

Abstract
The three-dimensional folding of chromosomes compartmentalizes the genome and and can bring distant functional elements, such as promoters and enhancers, into close spatial proximity 2-6. Deciphering the relationship between chromosome organization and genome activity will aid in understanding genomic processes, like transcription and replication. However, little is known about how chromosomes fold. Microscopy is unable to distinguish large numbers of loci simultaneously or at high resolution. To date, the detection of chromosomal interactions using chromosome conformation capture (3C) and its subsequent adaptations required the choice of a set of target loci, making genome-wide studies impossible 7-10.
We developed Hi-C, an extension of 3C that is capable of identifying long range interactions in an unbiased, genome-wide fashion. In Hi-C, cells are fixed with formaldehyde, causing interacting loci to be bound to one another by means of covalent DNA-protein cross-links. When the DNA is subsequently fragmented with a restriction enzyme, these loci remain linked. A biotinylated residue is incorporated as the 5' overhangs are filled in. Next, blunt-end ligation is performed under dilute conditions that favor ligation events between cross-linked DNA fragments. This results in a genome-wide library of ligation products, corresponding to pairs of fragments that were originally in close proximity to each other in the nucleus. Each ligation product is marked with biotin at the site of the junction. The library is sheared, and the junctions are pulled-down with streptavidin beads. The purified junctions can subsequently be analyzed using a high-throughput sequencer, resulting in a catalog of interacting fragments.
Direct analysis of the resulting contact matrix reveals numerous features of genomic organization, such as the presence of chromosome territories and the preferential association of small gene-rich chromosomes. Correlation analysis can be applied to the contact matrix, demonstrating that the human genome is segregated into two compartments: a less densely packed compartment containing open, accessible, and active chromatin and a more dense compartment containing closed, inaccessible, and inactive chromatin regions. Finally, ensemble analysis of the contact matrix, coupled with theoretical derivations and computational simulations, revealed that at the megabase scale Hi-C reveals features consistent with a fractal globule conformation.
Keywords: Cellular Biology, Issue 39, Chromosome conformation capture, chromatin structure, Illumina Paired End sequencing, polymer physics.
Protocol
This method was used in the research reported in Lieberman-Aiden et al., Science 326, 289-293 (2009).
I. Crosslinking, Digestion, Marking of DNA Ends, and Blunt-end Ligation
Hi-C begins with crosslinking of cells, which is a common thread among all 3C-based methods. To begin, grow between 2 x 107 and 2.5 x 107 mammalian cells, either adherent or in suspension, and crosslink the cells. (For details on crosslinking of cells, please see: 11
Lyse the cells in 550 μl lysisbuffer (500 μl 10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% Igepal CA-630 and 50 μl protease inhibitors) using a homogenizer. Spin the chromatin at 5,000 rpm and wash the pellet twice with 500 μl 1x NEBuffer 2.
Resuspend the chromatin in 1x NEBuffer 2, aliquot into 5 numbered tubes and add 1x NEBuffer 2 to a final volume of 362 μl. Add 38 μl 1% SDS, mix carefully and incubate at 65 °C for 10 minutes. Place tubes back on ice immediately after incubation.
Quench the SDS by adding 44 μl Triton X-100 and mix carefully. Digest the chromatin by adding 400 Units of HindIII and incubate at 37 °C overnight while rotating.
The next steps are Hi-C specific and include marking the DNA ends with biotin and performing blunt-end ligation of crosslinked fragments. This step will allow ligation junctions to be purified later. Tube 1 should not undergo the biotinylation step and should instead be kept separate and serve as a 3C control to ensure that digestion, and ligation conditions were optimal.
To fill in the restriction fragment overhangs and mark the DNA ends with biotin in the remaining 4 tubes, add 1.5 μl 10 mM dATP, 1.5 μl 10 mM dGTP, 1.5 μl 10 mM dTTP, 37.5 μl 0.4 mM biotin-14-dCTP, and 10 μl 5U/μl Klenow to tubes 2-5. Mix carefully and incubate for 45 minutes at 37°C.
Place the tubes on ice. To inactivate the enzymes, add 86 μl 10% SDS to tubes 1-5. Incubate the tubes at 65°C for exactly 30 minutes and place them on ice immediately afterwards.
