (Ankyrin-G) | 6 | 2 | ↑ | 2.37 | 2.69 | 1.71 | 1.92 | |
| CDC2 | Cell division control protein 2 homologue | 12 | 6 | ↓ | 2.85 | 2.76 | 2.20 | 2.28 | |
| CDKN1A | cyclin-dependent kinase inhibitor 1 (p21) | 2 | 1 | ↑ | 1.56 | 1.62 | 1.43 | 1.46 | |
| EGFR | Epidermal growth factor receptor | 42 | 20 | ↓ | 2.05 | 2.28 | 2.42 | 2.51 | Yes |
| FABP5 | Fatty acid-binding protein 5 | 7 | 3 | ↑ | 2.07 | 2.17 | 1.68 | 1.48 | |
| FASN | Fatty acid synthase | 54 | 53 | ↓ | 1.37 | 1.36 | 1.50 | 1.49 | |
| FDFT1 | Squalene synthetase | 8 | 2 | ↓ | 1.47 | 1.74 | 1.50 | 1.73 | |
| FDPS | Farnesyl pyrophosphate synthetase | 8 | 2 | ↓ | 1.61 | 1.48 | 1.87 | 1.59 | |
| GLUL | Glutamine synthetase | 5 | 2 | ↓ | 1.95 | 1.75 | 1.57 | 1.50 | |
| HADHA | Trifunctional enzyme subunit alpha, mitochondrial | 24 | 10 | ↑ | 1.63 | 1.62 | 1.48 | 1.44 | |
| HADHB | Trifunctional enzyme subunit beta, mitochondrial | 14 | 4 | ↑ | 1.65 | 1.74 | 1.44 | 1.49 | |
| HMGCS1 | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | 12 | 7 | ↓ | 2.26 | 2.48 | 2.04 | 2.03 | |
| IRF6 | Interferon regulatory factor 6 | 7 | 4 | ↑ | 2.40 | 2.36 | 1.58 | 1.67 | |
| KRT17 | Keratin 17 | 23 | 12 | ↑ | 2.52 | 3.08 | 1.41 | 1.42 | |
| LGALS7 | Galectin-7 | 8 | 4 | ↑ | 5.44 | 5.42 | 1.41 | 1.41 | Yes |
| MAT2A | Methionine adenosyltransferase IIα | 8 | 2 | ↓ | 1.92 | 1.43 | 1.88 | 1.45 | Yes |
| MCM2 | DNA replication licensing factor MCM2 | 6 | 2 | ↓ | 2.08 | 2.18 | 1.82 | 2.79 | |
| MCM6 | DNA replication licensing factor MCM6 | 5 | 2 | ↓ | 2.25 | 2.34 | 1.70 | 2.50 | |
| MTA2 | Metastasis-associated protein MTA2 | 5 | 1 | ↓ | 1.57 | 1.39 | 1.81 | 1.68 | |
| PCNA | Proliferating cell nuclear antigen | 11 | 6 | ↓ | 1.96 | 1.91 | 1.52 | 1.69 | |
| PHLDA1 | Pleckstrin homology-like domain family A member 1 | 3 | 2 | ↓ | 2.05 | 1.71 | 1.50 | 1.44 | |
| PHLDA2 | Pleckstrin homology-like domain family A member 2 | 11 | 3 | ↓ | 2.10 | 2.06 | 2.09 | 2.16 | |
| PHLDA3 | Pleckstrin homology-like domain family A member 3 | 9 | 3 | ↓ | 1.06 | 1.13 | 1.40 | 1.46 | |
| PTGES3 | Prostaglandin E synthase 3 | 2 | 1 | ↓ | 1.46 | 1.52 | 1.42 | 1.69 | |
| RFC5 | Replication factor C subunit 5 | 2 | 1 | ↓ | 1.61 | 1.73 | 1.42 | 1.51 | |
| SERPINB5 | Serpin B5 (Maspin) | 6 | 4 | ↑ | 1.97 | 1.91 | 1.52 | 1.43 | |
| SFN | 14–3–3 sigma (Stratifin) | 23 | 16 | ↑ | 1.95 | 1.94 | 1.46 | 1.45 | |
| TK1 | Thymidine kinase, cytosolic | 2 | 2 | ↓ | n/ai | 4.31 | n/a | 3.19 | Yes |
| TPPP3 | Tubulin polymerization-promoting protein family member 3 | 3 | 2 | ↑ | 3.99 | 4.06 | 1.61 | 1.62 | |
| VCL | Vinculin | 33 | 16 | ↓ | 1.55 | 1.49 | 1.56 | 1.42 | |
Unique peptide identified.
