

Abstract
We report on the design and evaluation of novel cyclic peptides targeting the N-terminal domain of the protein tyrosine phosphatase YopH from Yersinia. Cyclic peptides have been designed based on a short sequence from the protein SKAP-HOM [DE(pY)DDPF (pY = phosphotyrosine)], and they all contain the motif DEZXDPfK (where Z is a phosphotyrosine or a non-hydrolyzable phosphotyrosine mimetic, X is an aspartic acid or a leucine and f is a d-phenylalanine). These peptides present a ‘head to tail’ architecture, enabling cyclization through formation of an amide bond in between the side chains of the first aspartic acid and the lysine residues. Chemical shift perturbation studies have been carried out to monitor the binding of these peptides to the N-terminal domain of YopH. Peptides containing a phosphotyrosine moiety exhibit binding affinities in the low micromolar range; substitution of the phosphotyrosine with one of its non-hydrolyzable derivatives dramatically reduces the binding affinities. These preliminary studies may pave the way for the discovery of more potent and selective peptidebased ligands of the YopH N-terminal domain which could be further investigated for their ability to inhibit Yersiniae infections.
Keywords: cyclic peptides, NMR, protein tyrosine phosphatase, Yersinia, Yersinia outer protein H
The Gram-negative bacterium Yersinia includes three species (Yersinia pestis, Yersinia pseudoturbecolosis, and Yersinia enterocolitica) that are extremely pathogenic to humans (1,2). Yersiniae make use of a type III secretion system to inject six effector proteins in the cytosol of the infected cells and thus start to interfere with crucial signaling pathways of the host organism (3–5). Yersinia outer protein H (YopH) is one of Yersiniae effectors; it is a potent phosphotyrosine phosphatase (PTPase) that is made up of two distinctly folded domains; the PTPase catalytic site resides in the C-terminal part (aa 206–468) (2,6–8). YopH phosphatase activity is crucial for all types of Yersiniae infections (6,9).
YopH N-terminal domain (aa 1–129) has a tertiary structure consisting of two double-stranded beta sheets and four helices (10–12) (Figure 1B). This domain is able to bind the chaperone SycH (13) and play a role in the translocation of YopH in the host cells, but it can also interact with other phosphotyrosine containing substrates such as Cas and paxillin (2). It has been demonstrated that the N-terminal domain is important for efficient substrate dephosphorylation by YopH (2,6); the latter observation makes it of interest as a potential target to develop novel YopH inhibitors.
Figure 1.

(A) Comparison of [1H-15N]-HSQC spectra of 15N-labeled Yoph N-terminal domain in its unbound form (70 μm concentration, red) and after the addition of the Cyclo-DE(pY)DDPfK peptide (250 μm concentration, cyan). (B) Ribbon drawing of one representative conformer of the NMR solution structure of the Yoph N-terminal domain (yellow) in complex with the linear DE(pY)DDPF peptide [green, pdb code: 1 M0V, (12)]. The side chains of the phosphotyrosine together with Lysine 26 and Arginine 28 that could be involved in electrostatic interactions are reported in neon representation. The residues that undergo significant chemical shifts variations as indicated in panel A have been colored in magenta. (C) Binding curve, showing the 1H normalized chemical shift deviations for a residue in the putative binding site (Alanine 32) as function of the peptide concentration. The dissociation constant has been estimated as described in the Material and Methods.
The binding of small linear peptides to the N-terminal domain of YopH has already been characterized (12,14). In particular, NMR studies have been previously carried out, and the solution structure of the complex between the small peptide DE(pY)DDPF from the protein SKAP-HOM and the YopH N-terminal domain has been reported (12).
These structural studies indicate a shallow, positively charged peptide-binding site; the SKAP-HOM peptide binds the N-terminal domain of YopH with a dissociation constant Kd of about 0.1 μm (12), that is 100 times lower with respect to the one measured for the peptide DADE(pY)L, derived from a sequence of the epidermal growth factor receptor (EGFR), which has also been previously described as a good substrate for the YopH PTPase domain (15).
