

Abstract
We present a protocol for in vivo imaging of cortical tissue using a deep-brain imaging probe in the shape of a microprism. Microprisms are 1-mm in size and have a reflective coating on the hypotenuse to allow internal reflection of excitation and emission light. The microprism probe simultaneously images multiple cortical layers with a perspective typically seen only in slice preparations. Images are collected with a large field-of-view (~900 μm). In addition, we provide details on the non-survival surgical procedure and microscope setup. Representative results include images of layer V pyramidal neurons from Thy-1 YFP-H mice showing their apical dendrites extending through the superficial cortical layer and extending into tufts. Resolution was sufficient to image dendritic spines near the soma of layer V neurons. A tail-vein injection of fluorescent dye reveals the intricate network of blood vessels in the cortex. Line-scanning of red blood cells (RBCs) flowing through the capillaries reveals RBC velocity and flux rates can be obtained. This novel microprism probe is an elegant, yet powerful new method of visualizing deep cellular structures and cortical function in vivo.
Protocol
Part 1: Microprism Optics and Microscope Setup
The microprisms we use are 1-mm, right-angle prisms made from BK7 glass purchased from the Optosigma Corporation (Figure 1). Microprisms are handled using forceps whenever possible.
The hypotenuse of the microprism is coated with enhanced silver to provide a reflectivity of greater than 97% from 400 – 2000 nm. This provides internal reflection of both the laser excitation light and the emitted fluorescence.
Microprisms are always handled using forceps when possible. Gloves are worn at all times to keep the prism clean.
We use a custom-built two-photon microscope capable of small-animal imaging. The animal, attached to the stereotactic device, is placed on a 3-axis motorized stage.
The microscope is connected to a Windows PC running ScanImage software. ScanImage is image acquisition software written in Matlab by Pologruto et al1.
For microprism imaging, we collect images at a resolution of 1024 x 1024 or 512 x 512 pixels. In addition, we typically scan at 2 ms/line or 4 ms/line.
- Two types of objectives are used in the experiments:
- A lower numerical aperture (NA) air objective for wide field-of-view imaging (see Table of Reagents and Equipment).
- A higher NA air objective with a glass correction collar capable of minimizing spherical aberrations induced by 0 – 2 mm of glass (see Table of Reagents and Equipment).
When the animal is ready for imaging under a low magnification objective, align the square raster scanning pattern of the laser with the top of the microprism. This will help orient the image and make efficient use of the laser power.
Using a higher NA objective with a glass correction collar, set the correction collar to correct for approximately 1 mm of glass. Once a fluorescent image appears during the experiment, one can carefully reach inside the microscope to optimize the correction collar to maximize the imaging resolution.
Part 2: Non-survival Animal Surgery and Microprism Insertion
The entire procedure length, from the initial incision until completion of imaging, depends on the objectives of the experiment. However, the non-survival surgical procedure and microprism placement can be expected to take approximately 30 – 40 minutes to complete.
Mice (P30-P60) are weighed and anesthetized appropriately using an I.P. injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) to a plane suitable for surgical procedures.
Throughout the entire experiment, the animal should be checked every 15-20 minutes to assess its level of anesthesia. If the animal reacts to a firm toe-pinch, a supplemental dose of ketamine/xylazine is required. Typically a supplemental dose is one-third of the initial dose and can be prepared ahead of time in a syringe for immediate I.P. injection if needed.
Once the mouse is properly anesthetized, the majority of the hair on the animal’s head is shaved off using a trimmer with a #40 blade.
Nair® is applied to the remaining hairs on the head to help create a surface free of hairs that may get in the way of the microprism during imaging. After 5 minutes, the Nair® is cleaned off using a cotton-tipped swab.
The anesthetized animal is properly placed into a mouse stereotactic device. During the surgical procedures and imaging sessions the animal is kept on a water-filled heating pad set at 36 degrees Celsius.
With the animal’s head properly fixed in place, one clean incision is made using a scalpel down the medial line of the skull to separate the skin.
A small amount of 3% hydrogen peroxide is placed on a cotton-tipped swab and applied to the skull to clear away membranes between the skull and the skin.
The hydrogen peroxide also helps to reveal bregma, a major landmark in knowing where to perform the craniotomy and insert the microprism.
Before the craniotomy is started, the ear bars and bite bar are properly adjusted to tilt the exposed skull such that it is basically level with the table. This provides a better platform for the craniotomy. In addition, this makes the top surface of the prism perpendicular with the incoming excitation light. This will help minimize optical aberrations while imaging.
