

Abstract
Nucleotide analogues represent a major class of anti-cancer and anti-viral drugs, and provide an extremely powerful tool for dissecting the mechanisms of DNA and RNA polymerases. While the basic assays themselves are relatively straight-forward, a key issue is to appropriately design the studies to answer the mechanistic question of interest. This article addresses the major issues involved in designing these studies, and some of the potential difficulties that arise in interpreting the data. Examples are given both of the type of analogues typically used, the experimental approaches with different polymerases, and issues with data interpretation.
Keywords: polymerase, nucleotide, DNA, RNA, kinetics
Nucleotide analogues play two important roles biochemically. First, DNA and RNA polymerases represent one of the most important classes of enzymes targeted by both anti-cancer and anti-viral chemotherapeutics. Over a dozen clinically useful nucleotide analogues target viral RNA and DNA polymerases and at least 8 clinically useful anticancer agents exert their antiproliferative effects via their interactions with cellular DNA polymerases (Figure 1) (Burton and Everson, 2009, Parker, 2009, Villarreal, 2001, Vivet-Boudou, et al., 2006). Almost all of these analogues consist of a normal or slightly modified base conjugated to a modified sugar. Second, nucleotide analogues have provided a means of obtaining critical mechanistic insights into the functioning of DNA and RNA polymerases (Berdis and McCutcheon, 2007, Cavanaugh, et al., 2009, Henry, et al., 2004, Hwang and Romesberg, 2008, Kim, et al., 2005, Kincaid, et al., 2005, Kool, 2002, Leconte, et al., 2008, Lee and Berdis, 2009, Lee, et al., 2008, Matsuda, et al., 2003, Meyer, et al., 2004, Morales and Kool, 1998, Morales and Kool, 2000, Ogawa, et al., 2000, Patro, et al., 2009, Ramirez-Aguilar and Kuchta, 2004, Sintim and Kool, 2006, Thompson and Kuchta, 1995, Washington, et al., 2003, Zhang, et al., 2005). This includes, for example, how these enzymes differentiate ribose and 2′-deoxyribose, and how the enzymes discriminate between right and wrong (d)NTPs.
Figure 1.


Examples of clinically useful anticancer and antiviral nucleosides that involve DNA polymerase activity. Panel A. Antiviral agents. Panel B. Cancer chemotherapeutics.
This review will focus on using nucleotide analogues to better understand the mechanism of DNA/RNA polymerases. Two different classes of nucleotide analogues are routinely used to study polymerases – sugar analogues and base analogues – and both will be discussed. The results of this type of study potentially have two other very practical applications. As alluded to above, since multiple nucleoside-based chemotherapeutics obtain their efficacy due to effects on DNA and RNA polymerases, the results of these studies might eventually lead to novel chemotherapeutics. Additionally, a long-standing goal of molecular genetics researchers has been the expansion of the genetic code via the development of an additional base pair(s) that an organism can maintain and replicate with high efficiency and fidelity. While progress has been made in this direction (Hirao, et al., 2006, Hirao, et al., 2007, Hwang and Romesberg, 2008, Leconte, et al., 2008), substantial improvements in current novel base-pairs must be made.
Sugar Analogues.
As suggested in Figure 1, the synthesis of nucleotides containing sugar analogues and their interactions with polymerases has primarily been driven by the overriding goal of developing new anti-cancer or anti-viral chemotherapeutics. Reported modifications include; i) replacing the heteroatoms at the 2′ and 3′ carbons with hydrogen, another heteroatom or an alkyl group (R2, R2′, and R3 in Figure 2); ii) replacing the H’s at the 2′ carbon with a heteroatom or alkyl group; iii) replacing the 2′ and 3′ carbons with a heteroatom (Y and Z), most commonly S or O; iv) removing the 2′ and/or 3′ carbons to generate acyclic sugars; v) replacing the 4′-OH with N, S, or an alkyl group (X); vi) adding alkyl groups to the 4′-carbon (R4); vii) replacing the 5′-hydroxyl with N or a phosphonate (R1), and; viii) interconversion of both the sugar stereochemistry (D vs. L) and anomeric configuration (α vs. β) (Lee and Berdis, 2009, Parker, 2009). Thus, a huge number of nucleotides containing modified sugars exist for probing how a polymerase interacts with the sugar moiety of a nucleotide.
Figure 2.
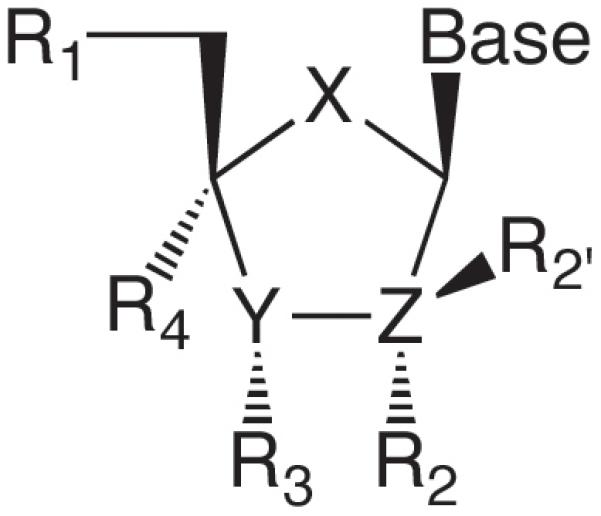
Reported modifications of the sugar of a nucleoside.
After synthesis of the sugar, the nucleosides are typically generated via relatively standard synthetic methods. In some cases the sugar modification is installed after generation of the glycosidic bond (Ex., AZT (Horwitz, et al., 1964, Lin and Mancini, 1983)). Conversion to the corresponding nucleoside triphosphate or phosphoramidite also typically involves established procedures, although it should be noted that triphosphorylation is still a less-than-optimal reaction even though researchers have used if for more than 30 years (Ludwig, 1981). In some cases, however, phosphorylation may require some modification of the procedures used with (deoxy)ribose due to the presence of additional heteroatoms in the sugar. For some modified nucleosides, chemoenzymatic syntheses have been developed to increase the yield of triphosphate. A number of reviews discuss nucleoside triphosphorylation in detail and can provide the interested reader with more detailed information (Burgess and Cook, 2000, Wu, et al., 2004).
Base Analogues
In contrast to the wide use of sugar-modified nucleosides as chemotherapeutics, very few nucleosides bearing highly modified bases are used as chemotherapeutics (Figure 1). This appears to largely reflect the frequent inability of cellular enzymes to convert the base-modified nucleoside into a nucleoside triphosphate, the actual inhibitor of DNA or RNA polymerases. The base analogues found in clinically useful therapeutics typically contain relatively subtle modifications, addition of a halogen at C-2 of adenine or at C-5 of a pyrimidine, replacement of N6 of adenine with oxygen, and replacement of O6 of guanine with S. Ribavirin, however, is an exception to this trend. Rather, developing an expanded genetic code and determining how polymerases identify the incoming (d)NTP as right or wrong has driven the synthesis and testing of most base-modified nucleotides.
