

SUMMARY
Many molecules that could manipulate cellular function are not practical due to their large size and concomitant undesirable pharmocokinetic properties. Here we describe a bioorthogonal, highly stable boronate ester (HiSBE) synthesis and use this reaction to synthesize a bioogically active molecule from smaller precursors in a physiological context. The rapid rate of HiSBE synthesis suggests that it may be useful for assembling a wide variety of biologically active molecules in physiological solutions.
INTRODUCTION
Synthetic reactions are typically performed using catalysts, temperatures, and organic solvents that are not compatible with living cells. However, certain chemical reactions can occur in biological solutions and physiological conditions, and are not affected by the complex mixture of small molecules and proteins that are characteristic of biological solutions. These reactions are referred to as "bioorthogonal," and provide the opportunity to probe cellular function with organic synthesis (Prescher and Bertozzi, 2005). Bioorthogonal reaction strategies involve pre-synthesized non-native chemical moieties that typically perform the final synthetic step in the cellular environment.
Certain reactions have been shown to exhibit bioorthogonality, such as copper-free “click” chemistry (Baskin et al., 2007), and the Staudinger ligation (Saxon and Bertozzi, 2000). These reactions are distinct from bioconjugation reactions, such as reactions of labeling agents with cysteine or tyrosine (Joshi et al., 2004; Sletten and Bertozzi, 2009), or the formation of oximes by reaction of aminoxy moieties with aldehydes or ketones (Sletten and Bertozzi, 2009), since the non-native chemical moieties involved in bioorthogonal reactions are non-perturbing to chemical functionalities found in living systems and demonstrate high selectivity (Prescher and Bertozzi, 2005; Sletten and Bertozzi, 2009). For example, copper-free click chemistry involves the reaction of an azide and an alkyne to form a triazole in a [3 + 2] cycloaddition (Baskin et al., 2007). The reactivity of the alkyne is promoted by ring strain and can be enhanced by halogen substitution, resulting in rapid reaction rates and an elimination of the need for copper, which is otherwise required for this class of click reactions (Baskin et al., 2007; Codelli et al., 2008). Bioorthogonal reactions have been used to develop chemical reporters for labeling and visualizing biomolecules in living cells (Prescher and Bertozzi, 2005). In these reactions, bioorthogonal functional groups are metabolically incorporated into biomolecules, and then probes that contain reactive moieties are covalently coupled by a spontaneous bioorthogonal reaction.
Current bioorthogonal reactions have limitations that affect their use. In the case of the Staudinger ligation, slow reaction rates and oxidation of reactants affect its usefulness (Lin et al., 2005). Although the copper-free click reaction exhibits significantly higher reaction kinetics (9.0 × 10−2 M−1 s−1) (Baskin et al., 2007; Codelli et al., 2008), the reaction rates are still slow relative to the time scales used for biological experiments, and the availablility of additional bioorthogonal reactions would provide additional versatility for chemical reactions in living cells.
Because of the limitations of existing bioorthogonal reactions, we sought to identify whether other previously described reactions could be used for bioorthogonal chemistry. Phenylboronic acid (PBA) interacts with D-glucose to form boronate esters that are readily hydrolyzed in water (Ferrier et al., 1965). However, recently Stolowitz and colleagues showed that PBA interacts with salicylhydoxamic acid (SHA) to form considerably more stable boronate esters (Stolowitz et al., 2001). The bioconjugate reactions of PBA and SHA have been used to purify biomolecules such as proteins and nucleic acids modified with SHA residues (Stolowitz et al., 2001). Unlike D-glucose, which interacts with boronic acid through vicinal diols (Ferrier et al., 1965), SHA interacts with boronic acid through both an oxygen and a nitrogen, which may account for the increased stability (Stolowitz et al., 2001). The boron-nitrogen bond has both ionic and covalent character (Plumley and Evanseck, 2007). These boronate esters exhibit pH-dependent hydrolysis, with instability at low pHs (pH < 5) (Stolowitz et al., 2001). Based on the increased resistance of these boronate esters to hydrolysis at neutral pHs, we refer to this reaction as highly stable boronate ester (HiSBE) synthesis. Due to its success in bioconjugation reactions, we considered the possibility that HiSBE chemistry would exhibit bioorthogonality.