A small dental burr (~0.8 mm head size) is connected to a Dremel® tool using a flexible extension arm. For craniotomies, we set the speed to approximately 7,000 to 10,000 rpms.
The dental burr is skimmed along the skull until a square groove is established in the bone. The sides of the square are approximately 2-3 mm in length and centered 1.5 mm caudal and 1 mm lateral to bregma.
Continuing thinning the bone until a small opening is made through the skull. A pair of forceps can lift the bone flap away from the skull.
The dura needs to be removed before the microprism can be inserted into the cortex. Bleeding is best controlled by letting the blood coagulate on the surface for several minutes. Afterwards, the coagulated blood is removed using a surgical sponge before inserting the microprism.
To help assist the alignment of the prism with the cortex, the vertical face of the microprism is lined up parallel to the handles on the forceps.
With the prism properly held, the entire microprism is inserted until the top of the prism is flush with the neocortical surface.
When inserted correctly, the microprism should remain in the proper position. Any bleeding that results is minimal and can be absorbed with a small surgical sponge. Once the prism is in place the animal is ready for imaging.
After a microprism has been used in an animal, it can be re-used several times. However, it does need to be properly cleaned to remove any remaining biological material. This can be accomplished by dipping the prism into a small volume of HCl acid, followed by a dip into methanol. The cleaned microprism can then be wrapped in lens paper until future use.
Part 3: Representative Results
 Figure 1: Images of the right-angle microprism. Prisms used in this experiment are one-millimeter, BK7 glass prisms with an enhanced silver coating on the hypotenuse to allow internal reflection of excitation and emission light.
Figure 1: Images of the right-angle microprism. Prisms used in this experiment are one-millimeter, BK7 glass prisms with an enhanced silver coating on the hypotenuse to allow internal reflection of excitation and emission light.
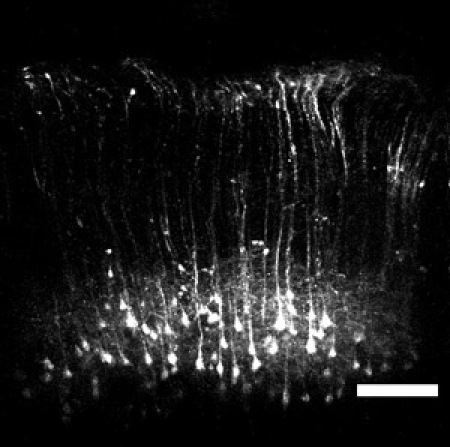 Figure 2: Image of layer V YFP pyramidal neurons. Wide field-of-view imaging with the microprism reveals a large population of pyramidal cell bodies ~800 – 900 μm deep from the cortical surface. In addition, apical dendrites extending up towards layer I can also be resolved. Scale bar = 200 μm.
Figure 2: Image of layer V YFP pyramidal neurons. Wide field-of-view imaging with the microprism reveals a large population of pyramidal cell bodies ~800 – 900 μm deep from the cortical surface. In addition, apical dendrites extending up towards layer I can also be resolved. Scale bar = 200 μm.
 Figure 3: A higher numerical aperture objective used in conjunction with the microprism provides spatial resolution sufficient to resolve dendritic spines on the apical dendrites on layer V pyramidal neurons. Scale bar = 10 μm.
Figure 3: A higher numerical aperture objective used in conjunction with the microprism provides spatial resolution sufficient to resolve dendritic spines on the apical dendrites on layer V pyramidal neurons. Scale bar = 10 μm.
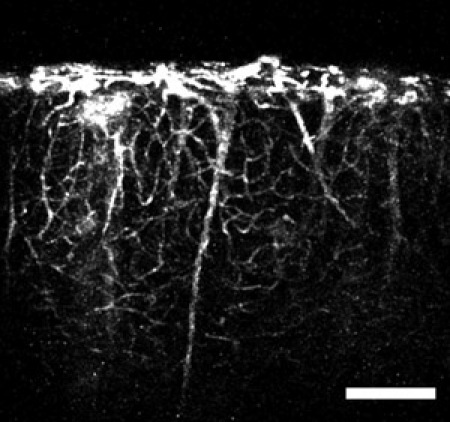 Figure 4: An image obtained using the microprism and a tail-vein injection of fluorescent dye. The image shows vertical, large caliber vessels extending from deep brain regions and smaller capillaries branching off through the cortical volume. Scale bar = 200 μm.
Figure 4: An image obtained using the microprism and a tail-vein injection of fluorescent dye. The image shows vertical, large caliber vessels extending from deep brain regions and smaller capillaries branching off through the cortical volume. Scale bar = 200 μm.