The ligation is performed under extremely dilute conditions in order to favor ligation events between crosslinked fragments. Working on ice, add 7.61 ml ligation mix [745 μl 10% Triton X-100, 745 μl 10x ligation buffer (500 mM Tris-HCl pH 7.5, 100 mM MgCl2, 100 mM DTT), 80 μl 10 mg/ml BSA, 80 μl 100 mM ATP and 5.96 ml water] to each of five numbered 15 ml tubes. Transfer each digested chromatin mixture to a corresponding 15 ml tube.
For regular 3C ligation, add 10 μl 1U/μl T4 DNA ligase to tube 1. For blunt-end Hi-C ligation, add 50 μl 1U/μl T4 DNA ligase to tubes 2-5. Mix by inverting the tubes and incubate all 5 tubes for 4 hours at 16°C.
Crosslinks are reversed and protein is degraded by adding 50 μl 10 mg/ml proteinase K per tube and incubating the tubes overnight at 65°C. Add an additional 50 μl 10 mg/ml proteinase K per tube the next day and continue the incubation at 65°C for another 2 hours.
Cool the reaction mixtures to room temperature and transfer them to five 50 ml conical tubes. Purify the DNA in these tubes by performing a phenol extraction. Add 10 ml phenol pH 8.0 and vortex for 2 minutes. Spin the tubes for 10 minutes at 3,500 rpm and carefully transfer as much of the aqueous phase as possible to a new 50 ml tube.
Repeat the extraction using phenol pH 8.0:chloroform (1:1) and precipitate the DNA using ethanol. (For details on DNA purification, please see: 11
After centrifugation of the ethanol precipitated DNA, dissolve each DNA pellet in 450 μl 1x TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Transfer the DNA mixture to a 1.7 ml centrifuge tube.
Another round of purification is performed by doing 2 phenol:chloroform extractions. Add 500 μl phenol pH 8.0:chloroform (1:1) and vortex for 1 minute. Centrifuge the tubes for 5 minutes at 14,000 rpm and transfer the aqueous phase to a new tube. After the second extraction, precipitate the DNA by adding 0.1x volume of NaOAc, 2x volume of 100% ethanol and incubate 30 minutes at -80 °C.
After spinning down the precipitated DNA, wash each DNA pellet with 70% ethanol and resuspend each DNA pellet in 25 μl 1x TE. Degrade any RNA that might be present by adding 1 μl 1 mg/ml RNAse A per tube and incubating the tubes for 30 minutes at 37°C. Pool the Hi-C contents of tubes 2-5, still keeping tube 1 separate as a 3C control.
- Now is a good opportunity to examine the Hi-C marking and ligation efficiency. These controls are excellent indicators of whether a Hi-C library is going to be successful.
- To check the quality and quantity of the libraries, run 2 μl and 6 μl aliquots of 1:10 dilutions from both 3C and Hi-C libraries on a 0.8% agarose gel. (See Figure 2A)
- Hi-C marking and Hi-C ligation efficiency is verified by a PCR digest assay. Successful fill-in and ligation of a HindIII site (AAGCTT) creates a site for the restriction enzyme NheI (GCTAGC). A particular ligation product formed from two nearby restriction fragments is amplified with PCR (as in 3C 11 using 0.2 μl of each library as template. The PCR products are subsequently digested with HindIII, NheI or both. After running the samples on a 2% gel, the relative number of 3C and Hi-C ligation events can be estimated by quantifying the intensity of the cut and uncut bands (See Figure 2B).
- Some fragments will not have been ligated: to avoid pulling them down later, remove biotin from these unligated ends using the exonuclease activity of T4 DNA polymerase.
- Biotin-14-dCTP at non-ligated DNA ends is removed with the exonuclease activity of T4 DNA polymerase. Mix 5 μg of Hi-C library with 1 μl 10 mg/ml BSA, 10 μl 10x NEBuffer 2, 1 μl 10 mM dATP, 1 μl 10 mM dGTP and 5 Units T4 DNA polymerase in a total volume of 100 μl and incubate the mixture at 12°C for 2 hours. If possible, multiple 5 μg reactions are performed.
- The reaction is stopped by adding 2 μl 0.5 M EDTA pH 8.0.
- To purify the DNA, a phenol pH 8.0:chloroform (1:1) extraction is done followed by ethanol precipitation.
- The supernatant is discarded and the DNA pellets are resuspended and pooled in a total volume of 100 μl water.