RESULTS AND DISCUSSION
HiSBE synthesis between SHA and PBA
We first asked if the HiSBE reaction was affected by biological nucleophiles. To measure boronate ester formation between SHA with PBA, we incubated SHA with PBA immobilized on agarose at pH 7.4. After incubation, the complexed SHA was eluted with low pH buffer (pH 4.5), which hydrolyzes SHA-PBA complexes (Stolowitz et al., 2001). Eluted SHA was quantified by absorption spectrophotometry. A linear increase in boronate ester formation was observed during the first 1 min of incubation, with saturation observed after 5 min (Fig. 1a). To determine the effect of common biological nucleophiles such as serine, cysteine or lysine, as well as D-glucose, on HiSBE synthesis, we measured the effect of these compounds on boronate ester synthesis measured at 5 min. None of these treatments significantly reduced the ester formation (Fig. 1b). Similarly, tissue culture media, which contains diverse small molecule cofactors, amino acids, and ions found in biological solutions, did not affect the interaction of PBA and SHA (Fig. 1b). Together, these data indicate that the reaction of PBA and SHA is not affected by molecules commonly found in biological solutions.
Figure 1. HiSBE synthesis is facile and orthogonal to common biological nucleophiles.
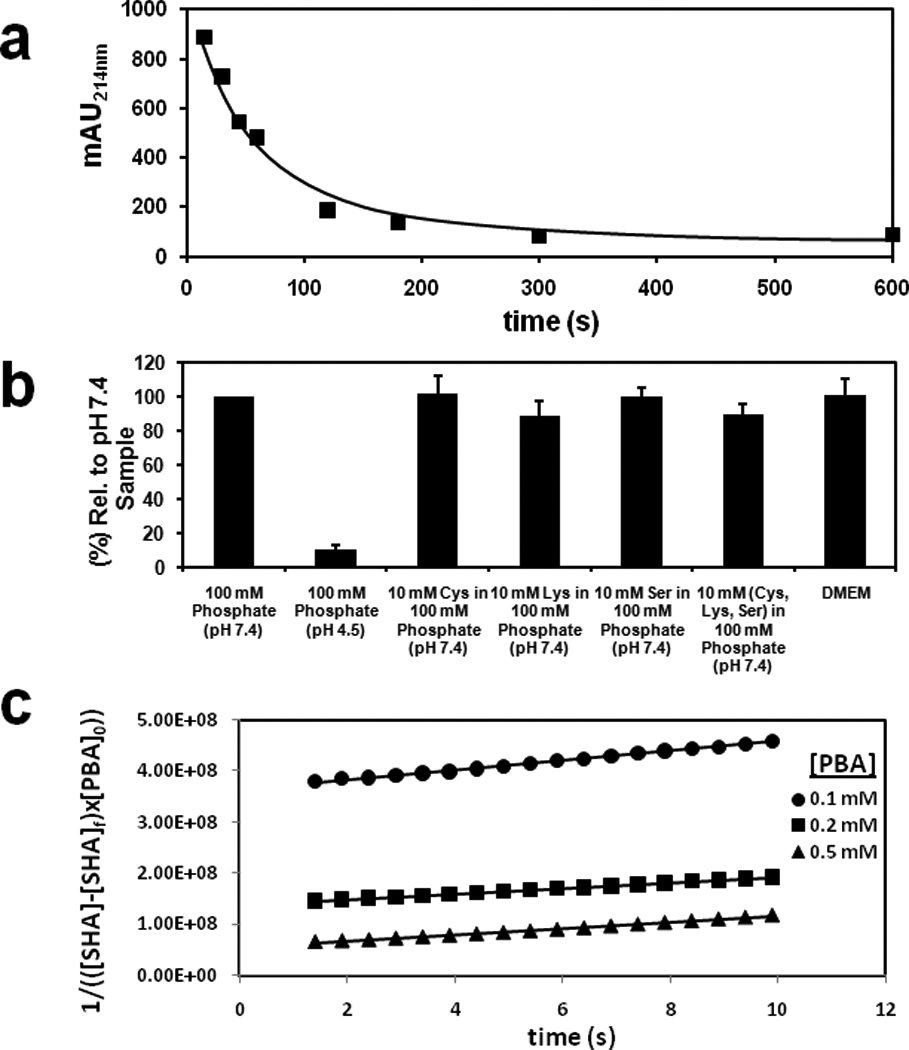
(a) Time dependent boronate ester formation between SHA and PBA immobilized on agarose in 100 mM phosphate buffer at pH 7.4. Reaction solutions containing 6 mM SHA (0.0036 mmoles, 0.6 mL) were incubated with PBA agarose (0.1 ml, 0.002 mmole of immobilized PBA). The SHA concentration in the supernatant was quantified by absorption spectrophotometry at 214 nm.