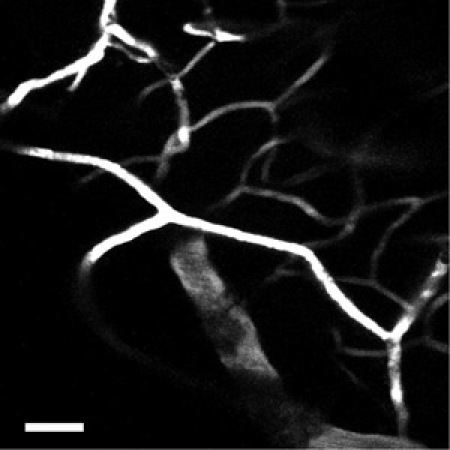 Figure 5: Using a higher numerical aperture objective, the network of capillaries in the neocortex can be imaged with greater detail. Scale bar = 50 μm.
Figure 5: Using a higher numerical aperture objective, the network of capillaries in the neocortex can be imaged with greater detail. Scale bar = 50 μm.
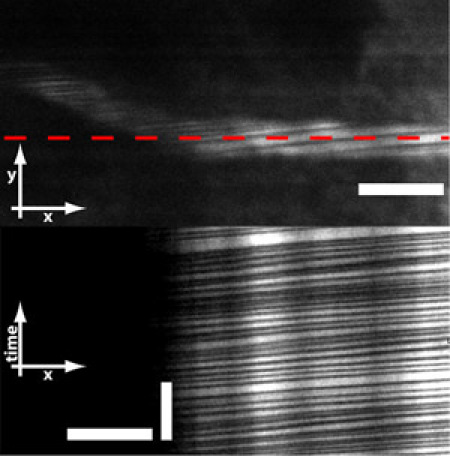 Figure 6: Microprisms also allows for imaging red blood cells (RBCs) flowing through vessels. RBCs do not absorb the fluorescent dye and therefore are seen as dark stripes across the vessel. By line-scanning across the vessel, one can obtain measurements of RBC flux and velocity. Scale bar = 10 μm (horizontal bar in top and bottom image) and 250 ms (vertical bar in bottom image).
Figure 6: Microprisms also allows for imaging red blood cells (RBCs) flowing through vessels. RBCs do not absorb the fluorescent dye and therefore are seen as dark stripes across the vessel. By line-scanning across the vessel, one can obtain measurements of RBC flux and velocity. Scale bar = 10 μm (horizontal bar in top and bottom image) and 250 ms (vertical bar in bottom image).
Discussion
Using a microprism in an imaging experiment is relatively straightforward and offers many advantages for in vivo studies on the neocortex. Representative examples using this technique include images taken from transgenic mice expressing yellow fluorescent protein in layer V cortical neurons. Using microprisms one can see a collection of large pyramidal cell bodies in layer V, nearly 1 mm below the cortical surface. Also seen are the apical dendrites that extend through all the superficial layers before diverging into tufts (Figure 2). The mice used for these experiments are transgenic animals designed to express YFP only in layer V neurons (YFP-H2). Using these YFP-H mice and a higher numerical aperture objective with a glass correction collar it is also possible to image the dendrites with greater detail and resolve dendritic spines (Figure 3).
One can also label the cortical blood vessels with a fluorescent dye such as fluorescein-dextran using established tail-vein injection techniques. Typically, for adult mice a 100 ul bolus injection of fluorescein-dextran at 20 mg/ml in physiological saline is appropriate. The best results come from injecting the dye after the microprism has been inserted into the neocortex (after step 2.16). Using this technique one can see the larger caliber vessels from deep layers extending towards the pia and branching off to form the network of microcapillaries (Figure 4). This intricate network of microcapillaries in the neocortex is best appreciated using a high numerical aperture objective (Figure 5).
In addition, it is possible to measure red blood cell velocity and flux from deep neocortical capillaries by line-scanning along the length of a vessel (Figure 6, red line in top image). Since red blood cells do not take up the fluorescent dye, they will appear as dark streaks. Images that result from a line-scan have a spatial dimension in one axes and a temporal dimension in the other axes (Figure 6, bottom image). From these images, one can calculate the flux and velocity of red blood cells through the capillary. In the case of figure 6, the RBC flux was measured to be 36 RBCs/second with a velocity of 0.45 mm/second. This is in agreement with results from the literature3.
Acknowledgments
We thank Anthony J. Koleske, PhD for providing the YFP mice.
References
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13–13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–41. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci U S A. 1998;95(26):15741–15741. doi: 10.1073/pnas.95.26.15741. [DOI] [PMC free article] [PubMed] [Google Scholar]