Whereas development of anti-cancer and anti-viral nucleosides has typically involved relatively minor modification of the base, studies to examine expanding the genetic code and elucidate how polymerases differentiate right from wrong (d)NTPs have involved both small and large base modifications (Berdis and McCutcheon, 2007, Cavanaugh, et al., 2009, Henry, et al., 2004, Hwang and Romesberg, 2008, Kim, et al., 2005, Kincaid, et al., 2005, Kool, 2002, Leconte, et al., 2008, Lee and Berdis, 2009, Lee, et al., 2008, Matsuda, et al., 2003, Meyer, et al., 2004, Morales and Kool, 1998, Morales and Kool, 2000, Ogawa, et al., 2000, Patro, et al., 2009, Ramirez-Aguilar and Kuchta, 2004, Sintim and Kool, 2006, Thompson and Kuchta, 1995, Washington, et al., 2003, Zhang, et al., 2005). Figure 3 provides a sampling of the modified bases that have been converted into nucleoside triphosphates and templating nucleotides, and the ability of DNA and/or RNA polymerases to replicate them probed. It should be noted that most of these base analogues have been performed with DNA polymerases and not RNA polymerases.
Figure 3.

Examples of modified bases that have been used to probe DNA and/or RNA polymerases. R = ribose or 2′-deoxyribose. This list is not meant to be comprehensive, and my apologies to those researchers and their compounds that were not included.
As one might expect from the large number and varied structures of the base analogues, a wide variety of synthetic schemes have evolved for both base synthesis and formation of the glycosidic bond, especially for C- versus N-glycosidic linkages. Typically, the reaction sequence involves synthesis of the base followed by glycosylation, and then either triphosphorylation or generation of the phosphoramidite for incorporation into DNA or RNA. In some cases, however, one generates the glycosidic bond and then either builds the entire base or just slightly modifies the base (Piccirilli, et al., 1990).
Interaction of modified nucleotides with DNA and RNA polymerases.
This review will consider the interaction of modified nucleotides with polymerases from two different aspects – what are the general assays that one uses to ask specific questions, and what are the limitations and precautions that one must consider when interpreting the results of kinetic assays. For simplicity, the reactions will generally be described in terms of DNA polymerases and dNTPs, although many of the concerns and approaches also apply to RNA polymerases. At the end of this section, some special considerations with RNA polymerases will be considered.
Potentially, one could use [α-32P]dNTP (analogues) to measure incorporation as shown in Figure 4. While feasible for the natural dNTPs since the radiolabeled forms are commercially available, this is not really convenient for the analogues since it would require synthesis of [α-32P]dNTP analogues. This approach is also disadvantageous in terms of accuracy. After performing the assay, the amount of [32P] incorporated into DNA requires separating unincorporated [α-32P]dNTP from [32P]-labeled DNA, typically either by gel electrophoresis or filter binding. Thus, this method requires accurate loading of either the filter or the gel. A much more accurate approach is to use 5′-[32P]-primer-template ([32P]DNAn) and perform the assay using unlabeled dNTP (analogue), and then separate the starting [32P]DNAn from the product [32P]DNAn+1 by gel electrophoresis (Figure 4). Now, the amount of product is given by the ratio of [32P]DNAn+1/([32P]DNAn+[32P]DNAn+1). Importantly, determining the amount of product ratio-metrically means that errors in loading the gel do not affect the accuracy of the measurement (Chiaramonte, et al., 2003, Ogawa, et al., 2000, Urban, et al., 2009).
Figure 4.

General methods to analyze dNTP polymerization onto a primer-template.
Depending upon the length of products one wants to analyze, different percentage acrylamide gels will suffice. For short products around 2-9 nucleotides long, 20% gels work well, while for longer products lower percentage gels work well (Ex., 15% acrylamide for separating 15- and 16-mers). The lower percentage gels are easier and faster to run, although this comes at the expense of resolution. The identity of the base at the 3′-terminus will alter the electrophoretic mobility of the DNA, especially shorter DNAs (less than around 20 nucleotides, depending on the base composition of the DNA). Thus, one can often differentiate between different nucleotides at the 3′-terminus of otherwise identical sequences (Kuchta and Willhelm, 1991, Ramirez-Aguilar, et al., 2005, Thompson and Kuchta, 1995, Urban, et al., 2009). Often, however, this will require the higher resolution obtained with higher percentage acrylamide gels.
Alternatively, one may find that the polymerase does not incorporate the dNTP analogue. In this case, one can only determine how tightly the dNTP analogue binds to the enzyme by measuring its ability to inhibit incorporation of a normal dNTP. Thus, one would measure the ability of different analogue dNTP concentrations to inhibit normal dNTP incorporation. This will give an IC50 for inhibition. If one assumes, and is generally the case, that the dNTP analogue inhibits activity competitively with respect to dNTP, one can obtain the Ki for the dNTP analogue from the KM for the dNTP and the Michaelis-Menten expression for competitive inhibition. Alternatively, one can measure inhibition at multiple substrate (dNTP) and multiple analogue concentrations to directly obtain the Ki (Segel, 1975).
Steady-state versus pre-steady-state kinetics. Two general methodologies can be used – steady-state and pre-steady-state. In general, steady-state analysis is easier and faster, while pre-steady-state analysis can provide more detailed information (Donlin, et al., 1991, Hsieh, et al., 1993, Kuchta, et al., 1987, Patel, et al., 1991, Wong, et al., 1991). The latter typically requires the experimenter to use rapid quench methods and consumes much larger quantities of enzyme. For studies using nucleotide analogues, one most commonly wants to determine how modifying the structure of the incoming dNTP or templating nucleotide affects the efficiency of dNTP incorporation. Both approaches are equally accurate for obtaining this information. Steady-state methods provide a kcat/KM whereas pre-steady-state methods provide a kcat/KD (Apparent).
While the specificity constants for different substrates can be interpreted and compared to those of other substrates in a straightforward manner, extreme care must be taken in interpreting the individual kinetic parameters that make up the second order rate constant. For processive polymerases, even mildly processive enzymes such as pol α or Klenow Fragment, the rate of DNA dissociation likely limits the rate of correct dNTP polymerization such that kcat (Vmax) reflects the rate of DNA dissociation (Kuchta, et al., 1988). For analogues, it is impossible to say a priori what step(s) it represents. Thus, by comparing Vmax’s for a normal dNTP and a dNTP analogue, one may end up comparing two different steps in the catalytic cycle, a relatively meaningless exercise. Likewise, the KM for the dNTP is equally hard to interpret. For both a normal dNTP and a dNTP analogue, it may include contributions from any and/or all steps of the reaction cycle, including the rate of DNA dissociation. In pre-steady-state studies where one examines a single turnover of the enzyme, kcat typically reflects all steps through dNTP polymerization. For many but probably not all polymerases, a conformational change prior to chemistry limits the overall rate (Dahlberg and Benkovic, 1991, Patel, et al., 1991, Showalter and Tsai, 2002, Tsai and Johnson, 2006). For incorporation of an analogue, however, the kcat might, or might not, be the rate of the conformational change step – this can only be ascertained through more extensive analysis. The KD (Apparent) might represent the true KD for dNTP binding (i.e., formation of the E-dNTP collision complex), but caution should be taken when interpreting it this way since there may also be fast, kinetically invisible steps involved as well.