II. Shearing and Size Selection
To make the biotinylated DNA suitable for high-throughput sequencing, the DNA must be sheared to a size of 300-500 basepairs with a Covaris S2 instrument (duty cycle 5, intensity 5, cycles/burst 200, time 60 secs for 4 cycles).
To repair the sheared DNA ends, add 14 μl 10x ligation buffer, 14 μl 2.5 mM dNTP mix, 5 μl T4 DNA polymerase, 5 μl T4 polynucleotide kinase, 1 μl Klenow DNA polymerase and 1 μl water. Incubate for 30 minutes at room temperature.
Following incubation, use a Qiagen MinElute column to purify the DNA according to manufacturer's recommendations. Elute the DNA twice with 15 μl 1x Tris-Low-EDTA (TLE: 10 mM Tris pH 8.0, 0.1 mM EDTA). Then, attach a dATP to the 3' ends of the end-repaired DNA by adding 5 μl 10x NEBuffer2, 10 μl 1 mM dATP, 2 μl water and 3 μl Klenow (exo-). Incubate the reaction for 30 minutes at 37°C.
To inactivate the Klenow fragment, incubate the reactions for 20 minutes at 65°C and subsequently cool the reactions on ice. Using a speedvac, reduce the reaction volumes to 20 μl.
Next, load the DNA in a 1.5% agarose gel with 1X TAE and run for 3.5 hours at 80-90V. After staining the gel with SYBR green, visualize the DNA on a DarkReader. Excise DNA fragments between 300 and 500 base pairs and purify them with a Qiagen gel extraction kit using 2-4 columns depending on the weight of the gel. Elute the DNA with 50 μl 1x TLE.
Combine the eluates from the Qiaquick columns and bring the final volume up to 300 μl with 1x TLE. Finally, determine the DNA concentration with the Quant-iT assay using the QuBit fluorometer and calculate the total amount of DNA.
III. Biotin Pull-down and Paired-end Sequencing
In this section of the protocol, ligation junctions are purified from the DNA pool, allowing for efficient identification of interacting chromatin fragments by paired-end sequencing. Perform all subsequent steps in DNA LoBind tubes.
- Prepare beads for biotin pull-down by washing 150 μl resuspended magnetic streptavidin beads twice with 400 μl Tween Buffer (TB: 5 mM Tris-HCl pH 8.0, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween). These and future washes consist of five steps:
- Add buffer to the beads
- Transfer the mixture to a new tube
- Rotate the sample for 3 minutes at room temperature
- Reclaim the beads using a magnetic particle concentrator
- Remove the supernatant
Resuspend the beads in 300 μl 2x No Tween Buffer (2x NTB: 10 mM Tris-HCl pH 8.0, 1 mM EDTA, 2 M NaCl) and combine with 300 μl Hi-C DNA. Allow the biotin labeled Hi-C DNA to bind to the streptavidin beads by incubating the mixture at room temperature for 15 minutes with rotation.
Reclaim the DNA bound streptavidin beads with the magnetic particle concentrator, and remove the supernatant. Wash the beads in 400 μl 1x NTB (5 mM Tris-HCl pH 8.0, 0.5 mM EDTA, 1 M NaCl), followed by 100 μl 1x ligation buffer. Resuspend the beads in 50 μl 1x ligation buffer and transfer the mixture to a new tube.
To prepare the DNA for Illumina Paired End sequencing, take the total amount of DNA used as input for the biotin pull-down, which was calculated earlier in step 2.6, and divide it by 20 to estimate the amount of Hi-C DNA that has been pulled down and is available for ligation. Add 6 picomoles of Illumina Paired End adapters per μg of Hi-C DNA available for ligation. Use 1200 Units T4 DNA Ligase to ligate the adapters to the DNA. Incubate for 2 hours at room temperature.
Remove non-ligated Paired End adapters by reclaiming the Hi-C DNA bound beads and washing the beads twice with 400 μl 1x TB.
Wash the beads with 200 μl 1x NTB, followed by 200 μl and then 50 μl 1x NEBuffer 2. After the last wash, resuspend the beads in 50 μl 1x NEBuffer 2 and transfer to a new tube.