Starting from the structural determinants responsible for the binding of linear peptides to the N-terminal domain and in an attempt to obtain peptide-based inhibitor of YopH with potentially improved pharmacokinetic properties, we have designed cyclic peptide mimetics targeting this domain. These peptides contain the pattern DEZXDPfK (where Z is a phosphotyrosine or a non-hydrolyzable phosphotyrosine derivatives, X is an aspartic acid or a leucine, and f is a d-phenylalanine) and are cyclized through an amide bond between the side chains of the first (Asp) and last residues (Lys). Among these peptides, those containing a phosphotyrosine moiety bind the YopH N-terminal domain with a dissociation constant in the micromolar range. Substitution of the phosphotyrosine with one of its non-hydrolyzable derivatives highly decreases the ability of the peptides to interact with the protein and also results in a very weak inhibition of the YopH catalytic activity.
Our work paves the way for the discovery of more potent and selective peptide ligand of the YopH N-terminal domain which could be further investigated for their ability to interfere with Yersiniae infections.
Methods and Materials
Protein expression procedures
The expression and purification protocols implemented for GST-tagged full-length YopH from Yersinia pestis and for the His-tag containing N-terminal domain of YopH from Yersinia Pseudotuberculosis have been previously reported (16–19).
To obtain 15N-labeled N-terminal domain of YopH, E. coli was grown on M9 minimal medium supported with 0.5 g/L of 15NH4Cl. Induction was achieved overnight at 20 °C at an OD600 = 0.6. The His-tagged protein was purified by affinity chromatography on a Ni-column (GE/Amersham Biosciences, Cardiff, UK) and dialyzed in a buffer at pH = 8.0 containing 30 mm Tris and 150 mm NaCl. GST-attached full-length YopH was instead dialyzed in 30 mm Tris, 150 mm NaCl, 1 mm DTT pH = 6.5.
NMR spectroscopy
NMR spectra were acquired with either a Bruker Avance 600 or an AvanceIII 700 MHz spectrometers (Bruker BioSpin Corporation, Billerica, MA, USA) both equipped with TCI cryoprobes. NMR spectra were processed with Bruker software (Topspin version 2.0) and analyzed with NEASY (20) as implemented in cara (http://www.nmr.ch/). Overlays of 2D spectra were generated with the program Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco).
The peptides DE(pY)DDPF (pY: phosphotyrosine), DE(pY)DDPfK (f = d-Phenylalanine); Cyclo-DE(pmp)DDPfK (pmp: phosphonomethylphenylalanine); Cyclo-DEYDDPfK; Cyclo-DE(pY)LDPfK; Cyclo-DE(FCOOH)LDPfK (FCOOH= phenylalanine with a carboxyl group at the para position) were purchased either from the MCW facility of the Wisconsin Medical College or from CPC Scientific (San José, CA, USA). Peptides contain an acetyl group at the N-termini, an amide group at the C-termini and are cyclized through an amide bond in between the side chains of the first residue (Asp) and the last residue (Lys).
For binding studies with the N-terminal domain of YopH, samples of 70–153 μm of 15N-uniformly labeled protein were used. [1H, 15N] HSQC spectra were acquired for the protein in its apo form and after addition of each peptide (concentration ranging from 50 to 1250 μm).
To calculate the dissociation constant (Kd), the chemical shift variations in the proton dimensions for several protein residues were plotted as function of the peptides concentration. Chemical shift variations were estimated with the relationship (δfree − δobs/δsat − δfree) where δobs is the measured value of the chemical shift at a given peptide concentration, δfree the value in the unbound state, and δsat the value at the last point of the titration (i.e.: saturation point). Because of the weakness of the binding, it was not possible to reach saturation in the case of the cyclo-DE(pmp)DDPfK peptide. Data were fitted to the equation:
where p is a constant equal to the protein concentration, and Kd, obtained from the fit, is the dissociation constant (21).
For all these binding experiments, the peptides were dissolved in deuterated DMSO to make concentrated stocks (100 mm concentration) and progressively diluted in the protein buffer (30 mm tris, 150 mm NaCl, pH = 8).
To assign the proton resonances of the Cyclo-DE(pY)DDPfK peptide, 2D [1H, 1H] NOESY spectra (22) (400 ms mixing times) and 2D [1H, 1H] TOCSY (23) (spin lock duration of 70 ms), were recorded at 700 MHz and 300 K and then compared (24). To prepare the NMR sample, starting from a concentrated stock in DMSO (100 mm concentration), the peptide was diluted in protein buffer to a final concentration of 1 mm. Spectra were acquired in the presence and absence of unlabeled N-terminal domain of Yoph at a concentration of 100–200 μm (90% H2O and 10% 2H2O).