In addition to measuring incorporation of a dNTP (analogue), one generally wants to know how fast the polymerase adds additional nucleotides onto this just-incorporated nucleotide (analogue). Two different approaches can be used. For example, let us suppose that one wants to measure how fast a polymerase polymerizes additional dNTPs onto a just incorporated nucleotide analogue (i.e., polymerization of dYTP in Figure 5). First, one can chemically synthesize the appropriate primer-template, and then measure dNTP polymerization using either steady-state or pre-steady-state approaches. This approach is advantageous in that one can use either steady-state or pre-steady-state methods to measure polymerization of additional dNTPs, and one can measure how incorporation of the analogue affects other properties of the DNA (Ex., how the analogue affects DNA binding). However, when the analogue is at the 3′-terminus of the primer strand, this approach is disadvantageous since one must synthesize the appropriate analogue precursor and load it onto the activated solid support. If one only wants to synthesize a small amount of the primer containing the analogue at the 3′ terminus, one can use an enzyme to incorporate the dNTP analogue into the DNA and then purify the desired product (Beckman, et al., 2007). Alternatively, one can use the running start methodology developed by Goodman and coworkers (Boosalis, et al., 1987, Cai, et al., 1993). Now, one starts with the original primer-template (DNAn in Scheme 2), and then includes the dNTP analogue (dXTP) along with varying concentrations of the next correct dNTP (dYTP). Initial polymerization of the dNTP analogue generates DNAn+1, which then has a choice of dissociating from the enzyme or being further elongated to DNAn+2. By measuring the relative amounts of DNAn+2 and DNAn+1 as a function of the dNTP concentration, one can obtain the efficiency with which the polymerase added the next correct dNTP onto the analogue. This latter approach is limited, however, to only providing information on the efficiency of elongation.
Figure 5.

Polymerization of two consecutive dNTPs by a DNA polymerase.
Typically, it is useful to examine polymerization of the next several nucleotides after a base pair containing an analogue since the analogue can affect polymerization events significantly downstream of the analogue. For example, incorporating 3-deazaadenine or 3-deazaguanine very strongly inhibits polymerization of the dNTP two nucleotides downstream by B family polymerases (Beckman, et al., 2007, Cavanaugh, et al., 2009, Hendrickson, et al., 2004), but has very mild effects on polymerization of the next correct dNTP. Similarly, incorporation of gemcitabine has only small effects on polymerization of the next correct dNTP, but potently inhibits polymerization of the following dNTP (Huang, et al., 1991, Plunkett, et al., 1996). Finally, incorporation of the anti-hepatitis B drug entecavir (Figure 6) by HIV reverse transcriptase results in strong chain termination 3 nucleotides later (Tchesnokov, et al., 2008).
Figure 6.
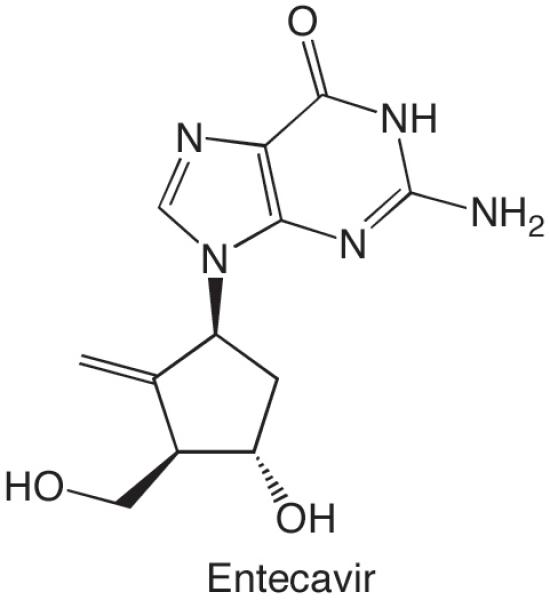
Structure of entecavir.
For those nucleotide analogues whose incorporation results in chain termination, an important question becomes whether or not the next correct dNTP can still bind. If it can bind, this result raises the possibility that this analogue may be a particularly potent inhibitor of the polymerase. The classic example of this is acyclovir triphosphate and herpes polymerase (Reardon, 1989, Reardon, 1990). If one only adds acyclovir triphosphate and an appropriate primer-template to herpes polymerase, the enzyme incorporates the acyclovir and then the exonuclease associated with the polymerase removes it. Under these conditions, acyclovir triphosphate is a very poor inhibitor of the polymerases. However, if one also includes the next correct dNTP (Figure 7), herpes polymerase incorporates the acyclovir to generate DNAn+ACV, and then the next correct dNTP binds to generate E-DNAn+ACV-dNTP locked in the polymerase active site. Under these conditions, acyclovir triphosphate very potently inhibits herpes polymerase, which is also the observed result in vivo. As implied by this discussion, one can identify dNTP analogues with this property via “induced inhibition”. If you assay your enzyme in the presence of just the dNTP analogue and a competing normal dNTP, the dNTP analogue weakly inhibits your polymerase. However, if you also include the second correct dNTP, now the dNTP analogue potently inhibits the enzyme.
Figure 7.

Inhibition of herpes DNA polymerase by acyclovir triphosphate (ACVTP).
Effects of DNA sequence.
The sequence of the DNA used for assaying polymerase activity can have remarkably large effects on the properties of the enzyme. The rates of misincorporation along a template vary dramatically, even for the same misincorporation event (Ex., misincorporation of dGTP opposite different templating T’s) (Bell, et al., 1997, Kunkel, 1985, Kunkel and Alexander, 1986, Lai and Beattie, 1988). The molecular mechanism by which DNA sequence affects polymerase fidelity remains unclear. While the effect of varying the DNA sequence on polymerization of nucleotide analogues has not been examined, the data for misincorporation of natural dNTPs suggests that polymerases will incorporate dNTP analogues at different rates in different sequence contexts. Thus, it is essential to use the same DNA sequence for all of the dNTP (analogues) tested.
3′-5′ Exonuclease Activity.