Peptide quantitated.
Regulation.
Normalized siEGFR/CON fold change.
“Heavy”/“Light”.
“Heavy”/“Heavy”.
Replicate 1.
Replicate 2.
Not applicable.
Western Blotting Validation
To further validate the SILAC results, Western blotting analysis was carried out for four regulated proteins whose antibodies were readily available. The proteins include EGFR, ALDH1A3, and MAT2A, which were significantly changed in both replicates, as well as TK1, which was remarkably downregulated in one replicate but not quantifiable in the other replicate where the peptide scores were lower than 33 (p < 0.05). As shown in Figure 3, the Western blotting results for ALDH1A3, MAT2A, and TK1 were consistent with their corresponding SILAC results. The verification of TK1 downregulation by Western blotting increased the number of significantly regulated proteins from 35 to 36. However, surprisingly, Western blotting analysis showed that the EGFR was much more dramatically (>6-fold) downregulated by siEGFR transfection than showed by the SILAC analysis (~2.5-fold). The SILAC ratios for EGFR are convincing because they were calculated based on 20 peptides. A possible explanation for the discrepancy is that siEGFR transfection might lead to certain post-translational modification(s) (e.g., phosphorylation or dephosphorylation) on the epitope for the anti-EGFR mAb (amino acids 1020 to 1046), resulting in a reduced affinity between the EGFR and the anti-EGFR mAb.
Figure 3.
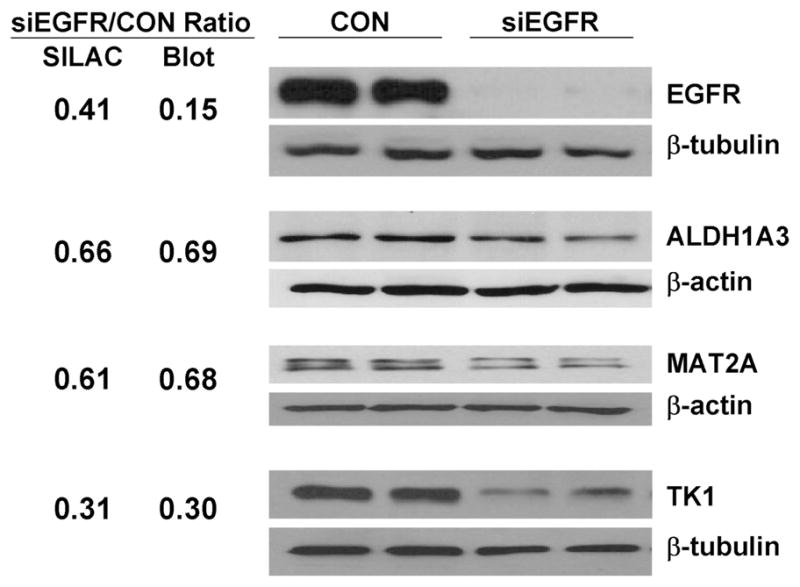
Western blotting validation of significantly changed proteins. The mean blot ratios for ALDH1A3, MAT2A, and TK1 are consistent with the mean SILAC ratios for the proteins. The discrepancy between the mean SILAC ratio and the mean blot ratio for EGFR might be caused by certain post-translational modifications on the epitope for anti-EGFR mAb.
Given that nontargeting siRNA transfection may have some effect on gene expression, which may compromise the accuracy of SILAC quantitation, we did not use siCON-transfected PCI-15B cells as a control for SILAC analysis. However, to exclude the possibility that it was transfection per se but not EGFR knockdown that regulated the significantly changed proteins, we compared the protein levels of LGALS7 and TK1 in PCI-15B cells transfected with siEGFR or siCON. As shown in Figure S7 (Supporting Information), the protein ratios determined by Western blotting analysis were consistent with those determined by SILAC quantitation, suggesting that the significantly changed proteins were actually regulated by EGFR knockdown and not by transfection per se.