Solution structure of the peptide Cyclo-DE(pY)DDPfK
Distance constraints for structure calculations were obtained from a 2D [1H, 1H] NOESY spectrum acquired with a mixing time of 400 ms (22). The program NEASY was used to manually integrate NOE cross-peaks. cyana (25) was implemented to transfer NOE intensities into upper distance limits according to an inverse sixth power peak volume-to-distance relationship and an inverse fourth power function for the backbone and side chains respectively. Starting from 100 random conformers, structures were calculated by means of the torsion angle dynamics protocol of the software cyana. Angle constraints were obtained by using the GRIDSEARCH module of cyana. A phosphotyrosine as well as a d-phenylalanine residues were included to the cyana standard residue library. At this aim, values of the atomic coordinates were obtained from Sybyl force field (Tripos, St. Loius, MO, USA) where the residues were built and energy minimized. The coordinates were then used to modify the standard entry for tyrosine and phenylalanine in the cyana residue library. Because the peptide is cyclized through an amide bond between the side chains of the first residue (Asp) and the last residue (Lys), a fixed distance constraint of 1.5 Å was set in between the NZ1 atom of the Lysine side chain and the CG atom of the Aspartic; the cyana ‘LYNK’ option was added to the sequence file as well. All colored Figures were generated with molmol (26).
Enzyme inhibition (fluorescence-based assay with DiFMUP)
The enzyme inhibition assay relies on the phosphatase-catalyzed hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMup; Invitrogen, Carlsbad, CA, USA) in the presence of a peptide at 25 °C (27,28). Enzyme inhibition was tested in a 96-well plate format with an assay volume of 100 μL and assay buffer: 30 mm Tris, 150 mm NaCl, 1 mM DTT, pH = 6.5. Peptides were dissolved in DMSO (5% final concentration). The protein (full-length GST-YopH) was used at a concentration of 3 nm. The concentration of substrate (DiFMup) was set at the Km value (50 μm).
Before the addition of the substrate, enzyme and peptides were pre-incubated for 10 min at room temperature. The initial reaction rate was measured using a fluorescence plate reader (VictorTM2 V; Perkin Elmer, Waltham, MA, USA) (excitation 358 nm, emission 455 nm). The non-enzymatic hydrolysis of the substrate was determined as control. The IC50 values were determined by plotting the percentage of inhibition versus the peptide concentration (logarithmic scale) using the software GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
Km evaluation (assay with the BIOMOL GREEN™ reagent)
The Michaelis-Menten (Km) constant for the Yoph-catalyzed dephosphorylation of the peptides DE(pY)DDPF, DE(pY)DDPfK and cyclo-DE(pY)DDPfK was determined by implementing an assay in a 96-well plate format and a reaction mixture volume of 50 μL. The reaction was started by addition of peptides (concentrations ranging from 125 to 0 μm) to full-length GST-Yoph at a concentration of 9 nm. Protein and peptides were incubated for 10 min at room temperature. After ten minutes, the reaction was quenched by the addition of 100 μL of BIOMOL GREEN reagent (Enzo Life Sciences International, Inc., Plymouth Meeting, PA, USA). Following further incubation for 30 min on a plate shaker for optimal color development, absorbance at 620 nm was measured with a Victor2 microplate reader (Perkin Elmer). The possible non-enzymatic hydrolysis of the peptides was evaluated by measuring the control (peptide in the absence of YopH).
The Michaelis and Menten constant (Km) was obtained by plotting the Absorbance [OD] as function of the peptide concentration [P] and fitting the data to the equation: [OD] = (Vmax*[P])/(Km + [P]) where Vmax is the maximum enzyme velocity, with Graph-Pad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
PAMPA assay
The parallel artificial membrane permeation assay (PAMPA) method (28,29) was implemented to estimate peptides' cell permeability. The permeation of a peptide through the artificial membrane layer is indicated by its log Pe value (See Table 1). The log Pe value of a given peptide was evaluated by keeping into account the concentration of the peptide in the acceptor compartment after 8 h and that of a reference well with the same concentration containing no membrane barrier.
Table 1.