(b) The efficiency of HiSBE synthesis in the presence of common biological nucleophiles. Reaction solutions containing 6 mM SHA (0.0036 mmoles, 0.6 mL) in 6% DMEM/100 mM phosphate buffer at the indicated pH and with the indicated nucleophiles were incubated with PBA agarose (0.1 ml, 0.002 mmole of immobilized PBA) for 5 min. After incubation, the resin was washed to remove uncomplexed SHA, and the complexed SHA was eluted with 100 mM phosphate buffer at pH 4.5. Eluted SHA was quantified by absorption spectrophotometry at 214 nm.
(c) Kinetic measurement of 0.1 mM SHA with 0.1 mM, 0.2 mM and 0.5 mM PBA in 100 mM phosphate buffer at pH 7.4. The reaction rates were modeled after third-order reaction kinetics and were determined by plotting 1/(([SHA]-[SHA]f) × [PBA]0)) versus time. The slopes were used to approximate the rate constants. k = (7.01 ± 2.04) × 106 M−2s−1.
We measured the association constants of PBA and SHA. The coordination state of the boron nucleus changes from trigonal to tertrahedral after complexation with SHA, which is detected by the chemical shift of boron in 11B-NMR (Beachell and Beistel, 1964; Stolowitz et al., 2001). To monitor association, we incubated 10 mM PBA with varying concentrations of SHA in phosphate buffer. The observed association constant was 17,800 M−1 at pH 7.4 and 4 M−1 at pH 4.5, indicating that the complexed form is highly favored at physiological pH. To measure the association rate constant, we used a UV spectrophotometry to measure the reaction of SHA with PBA with respect to time. Based on the linear relationship demonstrated in rate versus [SHA]2[PBA] (Fig. S1), the reaction of SHA and PBA was modeled with a third order rate constant. The plot of 1/([SHA][PBA]0) versus time was used to determine the kinetic rate of (7.01±2.04) × 106 M−2 s−1 (Fig. 1c). These data indicate that this reaction occurs considerably faster than previously described bioorthogonal chemical reactions.
Assembly of dimeric c-Mpl via HiSBE
We next wanted to determine if boronate ester synthesis can occur in a physiologically relevant setting. To test the bioorthogonality of this reaction, we sought to generate biologically active molecules in situ using HiSBE synthesis. Numerous receptors are activated by ligands which bind and dimerize their cognate receptors (Heldin, 1995). In the case of the thrombopoeitin (TPO) receptor c-Mpl, receptor dimerization and activation is triggered by binding TPO (Cwirla et al., 1997). Peptide ligands to c-Mpl, such as IEGPTLRQWLAARA, have been identified, but due to their monomeric nature exhibit only weak agonist activity (Cwirla et al., 1997). However, dimeric forms of the peptide act as agonists of the receptor. In one case, a c-Mpl-activating pseudosymmetric dimer was prepared on a lysine scaffold: the COOH-terminus of one IEGPTLRQWLAARA was coupled to the ε-amine of the lysine, while the COOH-terminus of another IEGPTLRQWLAARA was coupled to the α-amine of the lysine via a linker, resulting in a potent c-Mpl agonist (Cwirla et al., 1997).
To assess the bioorthogonality of HiSBE synthesis, we decided to determine if a dimeric c-Mpl agonist could be synthesized in situ, and if the synthesized compound could lead to the activation of c-Mpl signaling in cells. Rather than using a lysine linker, we decided to use a "dimerizer" comprising two SHA moieties connected by a linker (Fig. 2a). This dimerizer is expected to generate a dimeric c-Mpl ligand by interacting with two IEGPTLRQWLAARA, each synthesized with a PBA appended to the COOH-terminus. We reasoned that the c-Mpl-binding peptide would exhibit increased agonist activity if it became dimerized upon application to cells which contained the dimerizer.
Figure 2. Assembly of PBA on a dimerizing scaffold.