Molecular Network Construction
Genecards and PubMed were explored to obtain comprehensive and latest information about the significantly changed proteins. Following network modeling, these proteins were found to be primarily involved in long-chain fatty acid biosynthesis and β-oxidation, cholesterol biosynthesis, cell growth arrest, DNA replication, and apoptosis (Figure 4).
Figure 4.

Molecular networks regulated by EGFR level in HNC. (A) Cholesterol biosynthesis and long chain fatty acid biosynthesis were inhibited while the long chain fatty acid β-oxidation were enhanced. (B) Cell growth, DNA replication, and apoptosis were inhibited. The numbers near the protein names indicate fold changes.
Cell Cycle Analysis
Our network model (Figure 4B) suggested that EGFR knockdown inhibits the transition from G2 to M phase of PCI-15B cells. To validate this hypothesis, a flow cytometry analysis was performed. As shown in Figure 5, the percentage of G2/M phase cells was modestly (from 13.5 ± 6.9% to 17.4 ± 5.0%) but significantly (p)= 0.036) increased after EGFR knockdown, confirming that EGFR downmodulation retards G2/M phase progression in HNC cells.
Figure 5.

EGFR knockdown inhibits G2/M phase progression. (A) Flow cytometry plots of PCI-15B cells transfected with nontargeting control siRNA (siCON) or EGFR-specific siRNA (siEGFR). The data are representative of four independent experiments. (B) Comparison of the percentage of cells in the G0/G1, S, and G2/M phases after transfection of siCON and siEGFR. Student’s t-test indicated that G2/M phase progression was significantly (p = 0.036) inhibited by EGFR knockdown.
Discussion
EGFR antagonists have been clinically proved to be able to prolong the survival of a subset of cancer patients.6 However, the molecular mechanisms underlying the therapeutic resistance and response to EGFR targeting remain unclear. In the present study, unbiased quantitative proteomics analysis followed by Western blotting validation identified 36 proteins significantly regulated by EGFR levels in HNC cells. Network modeling revealed that these proteins are mainly involved in long-chain fatty acid synthesis and β-oxidation, cholesterol biosynthesis, cell growth arrest, DNA replication, and apoptosis (Figure 4).
SILAC-based quantitative proteomics analysis revealed that four enzymes (i.e., ACLY, ACAT2, ACACA, and FASN) involved in long-chain fatty acid synthesis were significantly downregulated, while three enzymes (i.e., ACADVL, HADHA, HADHB) involved in fatty acid β-oxidation and an fatty acid-binding protein (FABP5) were significantly upregulated (Figure 4A and Table 1). All these changes may lead to the dramatic downregulation of free long-chain fatty acids and fatty acyl-CoAs. Since long-chain fatty acyl-CoAs are the precursors of triglycerides and phospholipids—the main components of cellular membranes, the biogenesis of membranes may be severely impaired by EGFR knockdown. As a result, the cell growth and proliferation are inhibited (Figure 4B). In addition, long-chain fatty acyl-CoAs are substrates for protein acylation (e.g., S-palmitoylation and N-myristoylation). Because fatty acylation plays a key role in cancer development and progression,36 the reduced availability of long-chain fatty acyl-CoAs caused by EGFR knockdown may also suppress tumor progression through inhibiting protein fatty acylation (Figure 4B).