Membrane permeability data for the peptide series DEXDDPfK (X = phosphotyrosine or non-hydrolyzable phosphotyrosine mimetic). Values of logPe for the known drugs warfarin and verapamil are also reported for comparison purpose
| Peptide | Log (Pe) |
|---|---|
| DE(pY)DDPF | −5.89 |
| Cyclo-DE(pY)DDPfK | −5.24 |
| Cyclo-DEYDDPfK | −5.58 |
| Cyclo-DE(pmp)DDPfK | −5.78 |
| Warfarin | −4.95 |
| Verapamil | −4.42 |
Results and Discussion
YopH phosphotyrosine phosphatase activity is essential for all Yersiniae infections. However, for efficient substrates dephosphorylation by the C-terminal catalytic domain, the YopH N-terminal domain is also needed (2), suggesting that this domain is in principle suitable for drug discovery against Yersinae. Linear peptides binding to the YopH N-terminal domain have already been reported and consist of the SKAP-HOM-derived peptide DE(pY)DDPF and the EGFR-derived peptide DADE(pY)L (12).
Cyclic peptides have generally several advantages over their linear counterparts and are attractive in drug discovery (30,31). In fact, cyclic peptides present usually a more constrained architecture accompanied by more favorable pharmacokinetic properties, such as metabolism and excretion, and often exhibit increased binding affinities (30). A few cyclic and linear peptide-based inhibitors of phosphatases, containing phosphotyrosine or phosphotyrosine mimetics, have already been described (32–36). For example, potent inhibitors of the phosphatase PTP1B have been designed based on the EGFR sequence DADE(pY)L (33); the same sequence has also been used to design linear tripeptide inhibitors of YopH (34).
To design novel YopH inhibitors, we have used a similar approach starting from the sequence DE(pY)DDPF from the SKAP-HOM protein. Molecular modeling techniques and an accurate analysis of the NMR structure of the complex in between the YopH N-terminal domain and this linear peptide (Figure 1B) (12) drove out our design. The solution structure of this complex shows that the peptide ligand binds in a shallow pocket at the protein edge and that although it preserves a certain degree of conformational flexibility even when it is in complex with the protein, its backbone has a tendency to assume a curved structure in many of the NMR conformers (Figure 1B) (12). Hence, we have first tried to reduce the conformational flexibility of the DE(pY)DDPF linear peptide by constraining it to assume a turn-like structure, and at this aim a d-phenylalanine residue has been introduced in the primary sequence as a replacement for the l-phenylalanine, allowing for a proper geometry for the cyclization; second, a lysine residue has also been added at the C-terminus to facilitate the synthetic chemistry efforts and thus making possible the cyclization simply through formation of an amide bond in between the side chains of the first and last amino acid (i.e.: ‘head-to-tail’ architecture). Finally, to avoid the presence of free charges, an acetyl group and an amide group have also been used at the N- and C-termini respectively. The first peptide that we have designed and investigated is the Cyclo-DE(pY)DDPfK.
Chemical shift perturbation studies (37,38) have been carried out by using 15N-labeled N-terminal domain of YopH (Figure 1) and the previously published chemical shifts for this protein (17). [1H, 15N] HSQC spectra have been recorded in the absence and presence of increasing amount of the Cyclo-DE(pY)DDPfK peptide. Upon addition of the peptide, the chemical shifts of several residues change appreciably (Figure 1A). Mapping of these changes in the 3D structure of the protein in complex with the SKAP-HOM linear peptide [pdb id: 1M0V, (12)] indicates that the cyclic peptide is still targeting the same binding site of the parent linear peptide (Figure 1B). Residues affected by binding are located in fact mainly around the C-terminal portion of the α1 helix, the strands β2 and β1, the α2 helix and the adjacent loop region (Figure 1B). By following the chemical shift changes as function of the peptide concentration for the residue Ala32 that is located in this putative binding site, we have obtained an estimation of the dissociation constant Kd (value of 15 ± 6 μm) (Figure 1C). The Kd value has been further confirmed by similar binding curves evaluated for other two residues in the putative binding pocket: Arg49 and Glu52 that have provided Kd values corresponding respectively to 15 ± 4 and 18 ± 4 μm.