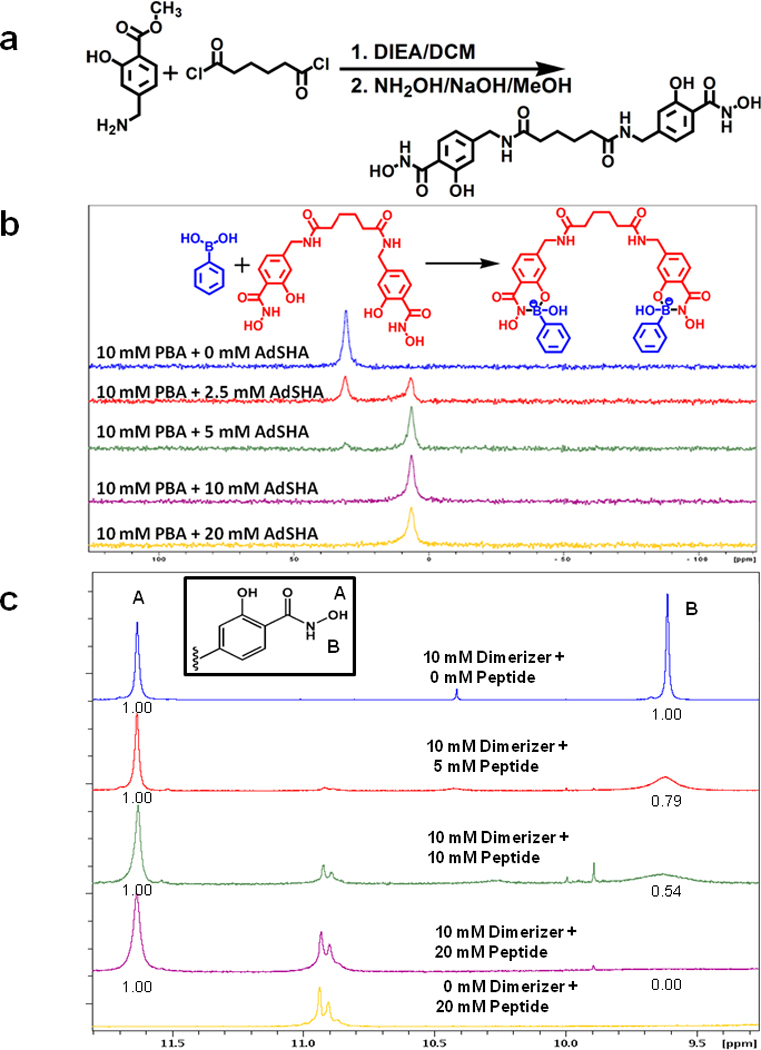
(a) Synthetic scheme of “dimerizer” synthesis.
(b) Titration of a dimerizer reveals stoichiometric complexation with PBA. 11B-NMR spectra of 10 mM PBA and 0 mM, 2.5 mM, 5 mM, 10 mM, and 20 mM of the dimerizer in 6% DMSO-d6/ 100 mM of deuterated phosphate buffer in D2O (pD = 7.4). PBA is largely complexed when present at a 2:1 ratio with dimerizer, as evidenced by the appearance of the tetrahedral coordination state of boron (6.7 ppm), and loss of the free PBA, which exhibits tetrahedral boron coordination geometry (30.8 ppm).
(c) 1H NMR spectra of dimerizer and PBA-modified c-Mpl-activating peptide at various relative concentrations in DMF-d7. The inset shows the structure of one of the two SHA present on the dimerizer. The hydroxamate O-H proton is labeled A and this proton does not participate in the complex formation. The hydroxamate N-H proton is labeled B and this proton is removed when the nitrogen atom forms a dative coordinate bond with boron in the peptide. The numbers below peaks A and B represent the relative area under each peak. With increasing concentrations of the peptide, the area under peak B is diminished, indicating the complex formation.
The peak assignments for protons A, and B were made based on the previous reports (Garcia et al., (2007).
We first confirmed that this dimerizer could simultaneously bind two PBA molecules. The dimerizer was titrated into a solution of 10 mM PBA in 100 mM phosphate buffer at pH 7.4, and the formation of PBA-SHA complexes was measured by the appearance of the tetrahedral boron hybridization state as measured by 11B-NMR. At a 2:1 PBA:dimerizer ratio, the boron was essentially completely observed to be in the tetragedral configuration (Fig. 2b), indicating that each dimerizer was simultaneously complexed to two PBA molecules.