To determine the number of cycles necessary to generate enough PCR product for sequencing, set up four test PCR reactions with 6, 9, 12 or 15 cycles. (For details of the PCR amplification, please see: 12. Determine the optimal cycle number by running the PCR reactions on a 5% polyacrylamide gel and staining with Sybr Green, ensuring the absence of spurious bands and the presence of a smear between 400-600 base pairs, which is the length of the sheared products after ligation to the adapters.
Amplify the rest of the Hi-C-library-bound streptavidin beads in a large-scale PCR with the optimal number of PCR cycles. Pool the PCR products from the separate wells and reclaim the beads. Keep 1% of the large scale PCR product separate to run on a gel and purify the remainder of the PCR product with 1.8x volume Ampure beads according to the manufacturer's recommendations.
- Elute the DNA with 50 μl 1x TLE buffer and compare 1% of the Ampure bead purified PCR product to the 1% aliquot of original PCR product on a 5% polyacrylamide gel, ensuring the successful removal of the PCR primers.
- We also recommend cloning 1 μl of the Hi-C library and determining the product of about 100 clones using Sanger sequencing. This will enable you to assess the relative number of alignable Hi-C reads in the PCR mixture. For typical results, see Figure 3B.
Sequence the Hi-C library with Illumina paired end sequencing. Align each end independently using Maq (http://maq.sourceforge.net/) to identify interacting chromatin fragments.
IV. Representative Hi-C Results
The following results are expected when the Hi-C protocol is executed technically well and can be considered quality control standards.
Quality control steps should reveal that both 3C and Hi-C libraries run as rather tight bands larger than 10 kb. A DNA smear indicates poor ligation efficiency. Typically, ligation efficiency is slightly lower in a Hi-C library as compared to a 3C template (See Figure 2A).
Hi-C marking and ligation efficiency can be estimated by digestion of a PCR product generated using 3C primers. 3C junctions are cut by HindIII and not by NheI. The reverse is true for Hi-C junctions. This PCR digest assay shows that 70% of Hi-C amplicons are cut by NheI and not by HindIII, confirming efficient marking of ligation junctions (See Figure 2B).
Analysis of the sequenced reads should show that reads from both intrachromosomal and interchromosomal interactions, indicated by the blue and red lines, align significantly closer to HindIII restriction sites as compared to randomly generated reads, shown in green (See Figure 3A).
In a successful experiment, 55% of the alignable read pairs represent interchromosomal interactions. Fifteen percent represent intrachromosomal interactions between fragments less than 20 kb apart and 30% are intrachromosomal read pairs that are more than 20 kb apart (See Figure 3B). This distribution may be sampled prior to high-throughput sequencing, as a form of quality control; cloning and Sanger sequencing of about 100 clones is usually sufficient.
The chromatin interactions can be visualized as a heatmap, where the x- and y- axes represent loci in genomic order and each pixel represents the number of observed interactions between them. Typically, DNA fragments that are very close to each other in the linear genome will have the tendency to interact frequently with each other. This is seen in the intrachromosomal heatmaps as a prominent diagonal (See Figure 4A).
The following results show different ways of analyzing the data to reveal various levels of genome organization. Plotting the contact probability versus genomic distance (See Figure 5A) shows that probability of contact decreases as a function of genomic distance, eventually reaching a plateau. At every distance, intrachromosomal interactions, shown in the solid line, are enriched relative to interchromosomal interactions, represented by the dashed lines. This directly implies the presence of chromosome territories.
Calculating the observed/expected number of interchromosomal contacts between all pairs of chromosomes reveals preferential association between particular chromosome pairs. Small gene-rich chromosomes preferentially interact with each other, indicated by the bright red color (See Figure 5B).
Individual chromosomes can also be examined. The raw heatmap can be adjusted using an expected heatmap to account for the genomic distance between pairs of loci, resulting in an observed/expected heatmap. Then, a correlation matrix can be produced by correlating the rows and columns of the observed/expected heatmaps. Using correlation analysis, it is demonstrated that the human genome segregates into two compartments. This is illustrated by the plaid-pattern in the correlation heatmaps (See Figure 4A-D). (For details of the Hi-C data analysis, please see: 1.
Using Hi-C data, new insights were gained into chromatin folding at the megabase scale. The classical model of polymer condensation suggests that chromatin packs into an equilibrium globule. Plotting contact probability as a function of distance illustrates that contact probability scales as a power law with genomic distance whose slope is approximately -1 (See Figure 6A). This is not consistent with the behavior of an equilibrium globule, but does match expectations for an alternative structure known as a fractal globule (See Figure 6B).