The EGFR knockdown also led to the significant downregulation of five enzymes (i.e., ACLY, ACAT2, HMGCS1, FDPS, and FDFT1) involved in cholesterol biosynthesis (Figure 4A and Table 1). Cholesterol is a neutral lipid that accumulates in certain membrane microdomains such as lipid rafts and tetraspanin-enriched microdomains, which serve as crucial signal transduction platforms and play important roles in cancer development and progression.37 Given that lipid rafts but not tetraspanin-enriched microdomains are sensitive to cholesterol depletion,37 it is plausible that only lipid rafts were significantly modulated by EGFR knockdown, consequently certain lipid raft-orchestrating signaling pathways governing cell growth and proliferation (e.g., the EGFR and Src pathways) were attenuated. In addition, farnesyl pyrophosphate, a metabolite in the cholesterol biosynthesis pathway, serves as a substrate for protein prenylation including farnesylation and geranylgeranylation.38 This type of lipid modification regulates the membrane association and functionality of various intracellular signaling proteins (e.g., Ras, Gγ subunits, and nuclear lamins).38 The inhibition of farnesyl pyrophosphate synthesis may result in the attenuation of certain cell signaling transmitted by these prenylated proteins, consequently inhibiting cancer progression.
Network modeling suggested that the significant upregulation of four proteins (i.e., CDKN1A, SFN, SERPINB5, LGALS7) may arise from the modulation of p53 (Figure 4B). However, SILAC analysis showed that p53 was unchanged at the protein level by EGFR knockdown (Table S2, Supporting Information). Nonetheless, the significant downregulation of MTA2, a component of histone deacetylase 1 (HDAC1)-containing complexes which deacetylate p53,39 implies that p53 may upregulate the four proteins through enhanced acetylation.40 Subsequently, the upregulation of CDKN1A (p21) and SFN (stratifin) resulted in the G1 and G2 phase arrest, respectively.41,42 The remarkable downregulation of cdc2 resulting from EGFR knockdown may further inhibit the G2 phase progression (Figure 4B and Figure 5).43
Maspin (SERPINB5) is a tumor suppressor which increases cell adhesion and apoptosis and decrease cell invasion, metastasis, and angiogenesis.44 The protein can be regulated via several mechanisms including p53 signaling.44 Maspin interacts with a diverse group of proteins, such as p53-deacetylating enzyme HDAC1 and transcription factor interferon regulatory factor 6 (IRF6).44 It directly binds and inhibits HDAC1,45 thereby executing a positive feedback effect on p53 acetylation and activity. Maspin also interacts with IRF6, which was upregulated by EGFR knockdown in this study, and augments its effect on promoting cell cycle arrest.46 Notably, two 2D-gel-based proteomic studies also showed that EGFR downmodulation47 or inhibition48 in different tumor cells led to significant upregulation of maspin, suggesting that maspin may play an important and common role in the anti-EGFR-induced antitumor effects. It has recently been reported that maspin is frequently mutated in cancer cells and tissues.49 It is likely that mutation(s) of maspin in HNC cells may reduce or abrogate its interactions with HDAC1 and/or IRF6, consequently leading to the HNC resistance to EGFR targeting.
Galectin-7 (LGALS7) is a β-galactoside-binding animal lectin whose expression is inducible by p53.50 The upregulation of galectin-7 by EGFR knockdown, probably through p53 acetylation, may promote tumor growth inhibition and apoptosis.51 Prostaglandin E synthase 3 (PTGES3), also called cytosolic prostaglandin E synthase (cPGES), catalyzes the conversion of prostaglandin H2 to prostaglandin E2 (PGE2) in cells. We previously reported that PGE2 stimulates HNC cell proliferation and invasion via transactivating EGFR.52 In the present study, the significant downregulation of PTGES3 may lead to the decreased synthesis of PGE2, consequently inhibiting cancer cell proliferation (Figure 4B).
In addition to the molecules involved in tumor growth inhibition, four proteins essential for DNA replication (i.e., MCM2, MCM6, RFC5, and PCNA) were found to be significantly downregulated in both biological replicates by SILAC quantitation. Three other proteins (i.e., TK1, DUT, and MCM3) were also found to be downregulated. However, they were only quantifiable in one biological replicate. Nevertheless, Western blotting analysis confirmed the remarkable downregulation of TK1 (Figure 3 and Figure S7, Supporting Information). Clearly, the downregulation of these proteins dramatically inhibited DNA replication, which is consistent with our observation that HNC cell proliferation was significantly reduced by EGFR knockdown (data not shown).