We next addressed whether the dimerizer could recruit two peptides to form a dimeric peptide ligand. When the PBA moiety on the peptide forms a complex with the SHA moiety on the dimerizer, the hydroxamate NH proton is removed and a N-B bond is formed. The formation of dimeric c-Mpl ligand was demonstrated by the complete disappearance of the hydroxamate NH proton NMR signal in a 2:1 mixture of PBA-modified IEGPTLRQWLAARA and dimerizer (Fig. 2c). We next sought to determine the mass of the complex. Standard MS approaches were not successful due to hydrolysis of the boronate ester by the acidic buffers used in ESI and the acidic matrices commonly used in MALDI. We therefore measured the size using 2D DOSY-NMR. The molecular size of the 2:1 ratio of PBA-modified IEGPTLRQWLAARA and the dimerizer measured by 2D DOSY NMR was consistent with the expected hydrodynamic size of a complex composed of two peptide molecules and a single dimerizer (Fig. S3a). Together, these data indicate that the dimerizer recruits two peptides to form a dimeric peptide complex.
Formation of a c-Mpl-activating ligand in situ
We next addressed whether HiSBE chemistry could be used to synthesize c-Mpl agonists in tissue culture. Consistent with previous studies (Kaushansky et al., 1994; Yamada et al., 1995), BaF3-cMpl cells, which overexpress c-Mpl, exhibit increased p42/44 MAPK phosphorylation in response to TPO (5 ng/ml), as measured by anti-p42/44 MAPK Western blotting (Fig. 3a,b). Application of the PBA-modified IEGPTLRQWLAARA also increased p42/44 MAPK activity, consistent with earlier data showing that this monomeric peptide exhibits weak agonist activity (Cwirla et al., 1997). However, inclusion of the dimerizer along with the peptide resulted in a doubling of p42/44 MAPK levels (Fig. 3a,b). This effect was mediated by c-Mpl, as BaF3 cells that did not express c-Mpl did not exhibit an increase in p42/44 MAPK upon TPO or peptide treatments, but remained responsive to IL-3, a cytokine that activates p42/44 MAPK through an independent pathway (Palacios and Steinmetz, 1985) (Fig. 3c). Furthermore, this effect was dependent on the dimerizer, as SHA, which functions as a monomeric control, did not elicit a statistically significant increase in p42/44 MAPK relative to peptide alone (Fig. 3d,e). Lastly, we examined the effect of a control peptide that, when dimerized, activates an unrelated receptor, TrkB (O'Leary and Hughes, 2003). Application of either the control peptide alone or the peptide in the presence of the dimerizer resulted in no detectable activation of p42/44 MAPK (Fig. 3f). Together, these data indicate that the dimerizer and the peptide react to form a c-Mpl-activating ligand in tissue culture media in the presence of cells.
Figure 3. Synthesis of an active c-Mpl ligand in situ.
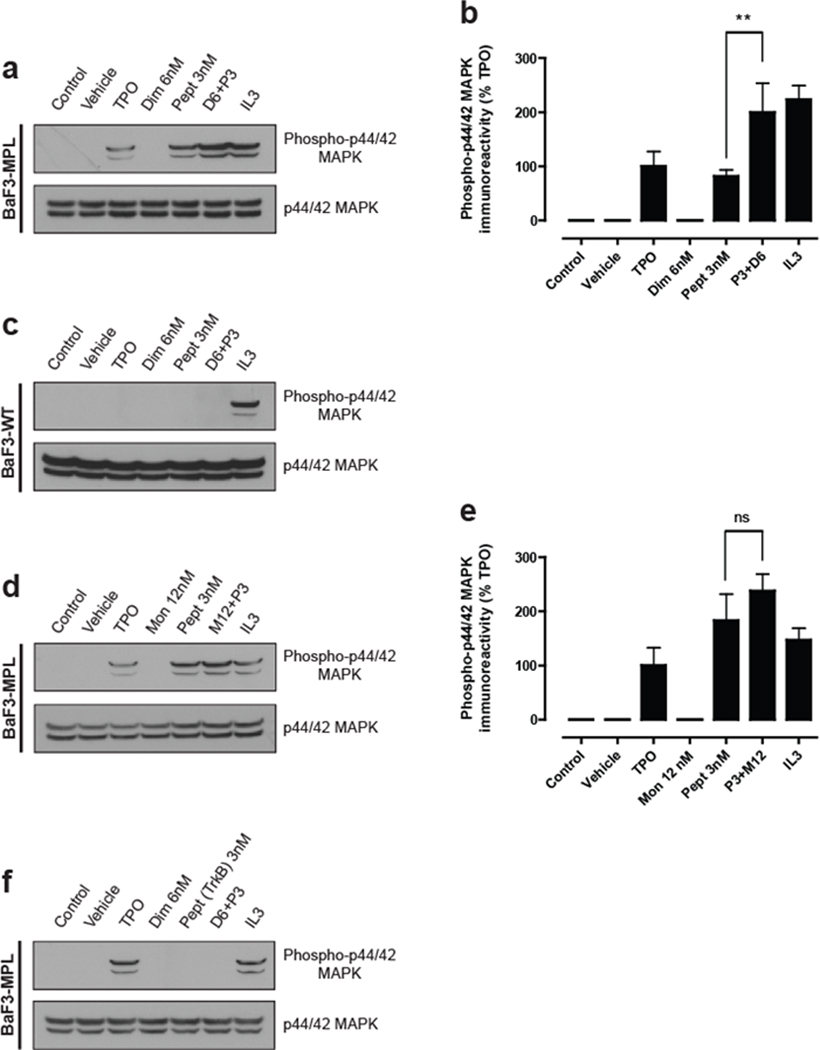
(a–f) BaF3-cMpl and BaF3-WT (parental) cells were transferred to media lacking IL-3, and cultured for 5 h. The cells were then stimulated with the indicated compounds for 10 min at 37°C and analyzed by immunoblot against phospho-p44/42 MAPK (upper panel) and total p44/42 MAPK (lower panel). In each experimental condition the immunoreactivity obtained was calculated as a percentage of the total signal obtained. Data are presented as mean ± s.e.m. of four independent experiments and each immunoblot is representative of four independent experiments.