Here, two globular structures are shown. Coloration corresponds to distance from one endpoint, ranging from blue to cyan, green, yellow, orange, and red (See Figure 6C top). Unlike equilibrium globules, fractal globules lack entanglements. In a fractal globule, loci that are nearby along the contour tend to be nearby in 3D, leading to the presence of monochromatic blocks (See Figure 6C middle). Such blocks are not found in the equilibrium globule (See Figure 6C bottom).
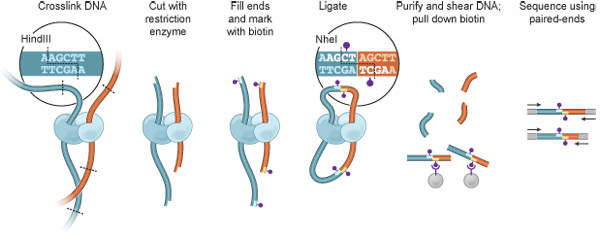 Figure 1. Hi-C overview. Cells are cross-linked with formaldehyde, resulting in covalent links between spatially adjacent chromatin segments (DNA fragments: dark blue, red; Proteins, which can mediate such interactions, are shown in light blue and cyan). Chromatin is digested with a restriction enzyme (here, HindIII; restriction site: dashed line, see inset). The resulting sticky ends are filled in with nucleotides, one of which is biotinylated (purple dot). Ligation is performed under extremely dilute conditions favoring intramolecular ligation events; the HindIII site is lost and an NheI site is created (inset). DNA is purified and sheared, and biotinylated junctions are isolated using streptavidin beads. Interacting fragments are identified by paired-end sequencing.
Figure 1. Hi-C overview. Cells are cross-linked with formaldehyde, resulting in covalent links between spatially adjacent chromatin segments (DNA fragments: dark blue, red; Proteins, which can mediate such interactions, are shown in light blue and cyan). Chromatin is digested with a restriction enzyme (here, HindIII; restriction site: dashed line, see inset). The resulting sticky ends are filled in with nucleotides, one of which is biotinylated (purple dot). Ligation is performed under extremely dilute conditions favoring intramolecular ligation events; the HindIII site is lost and an NheI site is created (inset). DNA is purified and sheared, and biotinylated junctions are isolated using streptavidin beads. Interacting fragments are identified by paired-end sequencing.
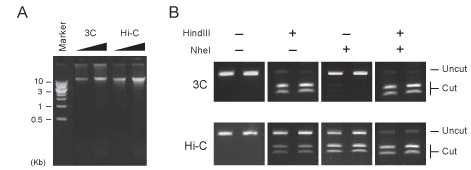 Figure 2. Hi-C library quality controls. (A) Increasing amounts of a 3C control and a Hi-C library were resolved on a 0.8% agarose gel. Both libraries run as a rather tight band larger than 10 kb. Typical ligation efficiency in a Hi-C library is slightly lower than what is observed in a 3C template, and is indicated by the smear in the Hi-C lanes. (B) PCR digest control. A ligation junction formed by two nearby fragments is amplified using standard 3C PCR conditions. Hi-C ligation products can be distinguished from those produced in conventional 3C by digestion of the ligation site. Hi-C junctions are cut by NheI, not HindIII; the reverse is true for 3C junctions. 70% of Hi-C amplicons were cut by NheI, confirming efficient marking of ligation junction. Two replicates were performed to ensure reliable quantification.
Figure 2. Hi-C library quality controls. (A) Increasing amounts of a 3C control and a Hi-C library were resolved on a 0.8% agarose gel. Both libraries run as a rather tight band larger than 10 kb. Typical ligation efficiency in a Hi-C library is slightly lower than what is observed in a 3C template, and is indicated by the smear in the Hi-C lanes. (B) PCR digest control. A ligation junction formed by two nearby fragments is amplified using standard 3C PCR conditions. Hi-C ligation products can be distinguished from those produced in conventional 3C by digestion of the ligation site. Hi-C junctions are cut by NheI, not HindIII; the reverse is true for 3C junctions. 70% of Hi-C amplicons were cut by NheI, confirming efficient marking of ligation junction. Two replicates were performed to ensure reliable quantification.