Many DNA polymerases contain a proofreading 3′-5′ exonuclease that can remove the 3′-terminal nucleotide from both single-stranded and double-stranded DNA. If one wants to just study the incorporation of a dNTP analogue, the exonuclease activity can seriously complicate the analysis – for incorporation of a single nucleotide, the true incorporation rate will be faster than the measured rate because the exonuclease will have hydrolyzed some of the incorporation product (DNAn+1) back to the starting material (DNAn). The most common solution to this problem is to use a form of the polymerase that lacks exonuclease activity (exo− Klenow Fragment, exo− herpes polymerase, etc.) generated by mutating the exonuclease active site.
Alternatively, one may want to know how fast the exonuclease hydrolyzes the 3′-terminal nucleotide on the primer strand due to the presence of a nucleotide analogue in either the template or primer strand. Assays are performed analogously to the polymerase assay, except now one starts with [32P]DNAn+1 and monitors the production of [32P]DNAn by denaturing gel electrophoresis.
Challenges of RNA polymerases.
Depending upon the RNA polymerase used, these enzymes can present a special challenge to examining incorporation of nucleotide analogues. In some cases, one can use a RNA primer-template, directly analogous to the assays with DNA polymerases (Anand and Patel, 2006, Castro, et al., 2005, Te Velthuis, et al., 2009). In other cases, however, this is not so easily accomplished. Many RNA polymerases produce single-stranded RNA as the product, and one cannot easily set up a RNA primer-template system to look at addition of single nucleotide. In this case, it may still be possible to obtain detailed kinetic data using a partitioning analysis (Choi, et al., 1996, Kuchta, et al., 1992, Ramirez-Aguilar, et al., 2005, Urban, et al., 2009). As depicted in Figure 8, the RNA polymerase initiates synthesis de novo and polymerizes NTPs until it reaches the site X, thereby generating RNAn. How one measures the polymerization efficiency of the NTP (analogue) opposite X will depend on what happens when the RNA polymerase encounters X. If the polymerase always terminates RNA synthesis upon producing RNAn. one can simply include varying concentrations of the analogue and measure the fraction of RNAn converted to RNAn+1 (Polymerization of YTP in Figure 8), analogous to the running start methodology described above. Alternatively, the RNA polymerase may polymerize one of the NTPs in the assay opposite X. Provided that one can differentiate the products due to incorporation of YTP from those due to incorporation of the NTP, one can measure the frequency with which the RNA polymerase incorporates the analogue as a function of analogue concentration. Comparing different NTP (analogues) under otherwise identical experimental conditions then allows one to quantify the effects of making a specific change (Urban, et al., 2009). Particularly for short RNA products up to around 6 nucleotides long, high percentage acrylamide gels (30-40%) will generally differentiate products that differ only as to whether the enzyme incorporated a natural or analogue nucleotide (Moore, et al., 2004, Ramirez-Aguilar, et al., 2005). The high acrylamide percentage precludes including urea in the gel. However, because the products are so short and the gel loading buffer contains EDTA to chelate any divalent metals, the products will not remain bound to the template. It should also be noted that these high percentage gels require much longer times to run, often up to 15 hours.
Figure 8.

Partitioning analysis of NTP polymerization by a RNA polymerase.
A second complication with many RNA polymerases, especially those involved in transcription, is that they have two modes of synthesis. During polymerization of the first few NTPs, the polymerases typically have low processivity and frequently abort synthesis (Hatoum and Roberts, 2008, Hieb, et al., 2006, Wang, et al., 2007). However, once they undergo “promoter escape”, synthtesis becomes highly processive. Thus, it may be important to consider the effects of the nucleotide analogue during both phases of transcription.
Data Interpretation.
Perhaps the biggest issue facing the use of analogues is that of data interpretation. A number of factors have conspired to make data interpretation particularly problematic, and each will be discussed in turn.
Kinetic Parameters.
As noted earlier, the kinetic parameter of greatest interest is the specificity constant, Vmax/KM or kpol/KD (Apparent). Comparing two dNTP (analogues) shows how a specific modification affects the efficiency of polymerization. The individual components of the specificity parameter, kcat or Vmax, and KD or KM, are notoriously difficult to rigorously interpret without experiments showing what step(s) each constant represents.
Different Polymerases.
Extending conclusions from one polymerase to other polymerases is extremely dangerous, especially if they are members of different evolutionary families. Polymerases form a number of evolutionarily families, including the A, B, C, X, Y, reverse transcriptase, bacterial primase, and eukaryotic/archael primase families (Burgers, et al., 2001, Kuchta and Stengel, 2009). Importantly, within these families, several distinct mechanisms by which the enzymes interact with the templating base and incoming (d)NTP exist, and different families do not use the same mechanisms for deciding whether or not to polymerize a (d)NTP. For example, A family DNA polymerases generally efficiently and accurately polymerize 2,4-difluorotoluene dNTP opposite a templating T (Morales and Kool, 1998, Morales and Kool, 2000, Moran, et al., 1997). In contrast, B family polymerases genrally do not efficiently incorporate 2,4-difluorotoluene dNTP (Morales and Kool, 2000). Thus, it is completely inappropriate to conclude that what one observes with, for example, the A family enzyme Klenow Fragment will also be true for the B family enzyme T4 DNA polymerase and vice versa. In some cases, even different members of the same evolutionary family may have different requirements. For example, most B family polymerase very efficiently add the next correct dNTP onto a primer containing either 3-deaza-dA or 3-deaza-dG at the primer terminus (DNAn + dNTP → DNAn+1), but only extremely poorly add the second correct dNTP (DNAn+1 + dNTP → DNAn+2 (Beckman, et al., 2007, Cavanaugh, et al., 2009, Hendrickson, et al., 2004)). In contrast, the B family polymerase Tli pol efficiently adds both the first and second dNTPs (Hendrickson, et al., 2004).
Identifying critical chemical features in the nucleotide.
One of the most daunting tasks of using nucleotide analogues is deconvoluting the data to identify the critical parameters. A simple inspection of a base reveals a number of potential parameters that a polymerase might recognize, including hydrophobicity, dipole moment, size, Watson-Crick hydrogen bonding capacity, hydrogen bonding capacity of other groups in the major and minor grooves (Major groove: N7 and N6/O6 of a purine, N4 of a pyrimidine; minor groove, N-3 of a purine and O2 of a pyrididine), and π electron density of the rings. Importantly, the electronic features of all of the atoms are interconnected due to the aromaticity of the bases. Thus, changing any one atom will result in electronic effects that reverberate throughout the structure. For example, adding a methyl group versus a halogen at C-2 of a purine will have very different effects since the methyl group is slightly electron donating whereas the halogens are strongly electron withdrawing. These changes in electronic character can have major effects on the ability of the various heteroatoms in the ring system to participate in hydrogen bonds. Thus, for example, if one converts dATP into 2-chloro-dATP and the change affects the efficiency of incorporation, the effects can be extraordinarily hard to interpret. Minimally, any effects of this simple change could be due to: i) steric effects, since Cl is much larger than H; ii) altered Watson-Crick hydrogen bonding, since the electron withdrawing effect of Cl will interfere with the ability of N-1 to form a Watson-Crick hydrogen bond with N-3 of uridine, and; iii) altered minor groove hydrogen bonding due to the electron withdrawing effects of the Cl on N-3.
