

Abstract
The protein transduction technique enables the direct delivery of biologically active material into mammalian cells [for review see 1,2]. For this one can make use of the translocating ability of so-called cell penetrating peptides (CPPs), also designated as protein transduction domains (PTDs). The TAT-CPP derived from the human immunodeficiency virus type 1 (HIV-1) Tat (trans-activator of transcription) protein has been widely used. The positively charged TAT promotes cell permeability thereby overcoming the barriers of the cellular membrane by endocytosis or/and direct membrane penetration2. In combination with a nuclear localization signal (NLS) fusion proteins are able to enter the nucleus exhibiting functionality. Our video presentation demonstrates, as an exemplification for the engineering of cell-permeable proteins, the construction, production and application of a cell-permeable version of the DNA-modifying enzyme Cre.
Cre is a site-specific recombinase that is able to recognize and recombine 34 base pair loxP sites in mammalian cells in vitro and in vivo. Therefore the Cre/loxP system is widely used to conditionally induce mutations in the genome of living cells3,4. The delivery of active Cre recombinase to cells, however, represents a limitation.
We describe the pSESAME vector system, which allows a direct insertion of the gene-of-interest and provides a platform to rapidly clone different domains and tags used within the vector in a convenient and standardized manner. Rearranging of the different tags has been shown to modify the biochemical properties of the fusion proteins providing a possibility to achieve higher yield and better solubility. We demonstrate how to express and purify recombinant cell-permeant proteins in and from E. coli. The functionality of the recombinant Cre protein is finally validated in cell culture by assessing its intracellular recombinase activity.
Keywords: Cellular Biology, Issue 34, Protein transduction, Cell penetrating peptide, Site-specific recombination, Stem cells, Protein purification
Protocol
Construction of expression vector and expression:
The pSESAME-Cre expression vector was constructed by inserting a Cre-encoding fragment into pSESAME via AvrII and NheI restriction sites using standard cloning methods. pSESAME encodes a fusion protein consisting of a histidine-tag, TAT-domain, NLS sequence and Cre, abbreviated HTNCre. For expression of HTNCre the pSESAME-Cre was transformed into TUNER (DE3) pLacI and used to prepare a glycerol stock.
An over-night culture was inoculated using a pipet tip coated with transformed bacteria from the glycerol stock. The over-night culture consisted of LB media supplemented with 0.5% glucose [v/v] and carbenicillin at a final concentration of 50 μg/mL and was allowed to grow at 37°C for 16 hours.
Next day the densely grown over-night culture was used to inoculate the expression culture at a ratio of 1 to 40 and was put in an incubator at 37°C. Expression culture consisted of TB media supplemented with 0.5% glucose [v/v] and ampicillin at a final concentration of 100 μg/mL.
At an OD595 of 1.5 the expression culture was induced with 0.5 mM IPTG for 1 h.
Subsequently bacteria were collected by centrifugation at 5000 rpm for 10 minutes in a SLA3000 rotor.
Bacteria pellets were stored at minus 20°C until purification.
Purification of cell-permeable protein:
Frozen bacteria pellets were resuspended in 10 mL lysis buffer per liter flask culture for 15 minutes at room temperature.
Suspension was then incubated with 1 mg/mL lysozyme for additional 15 minutes while mixing at room temperature.
25 U/mL benzonase was added afterwards and incubated while mixing for 15 minutes at room temperature.
After sonification on ice for 1.5 min with 0.5 s pulses at 45% of the power, 1 mL cold tartaric salt buffer (TSB) per mL suspension was carefully added while mixing and incubated for 5 min on ice. SDS-PAGE sample of lysate fraction (L) was taken.
Cleared lysate was obtained by centrifugation at 4°C for 30 min at 30,000g. SDS-PAGE samples of soluble (S) and insoluble fractions (I) were taken.
The supernatant was transferred into fresh 50 mL falcon tubes and was then gently mixed for 1 h at 4°C with 2 mL of 50% Ni-NTA slurry per liter of initial expression culture.
The suspension was packed into a gravity flow EconoPac column (SDS-PAGE sample of flow-through fraction (FT) was taken) and washed twice with 5 bed-volumes of washing buffer. SDS-PAGE samples of both washing fractions (W1 & W2) were collected.
HTNCre-containing fractions were eluted with 3 bed-volumes of elution buffer and sample of eluate fraction (E) for SDS-PAGE analysis was taken.
Imidazole was removed by dialyzing elution fraction against high salt buffer twice.
The protein solution was further concentrated by dialyzing against glycerol buffer twice. In all dialysis steps the ratio of buffer to sample was at least 50. This procedure resulted in a glycerol stock solution containing HTNCre at a usual concentration between 200 and 450 μM, i.e. 1 liter of expression culture will result in ~12 mg of protein. Sample of glycerol stock (GS) for SDS-PAGE analysis was collected. HTNCre stock solution can be stored at minus 20°C.