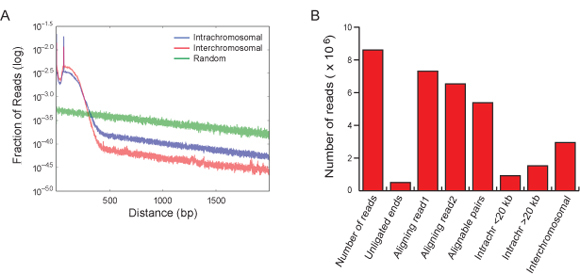 Figure 3. Hi-C read quality controls. (A) Reads from fragments corresponding to both intrachromosomal (blue) and interchromosomal (red) interactions align significantly closer to HindIII restriction sites as compared to randomly generated reads (green). Both the intrachromosomal reads and interchromosomal reads curves decrease rapidly as the distance from the HindIII site increases until a plateau is reached at a distance of ~500 bp. This corresponds to the maximum fragment size used for sequencing. (B) Typically, 55% of the alignable read pairs represent interchromosomal interactions. Fifteen percent represent intrachromosomal interactions between fragments less than 20 kb apart and 30% are intrachromosomal read pairs that are more than 20 kb apart. This distribution may be sampled prior to high-throughput sequencing, as a form of quality control; cloning and Sanger sequencing of about 100 clones is usually sufficient.
Figure 3. Hi-C read quality controls. (A) Reads from fragments corresponding to both intrachromosomal (blue) and interchromosomal (red) interactions align significantly closer to HindIII restriction sites as compared to randomly generated reads (green). Both the intrachromosomal reads and interchromosomal reads curves decrease rapidly as the distance from the HindIII site increases until a plateau is reached at a distance of ~500 bp. This corresponds to the maximum fragment size used for sequencing. (B) Typically, 55% of the alignable read pairs represent interchromosomal interactions. Fifteen percent represent intrachromosomal interactions between fragments less than 20 kb apart and 30% are intrachromosomal read pairs that are more than 20 kb apart. This distribution may be sampled prior to high-throughput sequencing, as a form of quality control; cloning and Sanger sequencing of about 100 clones is usually sufficient.
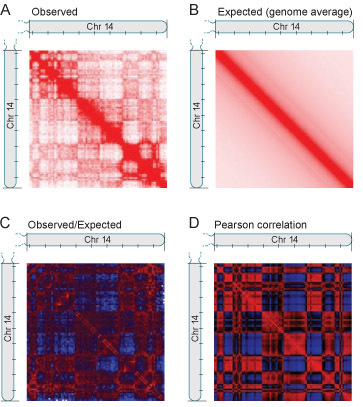 Figure 4. Correlation analysis demonstrates that the nucleus is segregated into two compartments. (A) Heatmap corresponding to intrachromosomal interactions on chromosome 14. Each pixel represents all interactions between a 1-Mb locus and another 1-Mb locus; intensity corresponds to the total number of reads (range: 0-200 reads). Tick marks appear every 10 Mb. The heatmap exhibits substructure in the form of an intense diagonal and a constellation of large blocks. (Chromosome 14 is acrocentric; the short arm is not shown.) Using the Hi-C dataset to compute the average contact probability for a pair of loci at a given genomic distance, an expectation matrix is produced (B) corresponding to what would be observed if there were no long-range structures. The quotient of these two matrices is an observed/expected matrix (C) where depletion is shown in blue and enrichment in red [range: 0.2 (blue) to 5 (red)]. The block pattern becomes more evident. The correlation matrix (D) illustrates the correlation [range: -1 (blue) to 1 (red)] between the intrachromosomal interaction profiles of every pair of loci along chromosome 14. The striking plaid pattern indicates the presence of two compartments within the chromosome.
Figure 4. Correlation analysis demonstrates that the nucleus is segregated into two compartments. (A) Heatmap corresponding to intrachromosomal interactions on chromosome 14. Each pixel represents all interactions between a 1-Mb locus and another 1-Mb locus; intensity corresponds to the total number of reads (range: 0-200 reads). Tick marks appear every 10 Mb. The heatmap exhibits substructure in the form of an intense diagonal and a constellation of large blocks. (Chromosome 14 is acrocentric; the short arm is not shown.) Using the Hi-C dataset to compute the average contact probability for a pair of loci at a given genomic distance, an expectation matrix is produced (B) corresponding to what would be observed if there were no long-range structures. The quotient of these two matrices is an observed/expected matrix (C) where depletion is shown in blue and enrichment in red [range: 0.2 (blue) to 5 (red)]. The block pattern becomes more evident. The correlation matrix (D) illustrates the correlation [range: -1 (blue) to 1 (red)] between the intrachromosomal interaction profiles of every pair of loci along chromosome 14. The striking plaid pattern indicates the presence of two compartments within the chromosome.