In the present study, ALDH1A3, an enzyme converting retinal to retinoic acid, was found to be modestly but significantly downregulated. It was reported that a high FABP5/CRABP-II (cellular retinoic acid binding protein-II) ratio inhibits retinoic acid-induced apoptosis.53 It is possible that the downregulation of ALDH1A3 and upregulation of FABP5 promoted cell survival (Figure 4B). In addition, although the upregulation of maspin54 and galectin-755 as well as the downregulation of MAT2A56 may promote apoptosis, the upregulation of p2157 and stratifin57 and the downregulation of pleckstrin homology-like domain family A member 1–3 (PHLDA1–3)58–60 may antagonize cell apoptosis. The fact that significantly changed anti-apoptotic molecules occurred more commonly than changes in the pro-apoptotic molecules is consistent with our observations that: (1) cell morphology was not significantly changed by EGFR knockdown; and (2) poly (ADP-ribose) polymerase (PARP), whose cleavage by caspase-3 from the native 116 to 85 kDa is a hallmark of apoptosis, was only reduced by 11% (see Table S2, Supporting Information).
In addition to the proteins involved in the above-mentioned molecular networks, several cytoskeletal proteins such as ankyrin-3 (ANK3), keratin-17 (KRT17), and tubulin polymerization-promoting protein family member 3 (TPPP3) were significantly upregulated while vinculin (VCL) was significantly downregulated. Currently, relatively little is known about their functions in cancer cells, but it is likely that they are involved in regulating cell motility.
In summary, the molecular networks regulated by EGFR levels in HNC cells were uncovered by quantitatively comparing the proteomes of PCI-15B cells containing high or low level of EGFR. The introduction of an internal standard considerably reduced the systematic errors introduced by differential SILAC labeling, which has often been overlooked in prior studies. Quantitative analysis showed that at least 36 proteins were significantly regulated by EGFR siRNA knockdown. Subsequent bioinformatic analysis revealed that the 12 upregulated proteins were primarily involved in long-chain fatty acid β-oxidation and cell cycle arrest while the 24 downregulated proteins were implicated in the biosynthesis of long-chain fatty acids and cholesterol as well as DNA replication. Notably, some proteins involved in the molecular networks, such as HMG-CoA reductase61 and HDAC1,62 have been proposed as therapeutic targets. Many other proteins, such as FASN,63 farnesyl transferase,64 palmitoyl acyltransferase,65 and maspin,66 represent potential anticancer targets. Accumulating evidence suggests that targeting the EGFR axis at multiple levels may improve the antitumor effects. In fact, we have recently shown that combined inhibition of EGFR and cyclooxygenase-252 or gastrin-releasing peptide receptor67 or Src33 significantly improved growth inhibition of HNC. Mutation or aberrant regulation of certain proteins (e.g., acetylated p53, maspin, MTA2) in this molecular network may lead to the resistance to EGFR targeting in HNC. These proteins may serve as biomarkers to screen out the patients who will barely benefit from EGFR-targeting alone. Combined targeting of these proteins in conjunction with EGFR blockade may significantly improve the treatment of HNC for this subset of cancer patients.
Supplementary Material
Acknowledgments
We thank Zachary Waldon and Yin Yin Lin for assistance with mass spectrometry.