(a) The agonist activity of the c-Mpl peptide is enhanced by the dimerizer. BaF3-cMpl cells were stimulated with TPO and IL-3, resulting in enhanced phospho-p44/42 MAPK levels as measured by Western blotting. The PBA-modified c-Mpl peptide (P) is a weak agonist of c-Mpl signaling, which is enhanced by treatment with the dimerizer (D). The dimerizer does not exert any agonist activity on its own.
(b) Quantification of results in (a). Groups were compared using a one-way ANOVA and Newman-Keuls post-test: **p<0.001 (n =4) of P alone relative to P + D. Data are presented as mean ± s.e.m. of four independent experiments.
(c) The effects of peptide-dimer complexes are mediated through c-Mpl. BaF3-WT cells, which are the parental line for the BaF3-cMpl cells, do not express c-Mpl. Under the same conditions as (a), the c-Mpl peptide and the dimer fail to activate c-Mpl signaling as assayed by phospho-p44/42 MAPK Western blotting.
(d) The c-Mpl-binding peptide is not significantly activated by a monomerizer. SHA, a monomeric control (M) to the dimerizer used in (a), was used to form complexes with the PBA-modified c-Mpl binding peptide. Treatment of cells with SHA along with the c-Mpl peptide failed to significantly increase phospho-p44/42 MAPK levels. These data support the idea that the dimerizer induces the formation of a dimeric complex which is the most efficacious ligand to activate c-Mpl signaling.
(e) Quantification of the results in (d). Groups were compared using a one-way ANOVA and Newman-Keuls post-test: n.s. = non significant (n = 4) of P alone relative to P + M. Data are presented as mean ± s.e.m. of four independent experiments.
(f) Activation of c-Mpl signaling requires a c-Mpl-binding peptide. When a PBA-modified peptide that targets TrkB was used, no phospho-44/42 MAPK activation was detected with either the peptide alone or in conjunction with the dimerizer. These results suggest that activation of c-Mpl signaling is mediated by a sequence-specific interaction with c-Mpl by a dimerized peptide.
To further examine the bioorthogonality of HiSBE synthesis, we assessed the effects of the reactants on cell proliferation and viability. Neither the dimerizer or the PBA-modified peptide reduced cell proliferation as measured by the MTT viability assay (Fig. 3b), consistent with previous data suggesting the compatibility of the boronic acid and salicylhydroxamic acid pharmacophores in living systems (Barnicoat et al., 1981; Dennis et al., 2003).
Our data suggest that HiSBE synthesis can be used to assemble bioactive compounds in a physiologic setting. Multivalent presentation of receptor ligands is a well-established approach to switch inactive or relatively inactive ligands to receptor agonists (Fournel et al., 2005), and combinatorial libraries of symmetric multivalent ligands as potential receptor agonists have attracted considerable attention (Boger et al., 1997). However, these molecules, and molecules with similarly high molecular weights are typically accompanied by poor solubility or undesirable pharmacokinetics (Lipinski, 2000). Synthesis of these compounds in situ provides a previously unexplored avenue to alleviate some of these problems, providing a strategy to facilitate the therapeutic use of multivalent ligands.