The solution structure of this peptide has been studied by NMR spectroscopy (Figure 2). Comparison of TOCSY (23) and NOESY (22) spectra has allowed to assign proton resonances (Table S1). The structure of the peptide has been calculated with the program cyana (Figure 2), and the 10 conformers with the lowest value of the cyana target function (Figure 2, middle panel) have been further analyzed. The structures are not relatively well defined however still contain significant less-ordered substructures, indicating that actually the peptide is still relatively flexible in spite of the cyclization. This is also reflected by the presence of additional spin systems in the NOESY and TOCSY spectra, presumably indicating multiple conformations in solution. In all the calculated conformers, the proline was fixed in a peptide-bond trans conformation as it is clearly indicated by the presence of the strong NOEs between the Asp6 proton α and the protons δ and δ' of the proline, and evidence of cis-trans isomerization could be revealed in the spectra (24). Five of ten conformers present an H-bond in between the HN of Glu2 and the OD1 of Asp5 and between the HN of Phe7 and the backbone carbonyl oxygen O of Asp5. Next, we have investigated the ability of this cyclic peptide to be dephosphorylated by YopH by means of an in vitro colorimetric enzymatic assay. In this assay, the free orthophosphate, derived from dephosphorylation of the Cyclo-DE(pY)DDPfK peptide by the full-length GST-YopH, is revealed by formation of a green complex with the Malachite Green and molibdate reagents and subsequently quantified by measuring absorbance at 620 nm. This assay allows evaluating the Michaelis and Menten (Km) constant for the enzymatic reaction, thus getting an indication of the affinity of this peptide for YopH. The Km value has resulted 42 ± 4 μm and is close to those obtained for the parent linear peptides DE(pY)DDPF and DE(PY)DDPfK (Figure S1).
Figure 2.
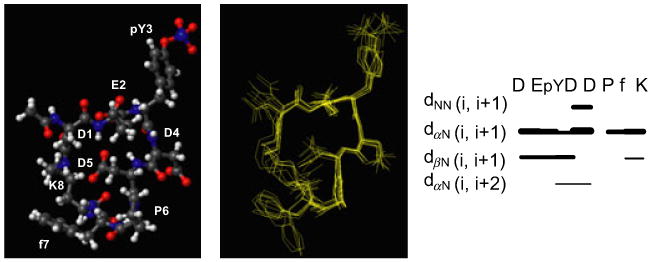
(Left panel) Ball and stick drawing of the first NMR conformer of the Cyclo-DE(pY)DDPfK peptide. (Middle panel) Overlay on the heavy atoms of ten conformers. RMSD = 1.01 Å. Structures were calculated by using 79 upper distance limits (37 intra, 29 short, 5 medium, 8 long range), and 29 angle constraints. The final structures show no violations of the experimental constraints. (Right panel) Pattern of short and medium range NOEs observed for the Cyclo-DE(pY)DDPfK peptide as generated with the program cyana (25). dxy(i, i + 1) indicates distances between protons x and y in the residues i and i + 1.
Based on these results, we have next replaced the phosphotyrosine in the Cyclo-DE(pY)DDPfK with a tyrosine (Y) and a phosphonomethyl-phenylalanine (pmp). The binding of the resulting peptides Cyclo-DEYDDPfK and Cyclo-DE(pmp)DDPfK to the N-terminal domain of YopH has been then investigated, as well as their abilities to inhibit the YopH phosphatase activity.
Chemical shift perturbation studies (Figure 3) indicate that the cyclic peptide containing the pmp moiety preserves some ability to bind the YopH N-terminal domain albeit with a rather reduced affinity with respect to the parent peptide containing a phosphotyrosine (Kd values higher than 300 μm). Chemical shift variations upon addition of this peptide to the N-terminal domain of YopH only affect a few residues located on the strands β1 and β2 and on the helix α1 thus indicating that the peptide is still targeting the same site (Figure 3). Similar NMR studies reveal that substitution of the phosphotyrosine with a tyrosine totally abolishes binding to the N-terminal domain of YopH and indicates the importance of the oxygen atoms of the phosphate groups in directing this interaction.
Figure 3.

(Left) Comparison of a region of the [1H-15N]-HSQC spectra of the 15N-labeled N-terminal domain of Yoph at a concentration of 70 μm in the absence (red) and presence of the Cyclo-DE(pmp)DDPfK peptide (1250 μm concentration) (blue). (Right) Ribbon representation of the Yoph N-terminal domain in complex with the DE(pY)DDPF peptide (NMR conformer number 1). The regions where most chemical shifts changes occur upon binding to the Cyclo-DE(pmp)DDPfK peptide are reported in red, the side chains of Lys26, Arg28 and the phosphotyrosine are shown as neon representation.