How, then, does one try to isolate the important parameters affected by the chlorine that are sensed by the polymerase? Ultimately, it requires looking at multiple analogues that test the same parameter as well as looking at the same modification within the context of multiple, similar bases. For example, if one suspects that the effects of the chlorine resulted from electronic effects on N-1 and consequent weaker hydrogen bonding to NH-3 of a template thymine, one could first examine how the enzyme interacts with 2-flouro-dATP and 2-methyl-dATP. Concomitant with this, one should examine a series of analogues that explicitly test the role of Watson-Crick hydrogen bonds during dNTP incorporation (Ex., 1-deaza-dATP opposite T). Unfortunately, even after examining extensive modifications, one may be left with a less than clear picture of exactly how a polymerase functions (see below).
Different chemical contexts.
Especially essential is to examine the same modification in different chemical contexts. The most extensive data on the effects of substrate modifications exists for Klenow Fragment (an A family enzyme), herpes and human DNA primase, and three B family polymerases (Pol α, herpes DNA pol, and T4 DNA pol) (Berdis and McCutcheon, 2007, Cavanaugh, et al., 2009, Henry, et al., 2004, Hwang and Romesberg, 2008, Kim, et al., 2005, Kincaid, et al., 2005, Kool, 2002, Leconte, et al., 2008, Lee and Berdis, 2009, Lee, et al., 2008, Matsuda, et al., 2003, Meyer, et al., 2004, Morales and Kool, 1998, Morales and Kool, 2000, Ogawa, et al., 2000, Patro, et al., 2009, Ramirez-Aguilar and Kuchta, 2004, Sintim and Kool, 2006, Thompson and Kuchta, 1995, Washington, et al., 2003, Zhang, et al., 2005). From these data, it is clear that the structural context of a mutation can dramatically alter the effects of modifying the base of a dNTP or NTP in a polymerase dependent manner. For the B family enzymes, similar modifications tend to have the similar effects within different structural contexts (Beckman, et al., 2007, Cavanaugh, et al., 2009). The A family enzyme Klenow Fragment typically shows similar effects, although notable exceptions exist. For example, Romesberg and coworkers examined the effects of converting furo[2,3-c]pyridin-7(6H)-one dNTP into furo[2,3-c]pyridine-7-thiol dNTP and converting furo[3,2-c]pyridin-4(5H)-one dNTP into furo[3,2-c]pyridine-4-thiol dNTP (Figure 9). Even though both conversions involve changing an oxygen into sulfur, the effect of this change varied by up to >3000-fold in the case of polymerization opposite a template dC (Henry, et al., 2003). The primases, however, often show radically different effects of the identical change in the base but within two different contexts (Urban, et al., 2009). For example, whereas removing N-3 of a templating A has minor effects on polymerization of UTP, removing O2 of a templating T or C severely impairs polymerization of ATP and GTP, respectively. Similarly, adding back N-1 to 1-deazapurine NTP greatly enhances incorporation of the resulting purine NTP, but adding back the chemically equivalent N-1 to 2-pyridone NTP greatly impairs polymerization of the resulting zubularine triphosphate. Thus, one clearly must exercise caution when interpreting the effects of a modification and extending the modification from one structural context to another.
Figure 9.
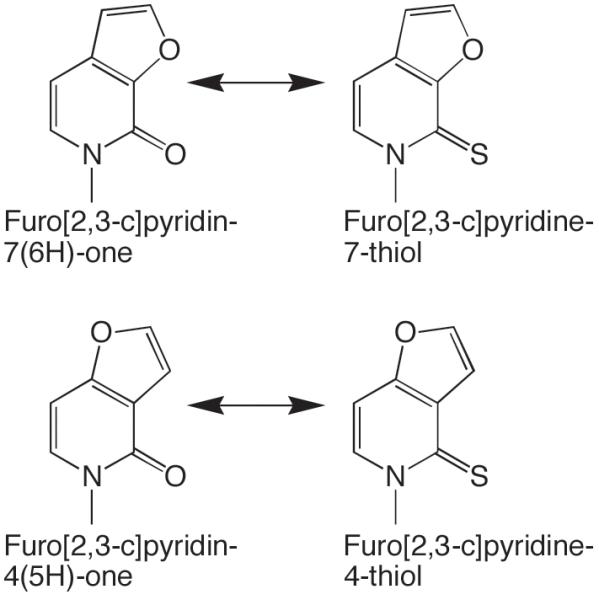
Two chemically similar interconversions that have dramatically different effects on incorporation of the resulting dNTP by Klenow Fragment.
In addition to structural context, whether the modification resides within the templating base or the incoming dNTP can dramatically affect the results. Indeed, this asymmetry has greatly impeded the development of novel base-pairs. Very frequently, a polymerase will generate a novel base-pair by efficiently polymerizing dXTP opposite a templating Y, but does not efficiently polymerize dYTP opposite a templating X (For examples, see (Chiaramonte, et al., 2003, Henry, et al., 2003, Lutz, et al., 1996, Piccirilli, et al., 1990)).
Examples of difficulty interpreting how a polymerase interacts with a nucleotide.
As suggested in this discussion, extreme care must be used when interpreting data as to how a polymerase interacts with a nucleotide. Two examples of this difficulty will be discussed – the role of Watson-Crick hydrogen bonds with herpes primase and the role of shape with Klenow Fragment.
Herpes primase.
From initial studies using 15 purine NTP analogues, herpes primase only efficiently incorporated those that could form Watson-Crick hydrogen bonds involving the purine N-1 and N6/O6 and the pyrimidine N-1 and N4/O4, respectively (Ramirez-Aguilar, et al., 2005). This led to the hypothesis that herpes primase requires the formation of Watson-Crick hydrogen bonds in order to polymerize a NTP. However, examination of a more extensive series of base analogues showed that while primase usually only efficiently incorporates NTPs whose bases can form Watson-Crick hydrogen bonds, several remarkable exceptions occur (Urban, et al., 2009). Most notably, herpes primase polymerizes 2-pyridone NTP and 4-methyl-2-pyridone NTP very efficiently across from the natural template bases, especially G, thereby indicating that primase does not absolutely require formation of Watson-Crick hydrogen bonds between the incoming NTP and templating base to efficiently polymerize the NTP.
Klenow Fragment.