Abbreviations
- CoA
coenzyme A
- CON
control
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- HDAC1
histone deacetylase 1
- HNC
head and neck cancer
- Lys0
12C614N2-lysine
- Lys8
13C615N2-lysine
- mAb
monoclonal antibody
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PGE2
prostaglandin E2
- SD
standard deviation
- siCON
nontargeting control siRNA
- siEGFR
EGFR-specific siRNA
- SILAC
stable isotope labeling with amino acids in cell culture
- siRNA
small interfering RNA
- TKI
tyrosine kinase inhibitor
Footnotes
Supporting Information Available: Supplemental Figures S1–7. Table S1: List of identified proteins and peptides. Table S2: List of quantitated proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Carvalho AL, Nishimoto IN, Califano JA, Kowalski LP. Trends in incidence and prognosis for head and neck cancer in the United States: a site-specific analysis of the SEER database. Int J Cancer. 2005;114:806–816. doi: 10.1002/ijc.20740. [DOI] [PubMed] [Google Scholar]
- 3.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–1584. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 5.Grandis JR, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 6.Kim S, Grandis JR, Rinaldo A, Takes RP, Ferlito A. Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck. 2008;30:667–674. doi: 10.1002/hed.20859. [DOI] [PubMed] [Google Scholar]
- 7.Grandis JR, Chakraborty A, Melhem MF, Zeng Q, Tweardy DJ. Inhibition of epidermal growth factor receptor gene expression and function decreases proliferation of head and neck squamous carcinoma but not normal mucosal epithelial cells. Oncogene. 1997;15:409–416. doi: 10.1038/sj.onc.1201188. [DOI] [PubMed] [Google Scholar]
- 8.Lai SY, Koppikar P, Thomas SM, Childs EE, Egloff AM, Seethala RR, Branstetter BF, Gooding WE, Muthukrishnan A, Mountz JM, Lui VW, Shin DM, Agarwala SS, Johnson R, Couture LA, Myers EN, Johnson JT, Mills G, Argiris A, Grandis JR. Intratumoral epidermal growth factor receptor antisense DNA therapy in head and neck cancer: first human application and potential antitumor mechanisms. J Clin Oncol. 2009;27:1235–1242. doi: 10.1200/JCO.2008.17.8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 10.Harari PM, Wheeler DL, Grandis JR. Molecular target approaches in head and neck cancer: epidermal growth factor receptor and beyond. Semin Radiat Oncol. 2009;19:63–68. doi: 10.1016/j.semradonc.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandis JR. Established and emerging concepts in epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2007;69:S22–24. doi: 10.1016/j.ijrobp.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 12.Egloff AM, Grandis JR. Improving Response Rates to EGFR-Targeted Therapies for Head and Neck Squamous Cell Carcinoma: Candidate Predictive Biomarkers and Combination Treatment with Src Inhibitors. J Oncol. 2009;2009:896407. doi: 10.1155/2009/896407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 14.Yang W, Steen H, Freeman MR. Proteomic approaches to the analysis of multiprotein signaling complexes. Proteomics. 2008;8:832–851. doi: 10.1002/pmic.200700650. [DOI] [PubMed] [Google Scholar]
- 15.Kruger M, Kratchmarova I, Blagoev B, Tseng YH, Kahn CR, Mann M. Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc Natl Acad Sci US A. 2008;105:2451–2456. doi: 10.1073/pnas.0711713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everley PA, Krijgsveld J, Zetter BR, Gygi SP. Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol Cell Proteomics. 2004;3:729–735. doi: 10.1074/mcp.M400021-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Everley PA, Bakalarski CE, Elias JE, Waghorne CG, Beausoleil SA, Gerber SA, Faherty BK, Zetter BR, Gygi SP. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J Proteome Res. 2006;5:1224–1231. doi: 10.1021/pr0504891. [DOI] [PubMed] [Google Scholar]
- 18.Liang X, Zhao J, Hajivandi M, Wu R, Tao J, Amshey JW, Pope RM. Quantification of membrane and membrane-bound proteins in normal and malignant breast cancer cells isolated from the same patient with primary breast carcinoma. J Proteome Res. 2006;5:2632–2641. doi: 10.1021/pr060125o. [DOI] [PubMed] [Google Scholar]