The ability of these two peptides (Cyclo-DEYDDPfK and Cyclo-DE(pmp)DDPfK) to inhibit YopH phosphatase activity has been monitored in an in vitro fluorescence-based assay (28,39) (see Methods and Materials); the two peptides are relatively weak YopH inhibitors (IC50 values around 100 μm) (see Figure S2).
We have also analyzed the membrane permeability of these peptides using the PAMPA method (Table 1 and Methods and Materials) (Table 1). Although all the peptides show lower membrane permeability with respect to the known drugs verapamil and warfarin (Table 1), cyclic peptides exhibit a significant improvement in membrane permeability compared with the parent linear peptide DE(pY)DDPF.
Next, in an attempt to make the peptides more similar to the EGFR sequence DE(pY)L, we have designed the Cyclo-DE(pY)LDPfK by the replacement of an aspartic acid residue with a leucine. We have performed chemical shift perturbation studies (Figure 4) that indicates that the peptide binds the YopH N-terminal domain in the usual site that is centered around the basic residues Lys26 and Arg28 (Figure 4A,B). The dissociation constant for the binding of this peptide to the N-terminal domain of YopH has been evaluated from the chemical shifts variations of the residue Ala32 and its value is 23 ± 4 μm. This value is comparable, within the experimental error, to that obtained for the analog Cyclo-DE(pY)DDPfK peptide (Figure 4C), thus indicating that substitution of an aspartic acid with a leucine residue does not greatly affect the binding affinity of the peptide for the YopH N-terminal domain. An analog of this peptide containing a non-hydrolyzable phosphotyrosine derivative (i.e.: Cyclo-DE(FCOOH)LDPfK in which FCOOH represents a phenylalanine with a carboxyl group in the para position) fails to bind the N-terminal domain of YopH and is a weak inhibitor of the C-terminal phosphatase activity (IC50 values higher than 100 μm) (Figure S2).
Figure 4.

(A) Chemical shift perturbation studies with the Cyclo-DE(pY)LDPfK peptide. The [1H, 15N] HSQC spectrum of the N-terminal domain of Yersinia outer protein H in its apo form (153 μm concentration, red) has been overlayed with the one recorded in the presence of the peptide (360 μm concentration, green). The residues with significant chemical shift variations are indicated with the single letter amino acid code and residue number. (C) The amino acids with significant chemical shift variations (reported in panel (A) with the one-letter code and residue numbers) have been colored in red. (B) Graph showing 1H normalized chemical shift deviations for the residue Alanine 32 as function of the peptide concentration. See text for details about Kd evaluation.
Conclusions
We have very recently described bi-dentate compounds as potent YopH inhibitors that target mainly the phosphatase catalytic C-terminal domain (28). In an attempt to obtain novel inhibitors of this protein tyrosine phosphatase from Yersinia, we have designed cyclic peptides targeting its N-terminal domain. These peptides are based on a short sequence derived from the protein SKAP-HOM [DE(pY)DDPF (pY = phosphotyrosine)] and the structural information previously gained by NMR studies (12). Among these peptides, those containing a phosphotyrosine moiety interact with the YopH N-terminal domain with a dissociation constant in the micromolar range. The structural and inhibition data gathered provide useful information for the design of more potent and selective peptide ligand of the YopH N-terminal domain that could be investigated for their ability to inhibit Yersiniae infections.
Supplementary Material
Figure S1. Km evaluation (assay with the BIOMOL GREEN™).
Figure S2. IC50 curves from the fluorescence-based phosphatase inhibition.
Table S1. Proton chemical shifts (ppm) of the Cyclo-DE(pY)DDPfK peptide evaluated at 300 K and 700 MHz.
Acknowledgments
Financial support was obtained thanks to NIH grants AI059572, AI070494, and AI081128 to MP.