Although our focus is the design of dimeric and multimeric receptor agonists that act on the cell surface, in situ ligand assembly could also conceivably be used to synthesize molecules within cells. In this approach, at least two cell-permeable molecules, one containing a PBA moiety and the other containing a SHA moiety would be applied to cells, allowing bioactive complexes to form in the cytoplasm. This approach may enable the formation of large and otherwise impermeable molecules to accumulate and exert effects in cells.
Assembly of molecules in situ requires rapid reaction kinetics in order to provide temporal control over ligand synthesis. One prominent feature of HiSBE synthesis is its rapid reaction rate. The copper-free click reaction utilizing difluorinated cyclooctyne (DIFO) exhibits rate constants (Baskin et al., 2007; Codelli et al., 2008; Lin et al., 2005) that are several orders of magnitude slower than the rate of HiSBE synthesis. The rapid rate of the HiSBE synthesis suggests that bioorthogonal boronate ester formation will be useful for temporally controlled, rapid assembly of molecules in vivo. Another feature of HiSBE synthesis is that the reaction is reversible by ester hydrolysis. This reversibility allows HiSBE-linked molecules to be hydrolyzed to lower molecular weight components, providing a route to temporally control the duration of action of HiSBE-linked molecules. Although the copper-free click reactions using DIFO exhibits slower reaction kinetics compared to the HiSBE reaction, it may also be suitable for in situ ligand synthesis, especially in applications where a nonhydrolyzable product is desired.
The major application of bioorthogonal chemistry thus far is for labeling biomolecules in vivo with reporter groups (Prescher and Bertozzi, 2005). In these experiments, the cellular metabolic machinery is exploited to incorporate bioorthogonal functional groups within biological molecules. Reporter probes are then covalently coupled to these functional groups by spontaneous bioorthogonal reactions. Copper-free click chemistry has been particularly useful for these reactions since alkynes and azides, the bioorthogonal fuctional groups in this reaction, are relatively small allowing modified glycans, lipids and amino acid precursors to be readily incorporated into biomolecules (Prescher and Bertozzi, 2005). HiSBE synthesis may also be suitable for bioorthogonal chemical reporters. PBA moieties have been swapped for aromatic rings in both nucleotides and amino acids, resulting in only mild perturbations of structure (Lin et al., 2007; Snyder et al., 1958). Additionally, HiSBE formation can easily be conducted with linkers that contain multiple SHA moieties, potentially leading to multivalent bioactive ligands. Combinations of PBA-modified ligands and SHA linkers raise the exciting possibility of rapidly generative diverse combinatorial libraries. Also, HiSBE formation leads to products containing tetrahedral boron coordination states, which can be imaged in living animals by 11B-magnetic resonance imaging (Kabalka et al., 1988), raising the possibility of monitoring bioorthogonal reactions in vivo.
SIGNIFICANCE.
We demonstrate that the reaction of phenyl boronic acid and salicylhydroxamic acid, HiSBE synthesis, exhibits bioorthogonality, and unusually rapid reaction kinetics. HiSBE synthesis leads to boronate esters that are acid labile, and which therefore can produce products that are expected to be biodegradable under cellular conditions. Because of the rapid reaction kinetics of HiSBE synthesis, we show that this reaction can be used to generate bioactive compounds from relatively inert precursors. Assembling ligands in situ using bioorthogonal chemistry provides a strategy to bypass problems of membrane permeability and poor phamacokinetics of many high molecular weight compounds by allowing these compounds to be formed in a cellular context.
Supplementary Material
ACKNOWLEDGEMENTS
The BaF3-cMPL expressing cell line was a kind gift of Dr. Kenneth Kaushansky (University of California, San Diego). We thank D. Gin (Sloan Kettering Institute) for helpful comments. This project was supported by the Starr Cancer Consortium Award I1-A42 and NIH R21AI068512 (S.R.J.), Training Grant T32CA062948 from the National Cancer Institute (S.Y.S.), and a postodoctoral fellowship from Fundação para a Ciência e Tecnologia, Portugal (R.D.A.).
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT
The authors have no competing financial interests.