Klenow Fragment, an A family DNA polymerase, has probably been probed with more nucleotide analogues than any other enzyme. A series of studies using 2,4-dihalotoluene dNTPs and 2,4-difluorotoluene templating bases, a group of analogues isosteric with T, showed that Klenow fragment very efficiently generated base-pairs between these analogues and A but did not generate base-pairs with the other 3 natural bases (Morales and Kool, 1998, Morales and Kool, 2000, Moran, et al., 1997, Sintim and Kool, 2006). Furthermore, altering the shape of the 2,4-dihalotoluene so that it no longer resembled T eliminated the preference for generation of base-pairs with A. These data led to the idea that Klenow fragment discriminates between right and wrong dNTPs based on the shape of the base-pair between the incoming dNTP and templating base. However, Klenow fragment will also very efficiently generate some very mis-shapen base-pairs (Ex., 5-OMethylbenzimidazole dNTP:G, 5-Nitroindole dNTP:A, Furo[2,3-c]pyridine-7-thiol dNTP:G, Furo[3,2-c]pyridine-4-thiol (Figure 10) (Chiaramonte, et al., 2003, Henry, et al., 2003, Kincaid, et al., 2005)).
Figure 10.

Unusually shaped base-pairs that Klenow Fragment generates with high efficiency.
These data raise several important questions relating to polymerase mechanisms and using nucleotide analogues. How many analogues should one examine to feel confident in ones conclusions? While the bulk of the data with both herpes primase and Klenow Fragment clearly support key roles for Watson-Crick hydrogen bonds and shape, respectively, as key parameters for efficient (d)NTP incorporation by each enzyme, any model posited should ideally accommodate all of the data. How does one account for these outliers within the context of these models and the other analogues? Do these results reflect an unexplored property of the outliers, or are these results idiosyncratic to the polymerase of interest? Regardless, of the answers to these questions, it is clear that data should be interpreted cautiously.
Acknowledgments
This work was supported by NIH grants GM54194 and AI59764 to R.D.K.
References
- Anand VS, Patel SS. Transient State Kinetics of Transcription Elongation by T7 RNA Polymerase. J. Biol. Chem. 2006;281:35677–35685. doi: 10.1074/jbc.M608180200. [DOI] [PubMed] [Google Scholar]
- Beckman J, Kincaid K, Hocek M, Spratt T, Engels J, Cosstick R, Kuchta RD. Human DNA Polymerase alpha Uses a Combination of Positive and Negative Selectivity to Polymerize Purine dNTPs with High Fidelity. Biochemistry. 2007;46:448–460. doi: 10.1021/bi061243s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JB, Eckert KA, Joyce CM, Kunkel TA. Base Miscoding and Strand Misalignment Errors by Mutator Klenow Polymerases with Amino Acid Substitutions at Tyrosine 766 in the O-Helix of the Fingers Subdomain. J. Biol. Chem. 1997;272:7345–7351. doi: 10.1074/jbc.272.11.7345. [DOI] [PubMed] [Google Scholar]
- Berdis AJ, McCutcheon D. The Use of Non-natural Nucleotides to Probe Template-Independent DNA Synthesis. Chembiochem. 2007;8:1399–1408. doi: 10.1002/cbic.200700096. [DOI] [PubMed] [Google Scholar]
- Boosalis MS, Petruska J, Goodman MF. DNA Polymerase Insertion Fidelity: Gel Assay for Site-Specific Kinetics. J. Biol. Chem. 1987;262:14689–14696. [PubMed] [Google Scholar]
- Burgers PM, Koonin EV, Bruford E, Blanco L, Burtis KC, Christman MF, Copeland WC, Friedberg EC, Hanaoka F, Hinkle DC, W. LC, Nakanishi M, Ohmori H, Prakash L, Prakash S, Reynaud CA, Sugino A, Todo T, Wang Z, Weill JC, Woodgate R. Eukaryotic DNA Polymerases: Proposal for a Revised Nomenclature. J. Biol. Chem. 2001;276:43487–43490. doi: 10.1074/jbc.R100056200. [DOI] [PubMed] [Google Scholar]
- Burgess K, Cook D. Syntheses of Nucleoside Triphosphates. Chem. Rev. 2000;100:2047–2060. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- Burton JR, Everson GT. HCV NS5B Polymerase Inhibitors. Curr. Liver Dis. 2009;13:453–465. doi: 10.1016/j.cld.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Cai H, Bloom LB, Eritja R, Goodman MF. Kinetics of Deoxyribonucleotide Insertion and Extension at Abasic Template Lesions in Different Sequence Contexts Using HIV-1 Reverse Transcriptase. J. Biol. Chem. 1993;268:23567–23572. [PubMed] [Google Scholar]
- Castro C, Arnold JJ, Cameron CE. Incorporation Fidelity of the Viral RNA-Dependent RNA Polymerase: a Kinetic, Thermodynamic and Structural Perspective. Virus Res. 2005;107:141–149. doi: 10.1016/j.virusres.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh NA, Urban M, Beckman J, Spratt T, Kuchta RD. Identifying the Features of Purine dNTPs that Allow Accurate and Efficient DNA Synthesis by Herpes Simplex Virus 1 DNA Polymerase. Biochemistry. 2009;48:3554–3564. doi: 10.1021/bi8022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Facile Polymerization of dNTPs Bearing Unnatural Base Analogues by DNA Polymerase alpha and Klenow Fragment (DNA Polymerase I) Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- Choi DJ, Roth RB, Liu T, Geacintov NE, Scicchitano DA. Incorrect Base Insertion and Prematurely Terminated Transcripts during T7 RNA Polymerase Transcription Elongation Past Benzo[a]pyrenediol Epoxide-Modified DNA. J. Mol. Biol. 1996;264:123–219. doi: 10.1006/jmbi.1996.0635. [DOI] [PubMed] [Google Scholar]
- Dahlberg ME, Benkovic SJ. Kinetic Mechanism of DNA Polymerase I (Klenow Fragment): Identification of a Second Conformational Change and Evaluation of the Internal Equilibrium Constant. Biochemistry. 1991;30:4835–4843. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- Donlin MJ, Patell SS, Johnson KA. Kinetic Partitioning between the Exonuclease and Polymerase Sites in DNA Error Correction. Biochemistry. 1991;30:538–547. doi: 10.1021/bi00216a031. [DOI] [PubMed] [Google Scholar]
- Hatoum A, Roberts J. Prevalence of RNA Polymerase Stalling at Escherichia coli Promoters after Open Complex Formation. Mol. Microbiol. 2008;68:17–28. doi: 10.1111/j.1365-2958.2008.06138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson CL, Devine KG, Benner SA. Probing Minor Groove Recognition Contacts by DNA Polymerases and Reverse Transcriptases Using 3-Deaza-2′-deoxyadenosine. Nuc. Ac. Res. 2004;32:2241–2250. doi: 10.1093/nar/gkh542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry AA, Olsen AG, Matsuda S, Yu C, Geierstanger BH, Romesberg FE. Efforts to Expand the Genetic Alphabet: Identification of a Replicable Unnatural DNA Self-Pair. J. Am. Chem. Soc. 2004;126:6923–6931. doi: 10.1021/ja049961u. [DOI] [PubMed] [Google Scholar]