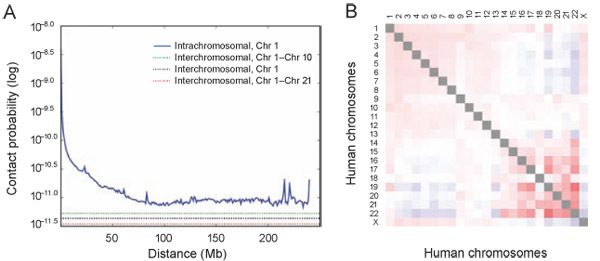 Figure 5. The presence and organization of chromosome territories. (A) Probability of contact decreases as a function of genomic distance on chromosome 1, eventually reaching a plateau at ~90Mb (blue). The level of interchromosomal contact (black dashes) differs for different pairs of chromosomes; loci on chromosome 1 are most likely to interact with loci on chromosome 10 (green dashes) and least likely to interact with loci on chromosome 21 (red dashes). Interchromosomal interactions are depleted relative to intrachromosomal interactions. (B) Observed/expected number of interchromosomal contacts between all pairs of chromosomes. Red indicates enrichment, and blue indicates depletion [range: 0.5 (blue) to 2 (red)]. Small, gene-rich chromosomes tend to interact more with one another.
Figure 5. The presence and organization of chromosome territories. (A) Probability of contact decreases as a function of genomic distance on chromosome 1, eventually reaching a plateau at ~90Mb (blue). The level of interchromosomal contact (black dashes) differs for different pairs of chromosomes; loci on chromosome 1 are most likely to interact with loci on chromosome 10 (green dashes) and least likely to interact with loci on chromosome 21 (red dashes). Interchromosomal interactions are depleted relative to intrachromosomal interactions. (B) Observed/expected number of interchromosomal contacts between all pairs of chromosomes. Red indicates enrichment, and blue indicates depletion [range: 0.5 (blue) to 2 (red)]. Small, gene-rich chromosomes tend to interact more with one another.
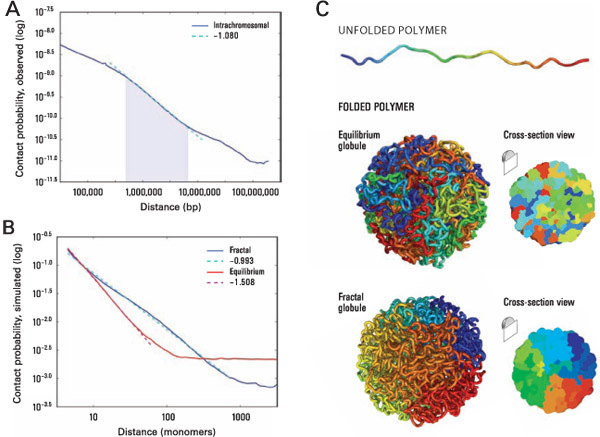 Figure 6. The local packing of chromatin is consistent with the behavior of a fractal globule. (A) Contact probability as a function of genomic distance, averaged across the genome (blue). A prominent power law scaling is seen between 500kb and 7Mb (shaded region) with a slope of -1.08 (fit shown in cyan). (B) Simulation results for contact probability as a function of distance for equilibrium (red) and fractal (blue) globules. The slope for a fractal globule is very nearly -1 (cyan), confirming our novel theoretical prediction 1. The slope for an equilibrium globule is -3/2, which matches prior theoretical expectations. The slope for the fractal globule closely resembles the slope observed in the Hi-C results, whereas the slope for an equilibrium globule is not seen in the Hi-C data. (C) Top: An unfolded polymer chain, 4000 monomers long. Coloration corresponds to distance from one endpoint, ranging from blue to cyan, green, yellow, orange, and red. Middle: Typical example of a fractal globule drawn from our ensemble. Fractal globules lack entanglements. Loci that are nearby along the contour tend to be nearby in 3D, leading to the presence of large monochromatic blocks that are apparent on the surface and in cross-section. Bottom: An equilibrium globule. The structure is highly entangled; loci that are nearby along the contour (similar color) need not be nearby in 3D.