- 19.Steiniger SC, Coppinger JA, Kruger JA, Yates J, 3rd, Janda KD. Quantitative mass spectrometry identifies drug targets in cancer stem cell-containing side population. Stem Cells. 2008;26:3037–3046. doi: 10.1634/stemcells.2008-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lund R, Leth-Larsen R, Jensen ON, Ditzel HJ. Efficient isolation and quantitative proteomic analysis of cancer cell plasma membrane proteins for identification of metastasis-associated cell surface markers. J Proteome Res. 2009;8:3078–3090. doi: 10.1021/pr801091k. [DOI] [PubMed] [Google Scholar]
- 21.Qiu H, Wang Y. Quantitative analysis of surface plasma membrane proteins of primary and metastatic melanoma cells. J Proteome Res. 2008;7:1904–1915. doi: 10.1021/pr700651b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Mi W, Cai J, Ying W, Liu F, Lu H, Qiao Y, Jia W, Bi X, Lu N, Liu S, Qian X, Zhao X. Quantitative proteomic signature of liver cancer cells: tissue transglutaminase 2 could be a novel protein candidate of human hepatocellular carcinoma. J Proteome Res. 2008;7:3847–3859. doi: 10.1021/pr800153s. [DOI] [PubMed] [Google Scholar]
- 23.Chen N, Sun W, Deng X, Hao Y, Chen X, Xing B, Jia W, Ma J, Wei H, Zhu Y, Qian X, Jiang Y, He F. Quantitative proteome analysis of HCC cell lines with different metastatic potentials by SILAC. Proteomics. 2008;8:5108–5118. doi: 10.1002/pmic.200800280. [DOI] [PubMed] [Google Scholar]
- 24.Heo DS, Snyderman C, Gollin SM, Pan S, Walker E, Deka R, Barnes EL, Johnson JT, Herberman RB, Whiteside TL. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989;49:5167–5175. [PubMed] [Google Scholar]
- 25.Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, Kamens J, Gooding WE, Grandis JR. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64:6166–6173. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 26.Yang W, Liu P, Liu Y, Wang Q, Tong Y, Ji J. Proteomic analysis of rat pheochromocytoma PC12 cells. Proteomics. 2006;6:2982–2990. doi: 10.1002/pmic.200500036. [DOI] [PubMed] [Google Scholar]
- 27.Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, Demichelis F, Freeman MR. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–5609. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam RM, Yang W, Di Vizio D, Mukhopadhyay NK, Steen H. Rapid preparation of nuclei-depleted detergent-resistant membrane fractions suitable for proteomics analysis. BMC Cell Biol. 2008;9:30. doi: 10.1186/1471-2121-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renard BY, Kirchner M, Monigatti F, Ivanov AR, Rappsilber J, Winter D, Steen JA, Hamprecht FA, Steen H. When less can yield more - Computational preprocessing of MS/MS spectra for peptide identification. Proteomics. 2009;9:4978–4984. doi: 10.1002/pmic.200900326. [DOI] [PubMed] [Google Scholar]
- 31.Schulze WX, Mann M. A novel proteomic screen for peptide-protein interactions. J Biol Chem. 2004;279:10756–10764. doi: 10.1074/jbc.M309909200. [DOI] [PubMed] [Google Scholar]
- 32.Mortensen P, Gouw JW, Olsen JV, Ong SE, Rigbolt KT, Bunkenborg J, Cox J, Foster LJ, Heck AJ, Blagoev B, Andersen JS, Mann M. MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J Proteome Res. 9:393–403. doi: 10.1021/pr900721e. [DOI] [PubMed] [Google Scholar]
- 33.Koppikar P, Choi SH, Egloff AM, Cai Q, Suzuki S, Freilino M, Nozawa H, Thomas SM, Gooding WE, Siegfried JM, Grandis JR. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:4284–4291. doi: 10.1158/1078-0432.CCR-07-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SK, Liao L, Kim JY, Yates JR., 3rd A computational approach to correct arginine-to-proline conversion in quantitative proteomics. Nat Methods. 2009;6:184–185. doi: 10.1038/nmeth0309-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonaldi T, Straub T, Cox J, Kumar C, Becker PB, Mann M. Combined use of RNAi and quantitative proteomics to study gene function in Drosophila. Mol Cell. 2008;31:762–772. doi: 10.1016/j.molcel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE. 2006;2006:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 37.Le Naour F, Andre M, Boucheix C, Rubinstein E. Membrane microdomains and proteomics: lessons from tetraspanin micro-domains and comparison with lipid rafts. Proteomics. 2006;6:6447–6454. doi: 10.1002/pmic.200600282. [DOI] [PubMed] [Google Scholar]