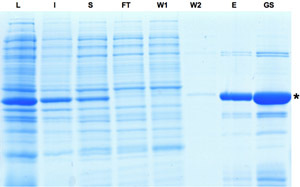 Figure 1: SDS-PAGE analysis of the samples collected during the purification process of Cre recombinase. Induction of Cre expression is indicated by dominant band in lysate fraction. Although a part of the protein is insoluble the Cre protein can be further enriched as seen in eluate and glycerol stock fractions. L: Lysate, I: Insoluble, S: Supernatant, FT: Flow-through, W: Washing, E: Eluate, GS: Gylcerol Stock. Please click here to see a larger version of figure 1.
Figure 1: SDS-PAGE analysis of the samples collected during the purification process of Cre recombinase. Induction of Cre expression is indicated by dominant band in lysate fraction. Although a part of the protein is insoluble the Cre protein can be further enriched as seen in eluate and glycerol stock fractions. L: Lysate, I: Insoluble, S: Supernatant, FT: Flow-through, W: Washing, E: Eluate, GS: Gylcerol Stock. Please click here to see a larger version of figure 1.
Protein transduction into murine embryonic stem (ES) cells:
ES cells carrying a conditional β-Galactosidase reporter5 construct were seeded as single cells using TrypLE™ Express for dissociation of adherent cells. After 4 to 6 hours the cells had re-attached and the medium was removed.
- Subsequently ES cells were incubated with HTNCre-containing medium for 16 hours.
- An appropriate amount of HTNCre protein (corresponding to 10 μM) out of the glycerol stock was diluted into ES medium and subsequently sterile filtrated (0.22 μm).
After protein transduction medium was changed back to normal growth medium.
After two days cells were washed with PBS and fixed with 4% Paraformaldehyde (PFA) for 10 minutes.
Two additional washing steps with PBS were executed before X-Gal staining was performed.
Fixed cells were covered with a layer of X-Gal staining solution6 and incubated over night at 37°C.
Representative Results:
Next day X-Gal staining solution was aspired and the cells were covered with a layer of PBS for microscopy analysis. 80 to 100% of recombined cells could be observed within the murine ES cells judged by β-Galactosidase activity.
Discussion
During the purification process of the Cre fusion protein it is important not to omit the addition of ice cold TBS buffer prior to centrifugation. Otherwise Cre recombinase tends to precipitate within the glycerol buffer.
If the eluate fraction appears to become turbid due to the high concentration of fusion protein additional elution buffer should be added until the solution has cleared again.
The application of 10 μM of Cre fusion protein typically results in a recombination efficiency of 80 to 100%. Fetal Calf Serum (FCS) being a major component of ES cell medium strongly inhibits protein transduction. Therefore high concentration of Cre recombinase had to be used. When working in serum-free conditions less protein (0.5 - 2 μM) can be used to achieve similar recombination efficiencies.
With the pSESAME vector system at hand one can apply the technique of protein transduction to other proteins including transcription factors such as Oct4 and Sox27 and Scl/Tal18.
Acknowledgments
We thank Oliver Brüstle and all members of the Stem Cell Engineering Group, University of Bonn, for support and valuable discussions. We thank Sabine Schenk for preparation of the SDS-PAGE and enduring support throughout the project. Nicole Russ and Anna Magerhans provided excellent technical support. Furthermore, we would like to thank Andreas Bär and Sheila Mertens for the production of the movie. This work was supported by grants from the Volkswagen Foundation (Az I/77864) and the German Ministry of Education and Research (BMBF, 01 GN 0813).
References
- Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med. 2007;13(10):443–448. doi: 10.1016/j.molmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Edenhofer F. Protein transduction revisited: novel insights into the mechanism underlying intracellular delivery of proteins. Curr Pharm Des. 2008;14(34):3628–3636. doi: 10.2174/138161208786898833. [DOI] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6(1):7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Nolden L. Site-specific recombination in human embryonic stem cells induced by cell-permeant Cre recombinase. Nat Methods. 2006;3(6):461–467. doi: 10.1038/nmeth884. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res. 1996;24(4):543–548. doi: 10.1093/nar/24.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitz M. Enhanced purification of cell-permeant Cre and germline transmission after transduction into mouse embryonic stem cells. Genesis. 2007;45(8):508–517. doi: 10.1002/dvg.20321. [DOI] [PubMed] [Google Scholar]
- Bosnali M, Edenhofer F. Generation of transducible versions of transcription factors Oct4 and Sox2. Biol Chem. 2008;389(7):851–861. doi: 10.1515/BC.2008.106. [DOI] [PubMed] [Google Scholar]
- Landry JR. Runx genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood. 2008;111(6):3005–3014. doi: 10.1182/blood-2007-07-098830. [DOI] [PubMed] [Google Scholar]