Figure 6. The local packing of chromatin is consistent with the behavior of a fractal globule. (A) Contact probability as a function of genomic distance, averaged across the genome (blue). A prominent power law scaling is seen between 500kb and 7Mb (shaded region) with a slope of -1.08 (fit shown in cyan). (B) Simulation results for contact probability as a function of distance for equilibrium (red) and fractal (blue) globules. The slope for a fractal globule is very nearly -1 (cyan), confirming our novel theoretical prediction 1. The slope for an equilibrium globule is -3/2, which matches prior theoretical expectations. The slope for the fractal globule closely resembles the slope observed in the Hi-C results, whereas the slope for an equilibrium globule is not seen in the Hi-C data. (C) Top: An unfolded polymer chain, 4000 monomers long. Coloration corresponds to distance from one endpoint, ranging from blue to cyan, green, yellow, orange, and red. Middle: Typical example of a fractal globule drawn from our ensemble. Fractal globules lack entanglements. Loci that are nearby along the contour tend to be nearby in 3D, leading to the presence of large monochromatic blocks that are apparent on the surface and in cross-section. Bottom: An equilibrium globule. The structure is highly entangled; loci that are nearby along the contour (similar color) need not be nearby in 3D.
Discussion
We present a method of studying the 3-dimensional architecture of the genome by mapping chromatin interactions in an unbiased, genome-wide manner. The most critical experimental step what sets this technology apart from previous work - is the incorporation of biotinylated nucleotides at the restriction ends of crosslinked fragments before blunt-end ligation. Performing this step successfully enables deep sequencing of all ligation junctions, and gives Hi-C its scope and power.
The number of reads will ultimately determine the resolution of the interaction maps. Here, a 1 Mb interaction map for the human genome is presented using ~30 million alignable reads. In order to increase 'all-purpose' resolution by a factor of n, the number of reads must be increased by a factor of n2.
The Hi-C technique may be readily combined with other techniques, such as hybrid capture after library generation (to target specific parts of the genome) and chromatin immunoprecipitation after ligation (to examine the chromatin environment of regions associated with specific proteins).
Disclosures
A provisional patent on the Hi-C method (no. 61/100,151) is under review.
Acknowledgments
We thank A. Kosmrlj for discussions and code; A. P. Aiden, X. R. Bao, M. Brenner, D. Galas, W. Gosper, A. Jaffer, A. Melnikov, A. Miele, G. Giannoukos, C. Nusbaum, A. J. M. Walhout, L. Wood, and K. Zeldovich for discussions; and L. Gaffney and B. Wong for help with visualization.
Supported by a Fannie and John Hertz Foundation graduate fellowship, a National Defense Science and Engineering graduate fellowship, an NSF graduate fellowship, the National Space Biomedical Research Institute, and grant no. T32 HG002295 from the National Human Genome Research Institute (NHGRI) (E.L.); i2b2 (Informatics for Integrating Biology and the Bedside), the NIH-supported Center for Biomedical Computing at Brigham and Women s Hospital (L.A.M.); grant no. HG003143 from the NHGRI, and a Keck Foundation distinguished young scholar award (J.D.). Raw and mapped Hi-C sequence data has been deposited at the GEO database (www.ncbi.nlm.nih.gov/geo/), accession no. GSE18199. Additional visualizations are available at http://hic.umassmed.edu.
References
- Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak ST, Groudine M. Form follows function: the genomic organization of cellular differentiation. Genes and Dev. 2004;18:1371–1384. doi: 10.1101/gad.1209304. [DOI] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Dekker J. Gene Regulation in the Third Dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- Sexton T, Schober H, Fraser P, Gasser SM. Gene regulation through nuclear organization. Nat Struct and Mol Biol. 2007;14:1049–1055. doi: 10.1038/nsmb1324. [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing Chromosome Conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A, Dekker J. Mapping Cis- and Trans Chromatin Interaction Networks Using Chromosome Conformation Capture (3C) Methods Mol Biol. 2009;464:105–121. doi: 10.1007/978-1-60327-461-6_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, Williams L, Young S, Nusbaum C, Jaffe DB. ALLPATHS 2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 2009;10:R103–R103. doi: 10.1186/gb-2009-10-10-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]