- 38.McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 40.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 42.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14–3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 43.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 44.Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJ. Biological functions of maspin. J Cell Physiol. 2006;209:617–624. doi: 10.1002/jcp.20782. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Yin S, Meng Y, Sakr W, Sheng S. Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Res. 2006;66:9323–9329. doi: 10.1158/0008-5472.CAN-06-1578. [DOI] [PubMed] [Google Scholar]
- 46.Bailey CM, Abbott DE, Margaryan NV, Khalkhali-Ellis Z, Hendrix MJ. Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Mol Cell Biol. 2008;28:2235–2243. doi: 10.1128/MCB.01866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu LR, Shao XX, Jiang WL, Xu D, Chang YC, Xu YH, Xia QC. Proteome alterations in human hepatoma cells transfected with antisense epidermal growth factor receptor sequence. Electrophoresis. 2001;22:3001–3008. doi: 10.1002/1522-2683(200108)22:14<3001::AID-ELPS3001>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Sung FL, Pang RT, Ma BB, Lee MM, Chow SM, Poon TC, Chan AT. Pharmacoproteomics study of cetuximab in nasopharyngeal carcinoma. J Proteome Res. 2006;5:3260–3267. doi: 10.1021/pr050452g. [DOI] [PubMed] [Google Scholar]
- 49.Jang HL, Nam E, Lee KH, Yeom S, Son HJ, Park C. Maspin polymorphism associated with apoptosis susceptibility and in vivo tumorigenesis. Int J Mol Med. 2008;22:333–338. [PubMed] [Google Scholar]
- 50.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 51.Saussez S, Kiss R. Galectin-7. Cell Mol Life Sci. 2006;63:686–697. doi: 10.1007/s00018-005-5458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, Gooding WE, Siegfried JM, Chan DC, Grandis JR. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–11839. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 53.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene. 2002;21:4089–4098. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- 55.Kuwabara I, Kuwabara Y, Yang RY, Schuler M, Green DR, Zuraw BL, Hsu DK, Liu FT. Galectin-7 (PIG1) exhibits pro-apoptotic function through JNK activation and mitochondrial cytochrome c release. J Biol Chem. 2002;277:3487–3497. doi: 10.1074/jbc.M109360200. [DOI] [PubMed] [Google Scholar]
- 56.Liu Q, Wu K, Zhu Y, He Y, Wu J, Liu Z. Silencing MAT2A gene by RNA interference inhibited cell growth and induced apoptosis in human hepatoma cells. Hepatol Res. 2007;37:376–388. doi: 10.1111/j.1872-034X.2007.00041.x. [DOI] [PubMed] [Google Scholar]
- 57.Janicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15:959–976. doi: 10.1038/cdd.2008.33. [DOI] [PubMed] [Google Scholar]
- 58.Neef R, Kuske MA, Prols E, Johnson JP. Identification of the human PHLDA1/TDAG51 gene: down-regulation in metastatic melanoma contributes to apoptosis resistance and growth deregulation. Cancer Res. 2002;62:5920–5929. [PubMed] [Google Scholar]
- 59.Qian N, Frank D, O’Keefe D, Dao D, Zhao L, Yuan L, Wang Q, Keating M, Walsh C, Tycko B. The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum Mol Genet. 1997;6:2021–2029. doi: 10.1093/hmg/6.12.2021. [DOI] [PubMed] [Google Scholar]
- 60.Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F, Taya Y. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell. 2009;136:535–550. doi: 10.1016/j.cell.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 61.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 62.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 63.Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents. Curr Pharm Biotechnol. 2006;7:483–493. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 64.Sousa SF, Fernandes PA, Ramos MJ. Farnesyltransferase inhibitors: a detailed chemical view on an elusive biological problem. Curr Med Chem. 2008;15:1478–1492. doi: 10.2174/092986708784638825. [DOI] [PubMed] [Google Scholar]
- 65.Tsutsumi R, Fukata Y, Fukata M. Discovery of protein-palmitoylating enzymes. Pflugers Arch. 2008;456:1199–1206. doi: 10.1007/s00424-008-0465-x. [DOI] [PubMed] [Google Scholar]
- 66.Khalkhali-Ellis Z. Maspin: the new frontier. Clin Cancer Res. 2006;12:7279–7283. doi: 10.1158/1078-0432.CCR-06-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Bhola NE, Lui VW, Siwak DR, Thomas SM, Gubish CT, Siegfried JM, Mills GB, Shin D, Grandis JR. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol Cancer Ther. 2007;6:1414–1424. doi: 10.1158/1535-7163.MCT-06-0678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.