- Henry AA, Yu C, Romesberg FE. Determinants of Unnatural Nucleobase Stability and Polymerase Recognition. J. Am. Chem. Soc. 2003;125:9638–9646. doi: 10.1021/ja035398o. [DOI] [PubMed] [Google Scholar]
- Hieb AR, Baran S, Goodrich JA, Kugel JF. An 8 Nt. RNA Triggers a Rate-Limiting Shift of RNA Polymerase II Complexes into Elongation. EMBO J. 2006;25:3100–3109. doi: 10.1038/sj.emboj.7601197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao I, Kimoto M, Mitsui T, Fujiwara T, Kawai R, Sato A, Harada Y, Yokoyama S. An Unnatural Hydrophobic Base Pair System: Site-Specific Incorporation of Nucleotide Analogs into DNA and RNA. Nat. Meth. 2006;3:729–735. doi: 10.1038/nmeth915. [DOI] [PubMed] [Google Scholar]
- Hirao I, Mitsui T, Kimoto M, Yokoyama S. An Efficient Unnatural Base Pair for PCR Amplification. J. Am. Chemical Soc. 2007;129:15549–15555. doi: 10.1021/ja073830m. [DOI] [PubMed] [Google Scholar]
- Horwitz JP, Chua J, Noel M, DaRooge MA. Nucleosides. IV. 1-(2-Deoxy-beta-D-lyxofuranosyl)-5-iodouracil. J. Med. Chem. 1964;7:385–386. doi: 10.1021/jm00333a045. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Zinnen S, Modrich P. Kinetic Mechanism of the DNA-dependent DNA Polymerase Activity of Human Immunodeficiency Virus Reverse Transcriptase. J. Biol. Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-Difluorocytidine on DNA Synthesis. Canc. Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- Hwang GT, Romesberg FE. Unnatural Substrate Repertoire of A, B, and X Family DNA Polymerases. J. Am. Chemical Soc. 2008;130:14872–14882. doi: 10.1021/ja803833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the Active Stie Tightness of DNA Polymerase in Subangstrom Increments. Proc. Nat. Acad. Sci. USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels J, Kuchta RD. Exploration of Factors Driving Incorporation of Unnatural dNTPs into DNA by Klenow Fragment (DNA Polymerase I) and DNA Polymerase alpha. Nuc. Acids Res. 2005;33:2620–2628. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool ET. Active Site Tightness and Substrate Fit in DNA Replication. Ann. Rev. Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- Kuchta RD, Benkovic P, Benkovic SJ. Kinetic Mechanism Whereby DNA Polymerase I (Klenow) Replicates DNA with High Fidelity. Biochemistry. 1988;27:6716–6725. doi: 10.1021/bi00418a012. [DOI] [PubMed] [Google Scholar]
- Kuchta RD, Ilsley D, Kravig KD, Schubert S, Harris B. Inhibition of DNA Primase and Polymerase Alpha by Arabinofuranosylnucleoside Triphosphates and Related Compounds. Biochemistry. 1992;31:4720–4728. doi: 10.1021/bi00134a027. [DOI] [PubMed] [Google Scholar]
- Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic Mechanism of DNA Polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- Kuchta RD, Stengel G. Mechanism and Evolution of DNA Primases. Biochim. Biophys. Acta. 2009 doi: 10.1016/j.bbapap.2009.06.011. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchta RD, Willhelm L. Inhibition of DNA Primase by 9-beta-D-Arabinofuranosyladenosine Triphosphate. Biochemistry. 1991;30:797–803. doi: 10.1021/bi00217a033. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. The Mutational Specificity of DNA Polymerase beta during in vitro DNA Synthesis. Production of Frameshift, Base Substitution, and Deletion Mutations. J. Biol. Chem. 1985;260:5787–5796. [PubMed] [Google Scholar]
- Kunkel TA, Alexander PS. The Base Substitution Fidelity of Eukaryotic DNA Polymerases. Mispairing Frequencies, Site Preferencs, Insertion Preferences, and Base Substitution by Dislocation. J. Biol. Chem. 1986;261:160–166. [PubMed] [Google Scholar]
- Lai MD, Beattie KL. Influence of DNA Sequence on the Nature of Mispairing during DNA Synthesis. Biochemistry. 1988;27:1722–1728. doi: 10.1021/bi00405a051. [DOI] [PubMed] [Google Scholar]
- Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE. Discovery, Characterization, and Optimization of an Unnatural Base Pair for Expansion of the Genetic Alphabet. J. Am. Chem. Soc. 2008;130:2336–2343. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Berdis AJ. Non-natural Nucleotides as Probes for the Mechanism and Fidelity of DNA Polymerases. Biochim. Biophys. Acta. 2009 doi: 10.1016/j.bbapap.2009.08.023. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Helquist SA, Kool ET, Johnson KA. Importance of Hydrogen Bonding for Efficiency and Specificity of the Human Mitochondrial DNA Polymerase. J. Biol. Chem. 2008;283:14402–14410. doi: 10.1074/jbc.M705007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TS, Mancini WR. Synthesis and Antineoplastic Activity of 3′-Azido and 3′-Amino Analogs of Pyrimidine Deoxyribonuclosides. J. Med. Chem. 1983;26:544–588. doi: 10.1021/jm00358a016. [DOI] [PubMed] [Google Scholar]
- Ludwig J. A New Route to Nucleoside 5′-Triphosphates. Acta Biochim. Biophys. Acad. Sci. Hung. 1981;16:131–135. [PubMed] [Google Scholar]
- Lutz MJ, Held HA, Hottinger M, Hubscher U, Benner SA. Differential Discrimination of DNA Polymerases for Variants of the Non-standard Nucleobase Pair between Xanthosine and 2,4-Diaminopyrimidine, Two Components of an Expanded Genetic Alphabet. Nuc. Ac. Res. 1996;24:1308–1313. doi: 10.1093/nar/24.7.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Henry AA, Schultz SS, Romesberg FE. The Effects of Minor-Groove Hydrogen-Bond Acceptors and Donors on the Stability and Replication of Four Unnatural Base Pairs. J. Am. Chem. Soc. 2003;125:6135–6139. doi: 10.1021/ja034099w. [DOI] [PubMed] [Google Scholar]
- Meyer AS, Blandino M, Spratt T. E. coli DNA Polymerase I (Klenow Fragment) Uses a Hydrogen-Bonding Fork from Arg668 to the Primer Terminus and Incoming dNTP to Catalyze DNA Replication. J. Biol. Chem. 2004;279:33043–33046. doi: 10.1074/jbc.C400232200. [DOI] [PubMed] [Google Scholar]