Abbreviations
- Cas
CrK-associated substrate
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- EGFR
epidermal growth factor receptor
- PAMPA
parallel artificial membrane permeation assay
- PMP
phosphonomethyl-phenylalanine
- PTPase
protein tyrosine phosphatases
- YopH
Yersinia outer protein H
Footnotes
Supporting Information: Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Ivanov MI, Stuckey JA, Schubert HL, Saper MA, Bliska JB. Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol Microbiol. 2005;55:1346–1356. doi: 10.1111/j.1365-2958.2005.04477.x. [DOI] [PubMed] [Google Scholar]
- 2.Montagna LG, Ivanov MI, Bliska JB. Identification of residues in the N-terminal domain of the Yersinia tyrosine phosphatase that are critical for substrate recognition. J Biol Chem. 2001;276:5005–5011. doi: 10.1074/jbc.M009045200. [DOI] [PubMed] [Google Scholar]
- 3.Cornelis GR. Molecular and cell biology aspects of plague. Proc Natl Acad Sci U S A. 2000;97:8778–8783. doi: 10.1073/pnas.97.16.8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Persson C, Nordfelth R, Holmstrom A, Hakansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995;18:135–150. doi: 10.1111/j.1365-2958.1995.mmi_18010135.x. [DOI] [PubMed] [Google Scholar]
- 5.Cheng LW, Schneewind O. Yersinia enterocolitica TyeA, an intracellular regulator of the type III machinery, is required for specific targeting of YopE, YopH, YopM, and YopN into the cytosol of eukaryotic cells. J Bacteriol. 2000;182:3183–3190. doi: 10.1128/jb.182.11.3183-3190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black DS, Montagna LG, Zitsmann S, Bliska JB. Identification of an amino-terminal substrate-binding domain in the Yersinia tyrosine phosphatase that is required for efficient recognition of focal adhesion targets. Mol Microbiol. 1998;29:1263–1274. doi: 10.1046/j.1365-2958.1998.01014.x. [DOI] [PubMed] [Google Scholar]
- 7.Black DS, Bliska JB. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J. 1997;16:2730–2744. doi: 10.1093/emboj/16.10.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black DS, Marie-Cardine A, Schraven B, Bliska JB. The Yersinia tyrosine phosphatase YopH targets a novel adhesion-regulated signalling complex in macrophages. Cell Microbiol. 2000;2:401–414. doi: 10.1046/j.1462-5822.2000.00061.x. [DOI] [PubMed] [Google Scholar]
- 10.Smith CL, Khandelwal P, Keliikuli K, Zuiderweg ER, Saper MA. Structure of the type III secretion and substrate-binding domain of Yersinia YopH phosphatase. Mol Microbiol. 2001;42:967–979. doi: 10.1046/j.0950-382x.2001.02711.x. [DOI] [PubMed] [Google Scholar]
- 11.Evdokimov AG, Tropea JE, Routzahn KM, Copeland TD, Waugh DS. Structure of the N-terminal domain of Yersinia pestis YopH at 2.0 A resolution. Acta Crystallogr D Biol Crystallogr. 2001;57:793–799. doi: 10.1107/s0907444901004875. [DOI] [PubMed] [Google Scholar]
- 12.Khandelwal P, Keliikuli K, Smith CL, Saper MA, Zuiderweg ER. Solution structure and phosphopeptide binding to the N-terminal domain of Yersinia YopH: comparison with a crystal structure. Biochemistry. 2002;41:11425–11437. doi: 10.1021/bi026333l. [DOI] [PubMed] [Google Scholar]
- 13.Wattiau P, Woestyn S, Cornelis GR. Customized secretion chaperones in pathogenic bacteria. Mol Microbiol. 1996;20:255–262. doi: 10.1111/j.1365-2958.1996.tb02614.x. [DOI] [PubMed] [Google Scholar]
- 14.Phan J, Lee K, Cherry S, Tropea JE, Burke TR, Jr, Waugh DS. High-resolution structure of the Yersinia pestis protein tyrosine phosphatase YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide. Biochemistry. 2003;42:13113–13121. doi: 10.1021/bi030156m. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, Dixon JE. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 16.Tautz L, Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods. 2007;42:250–260. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Khandelwal P, Keliikuli K, Smit CL, Saper MA, Zuiderweg ER. 1H, 15N and 13C assignments of the N-terminal domain of Yersinia outer protein H in its apo form and in complex with a phosphotyrosine peptide. J Biomol NMR. 2001;21:69–70. doi: 10.1023/a:1011971202626. [DOI] [PubMed] [Google Scholar]