REFERENCES
- Barnicoat AJ, van't Hoff WG, Morrison PJ, Rogers HJ. Observations on salicyl hydroxamic acid, an experimental trypanocide. Experientia. 1981;37:1290–1291. doi: 10.1007/BF01948366. [DOI] [PubMed] [Google Scholar]
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachell HC, Beistel DW. Nuclear magnetic Resonance Spectra of Phenylboronic Acids. Inorg Chem. 1964;3:1028–1032. [Google Scholar]
- Boger DL, Ozer RS, Andersson C-M. Generation of targeted C2-symmetrical compound libraries by solution-phase combinatorial chemistry. Bioorganic & Medicinal Chemistry Letters. 1997;7:1903–1908. [Google Scholar]
- Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cwirla SE, Balasubramanian P, Duffin DJ, Wagstrom CR, Gates CM, Singer SC, Davis AM, Tansik RL, Mattheakis LC, Boytos CM, et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science. 1997;276:1696–1699. doi: 10.1126/science.276.5319.1696. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Ferrier RJ, Hannaford AJ, Overend WG, Smith BC. Boric acid derivatives as reagents in carbohydrate chemistry: Part IV. The interaction of phenylboronic acid with hexopyranoid compounds Carbohydrate Res. 1965;1:38–43. [Google Scholar]
- Fournel S, Wieckowski S, Sun W, Trouche N, Dumortier H, Bianco A, Chaloin O, Habib M, Peter JC, Schneider P, et al. C3-symmetric peptide scaffolds are functional mimetics of trimeric CD40L. Nat Chem Biol. 2005;1:377–382. doi: 10.1038/nchembio746. [DOI] [PubMed] [Google Scholar]
- Garcia B, et al. Structural NMR and ab Initio Study of Salicylhydroxamic and p-Hydroxybenzohydroxamic Acids: Evidence for an Extended Aggregation. J. Org. Chem. 2007;72:7832–7840. doi: 10.1021/jo0709798. [DOI] [PubMed] [Google Scholar]
- Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Whitaker LR, Francis MB. A three-component Mannich-type reaction for selective tyrosine bioconjugation. J Am Chem Soc. 2004;126:15942–15943. doi: 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- Kabalka GW, Davis M, Bendel P. Boron-11 MRI and MRS of intact animals infused with a boron neutron capture agent. Magn Reson Med. 1988;8:231–237. doi: 10.1002/mrm.1910080214. [DOI] [PubMed] [Google Scholar]
- Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC, Forstrom JW, Buddle MM, Oort PJ, Hagen FS, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–571. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- Lin FL, Hoyt HM, van Halbeek H, Bergman RG, Bertozzi CR. Mechanistic investigation of the Staudinger ligation. J Am Chem Soc. 2005;127:2686–2695. doi: 10.1021/ja044461m. [DOI] [PubMed] [Google Scholar]
- Lin N, Yan J, Huang Z, Altier C, Li M, Carrasco N, Suyemoto M, Johnston L, Wang S, Wang Q, et al. Design and synthesis of boronic-acid-labeled thymidine triphosphate for incorporation into DNA. Nucleic Acids Res. 2007;35:1222–1229. doi: 10.1093/nar/gkl1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological & Toxicological Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- O'Leary PD, Hughes RA. Design of potent peptide mimetics of brain-derived neurotrophic factor. J Biol Chem. 2003;278:25738–25744. doi: 10.1074/jbc.M303209200. [DOI] [PubMed] [Google Scholar]
- Palacios R, Steinmetz M. Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 1985;41:727–734. doi: 10.1016/s0092-8674(85)80053-2. [DOI] [PubMed] [Google Scholar]
- Plumley JA, Evanseck JD. Covalent and ionic nature of the dative bond and account of accurate ammonia borane binding enthalpies. J Phys Chem A. 2007;111:13472–13483. doi: 10.1021/jp074937z. [DOI] [PubMed] [Google Scholar]
- Prescher JA, Bertozzi CR. Chemistry in living systems. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder HR, Reedy AJ, Lennarz WJ. Synthesis of Aromatic Boronic Acids. Aldehydo Boronic Acids and a Boronic Acid Analog of Tyrosine. J Am Chem Soc. 1958;80:835–838. [Google Scholar]
- Stolowitz ML, Ahlem C, Hughes KA, Kaiser RJ, Kesicki EA, Li G, Lund KP, Torkelson SM, Wiley JP. Phenylboronic acid-salicylhydroxamic acid bioconjugates. 1. A novel boronic acid complex for protein immobilization. Bioconjugate Chemistry. 2001;12:229–239. doi: 10.1021/bc0000942. [DOI] [PubMed] [Google Scholar]
- Yamada M, Komatsu N, Okada K, Kato T, Miyazaki H, Miura Y. Thrombopoietin induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in a human thrombopoietin-dependent cell line. Biochem Biophys Res Commun. 1995;217:230–237. doi: 10.1006/bbrc.1995.2768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.