- Moore CL, Zivkovic A, Engels J, Kuchta RD. Human DNA Primase Uses Watson-Crick Hydrogen Bonding Groups to Distinguish between Correct and Incorrect NTPs. Biochemistry. 2004;43:12367–12374. doi: 10.1021/bi0490791. [DOI] [PubMed] [Google Scholar]
- Morales JC, Kool ET. Efficient Replication between non-Hydrogen Bonded Nucleoside Shape Analogs. Nat. Struct. Biol. 1998;5:950–954. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- Morales JC, Kool ET. Varied Molecular Interactions at the Active Sites of Several DNA Polymerases: Nonpolar Nucleoside Isosteres as Probes. J. Am. Chem. Soc. 2000;122:1001–1007. doi: 10.1021/ja993464+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales JC, Kool ET. Varied Molecular Interactions at the Active Sties of Several DNA Polymerases: Nonpolar Nucleoside Isosteres as Probes. J. Am. Chem. Soc. 2000;122:1001–1007. doi: 10.1021/ja993464+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Ren RX-F, Rumney S, Kool ET. Difluorotoluene, a Nonpolar Isostere of Thymine, Codes Specifically and Efficiently for Adenine in DNA Replication. J. Am. Chemical Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa AK, Wu Y, McMinn DL, Liu J, Schultz PG, Romesberg FE. Efforts toward the Expansion of the Genetic Alphabet: Information Storage and Replcation with Unnatural Hydrophobic Base Pairs. J. Am. Chem. Soc. 2000;122:3274–3287. [Google Scholar]
- Parker W. Enzymology of Purine and Pyrimidine Antimetabolites in the Treatment of Cancer. Chem. Rev. 2009 doi: 10.1021/cr900028p. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Wong I, Johnson KA. Pre-Steady State Kinetic Analysis of Processive DNA Replication Including Complete Characterization of an Exonuclease Deficient Mutant. Biochemistry. 1991;30:511. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- Patro JN, Urban M, Kuchta RD. Role of the 2-Amino Group of Purines during dNTP Polymerization by Human DNA Polymerase alpha. Biochemistry. 2009;48:180–189. doi: 10.1021/bi801823z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic Incorporation of a New Base Pair into DNA and RNA Extends the Genetic Alphabet. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: Preclinical Pharmacology and Mechanisms of Action. Semin. Oncol. 1996;56:3–15. [PubMed] [Google Scholar]
- Ramirez-Aguilar KA, Kuchta RD. Herpes Simplex Virus 1 DNA Primase: A Polymerase with Extraordinarily Low Fidelity. Biochemistry. 2004;43:9084–9091. doi: 10.1021/bi049335+. [DOI] [PubMed] [Google Scholar]
- Ramirez-Aguilar KA, Moore CL, Kuchta RD. Herpes Simplex Virus I Primase Employs Watson-Crick Hydrogen Bonding to Identify Cognate NTPs. Biochemistry. 2005;44:15585–15593. doi: 10.1021/bi0513711. [DOI] [PubMed] [Google Scholar]
- Reardon JE. Herpes Simplex Virus Type 1 DNA Polymerase. Mechanism of Inhibition by Acyclovir Triphosphate. J. Biol. Chem. 1989;264:7405–7411. [PubMed] [Google Scholar]
- Reardon JE. Herpes Simplex Virus Type I DNA Polymerase: Mechanism-Based Affinity Chromatography. J. Biol. Chem. 1990;265:7112. [PubMed] [Google Scholar]
- Segel IH. Enzyme Kinetics. John Wiley and Sons; New York: 1975. [Google Scholar]
- Showalter AK, Tsai M-D. A Reexamination of the Nucleotide Incorporation Fidelity of DNA Polymerases. Biochemistry. 2002;41:10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- Sintim HO, Kool ET. Remarkable Sensitivity to DNA Base Shape in the DNA Polymerase Active Ste. Angew. Chem. Int. Ed. 2006;45:1974–1979. doi: 10.1002/anie.200504296. [DOI] [PubMed] [Google Scholar]
- Tchesnokov EP, Obikhod A, Schinazi RB, Gotte M. Delayed Chain Termination Protects the Anti-hepatitis B Virus Drug Entecavir from Excision by HIV-1 Reverse Transcriptase. J. Biol. Chem. 2008;283:34218–34228. doi: 10.1074/jbc.M806797200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Velthuis AJ, Arnold JJ, Cameron CE, van de Worm SJ, Snijder EJ. The RNA Polymerase Activity of SARS-coronavirus nsp12 Is Primer Dependent. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp904. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson H, Kuchta RD. Arabinofuranosyl Nucleotides Are Not Chain-Terminators during Initiation of New Strands of DNA by DNA Polymerase å-Primase. Biochemistry. 1995;34:11198–11203. doi: 10.1021/bi00035a027. [DOI] [PubMed] [Google Scholar]
- Tsai Y-C, Johnson KA. A New Paradigm for DNA Polymerase Specificity. Biochemistry. 2006;45:9675–9687. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M, Joubert N, Hocek M, Alexander RE, Kuchta RD. Herpes Simplex Virus-1 DNA Primase: A Remarkably Inacurate Yet Selective Polymerase. Biochemistry. 2009;48:10866–10881. doi: 10.1021/bi901476k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal EC. Current and Potential Therapies for the Treatment of Herpesvirus Infections. Progress in Drug Research. 2001:185–228. doi: 10.1007/978-3-0348-7784-8_5. [DOI] [PubMed] [Google Scholar]
- Vivet-Boudou V, Didierjean J, Isel C, Marquet R. Nucleoside and Nucleotide Inhibitors of HIV-1 Replication. Cell. Mol. Life Sci. 2006;63:163–186. doi: 10.1007/s00018-005-5367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tullius TD, Levin JR. Effects of Discontinuities in the DNA Template on Abortive Initiation and Promoter Escape by Escherichia coli RNA Polymerase. J. Biol. Chem. 2007;282:26917–26927. doi: 10.1074/jbc.M702473200. [DOI] [PubMed] [Google Scholar]
- Washington MT, Helquist SA, Kool ET, Prakash L, Prakash S. Requirement of Watson-Crick Hydrogen Bonding for DNA Synthesis by Yeast DNA Polymerase eta. Mol. Cell. Biol. 2003;23:5107–5112. doi: 10.1128/MCB.23.14.5107-5112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong I, Patel SS, Johnson KA. An Induced Fit Kinetic Mechanism for DNA Replication Fidelity: Direct Measurement by Single Turnover Kinetics. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- Wu W, Bergstrom DE, Jo Davisson V. Chemoenzymatic Preparation of Nucleoside Triphoshates. Curr. Protoc. Nucleic Acid Chem. 2004 May; doi: 10.1002/0471142700.nc1302s16. Chapter 13, Unit 13.12. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lee I, Berdis AJ. The Use of Nonnatural Nucleotides to Probe the Contribution of Shape Complementarity and Pi electron Surface Area during DNA Polymerization. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]