- 18.Alonso A, Bottini N, Bruckner S, Rahmouni S, Williams S, Schoenberger SP, et al. Lck dephosphorylation at Tyr-394 and inhibition of T cell antigen receptor signaling by Yersinia phosphatase YopH. J Biol Chem. 2004;279:4922–4928. doi: 10.1074/jbc.M308978200. [DOI] [PubMed] [Google Scholar]
- 19.Vazquez J, Tautz L, Ryan JJ, Vuori K, Mustelin T, Pellecchia M. Development of molecular probes for second-site screening and design of protein tyrosine phosphatase inhibitors. J Med Chem. 2007;50:2137–2143. doi: 10.1021/jm061481l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;5:11. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 21.Fielding L. NMR methods for the determination of protein-ligand dissociation constants. Curr Top Med Chem. 2003;3:39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 22.Kumar A, Ernst RR, Wuthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 23.Griesinger C, Otting G, Wüthrich K, Ernst RR. Clean Tocsy for H-1 spin system-identification in macromolecules. J Am Chem Soc. 1988;110:3. [Google Scholar]
- 24.Wuthrich K. NMR of Proteins and Nucleic Acids. New York, USA: John Wiley & Sons; 1986. [Google Scholar]
- 25.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 26.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 27.Tautz L, Bruckner S, Sareth S, Alonso A, Bogetz J, Bottini N, et al. Inhibition of Yersinia tyrosine phosphatase by furanyl salicylate compounds. J Biol Chem. 2005;280:9400–9408. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- 28.Leone M, Barile E, Vazquez J, Mei A, Guiney D, Pellecchia M. NMR-based design and evaluation of novel bidentate inhibitors of the protein tyrosine phosphatase YopH. Chem Biol Drug Des. 2010;76:10–16. doi: 10.1111/j.1747-0285.2010.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu B, Rega MF, Wei J, Yuan H, Dahl R, Zhang Z, et al. Discovery and binding studies on a series of novel Pin1 ligands. Chem Biol Drug Des. 2009;73:369–379. doi: 10.1111/j.1747-0285.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boddy CN. Sweetening cyclic peptide libraries. Chem Biol. 2004;11:1599–1600. doi: 10.1016/j.chembiol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 31.Horswill AR, Benkovic SJ. Cyclic peptides, a chemical genetics tool for biologists. Cell Cycle. 2005;4:552–555. doi: 10.4161/cc.4.4.1585. [DOI] [PubMed] [Google Scholar]
- 32.Combs AP, Yue EW, Bower M, Ala PJ, Wayland B, Douty B, et al. Structure-based design and discovery of protein tyrosine phosphatase inhibitors incorporating novel isothiazolidinone heterocyclic phosphotyrosine mimetics. J Med Chem. 2005;48:6544–6548. doi: 10.1021/jm0504555. [DOI] [PubMed] [Google Scholar]
- 33.Akamatsu M, Roller PP, Chen L, Zhang ZY, Ye B, Burke TR., Jr Potent inhibition of protein-tyrosine phosphatase by phosphotyrosine-mimic containing cyclic peptides. Bioorg Med Chem. 1997;5:157–163. doi: 10.1016/s0968-0896(96)00195-2. [DOI] [PubMed] [Google Scholar]
- 34.Lee K, Gao Y, Yao ZJ, Phan J, Wu L, Liang J, et al. Tripeptide inhibitors of Yersinia protein-tyrosine phosphatase. Bioorg Med Chem Lett. 2003;13:2577–2581. doi: 10.1016/s0960-894x(03)00481-5. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Durell SR, Feng H, Bai Y, Anderson CW, Appella E. Development of a substrate-based cyclic phosphopeptide inhibitor of protein phosphatase 2Cdelta, Wip1. Biochemistry. 2006;45:13193–13202. doi: 10.1021/bi061356b. [DOI] [PubMed] [Google Scholar]
- 36.Combs AP. Structure-based drug design of new leads for phosphatase research. IDrugs. 2007;10:112–115. [PubMed] [Google Scholar]
- 37.Pellecchia M, Sem DS, Wuthrich K. NMR in drug discovery. Nat Rev Drug Discov. 2002;1:211–219. doi: 10.1038/nrd748. [DOI] [PubMed] [Google Scholar]
- 38.Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, et al. Perspectives on NMR in drug discovery: a technique comes of age. Nat Rev Drug Discov. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, Vazquez-Rodriguez J, et al. Multidentate small-molecule inhibitors of vaccinia H1-related (VHR) phosphatase decrease proliferation of cervix cancer cells. J Med Chem. 2009;52:6716–6723. doi: 10.1021/jm901016k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Km evaluation (assay with the BIOMOL GREEN™).
Figure S2. IC50 curves from the fluorescence-based phosphatase inhibition.
Table S1. Proton chemical shifts (ppm) of the Cyclo-DE(pY)DDPfK peptide evaluated at 300 K and 700 MHz.