

Abstract
We describe a protocol for establishing mouse models of periventricular leukomalacia (PVL). PVL is the predominant form of brain injury in premature infants and the most common antecedent of cerebral palsy. PVL is characterized by periventricular white matter damage with prominent oligodendroglial injury. Hypoxia/ischemia with or without systemic infection/inflammation are the primary causes of PVL. We use P6 mice to create models of neonatal brain injury by the induction of hypoxia/ischemia with or without systemic infection/inflammation with unilateral carotid ligation followed by exposure to hypoxia with or without injection of the endotoxin lipopolysaccharide (LPS). Immunohistochemistry of myelin basic protein (MBP) or O1 and electron microscopic examination show prominent myelin loss in cerebral white matter with additional damage to the hippocampus and thalamus. Establishment of mouse models of PVL will greatly facilitate the study of disease pathogenesis using available transgenic mouse strains, conduction of drug trials in a relatively high throughput manner to identify candidate therapeutic agents, and testing of stem cell transplantation using immunodeficiency mouse strains.
Keywords: JoVE Neuroscience, Issue 39, brain, mouse, white matter injury, oligodendrocyte, periventricular leukomalacia
Protocol
Detailed procedures of creating neonatal brain injury in P6 mice:
Postnatal day 6 (P6) mice are anesthetized with indirect cooling on ice to the point of unresponsiveness to noxious stimulation (deep anesthesia).
Following cleaning the skin with alcohol, a midline ventral incision is made in the anterior neck.
Under a dissecting microscope, the bifurcation of the right common carotid artery is approached by gently retracting the omohyoid and sternocleidomastoid muscles.
The fascia around the carotid sheath is removed, and the proximal internal branch isolated from the nearby vagus nerve and sympathetic ganglia with a hook. The internal carotid artery is then cauterized using a cauterizing tip.
Following cauterization, the skin incision is sutured, and the animal is kept warm until fully awake and then returned to the dam.
One hour after ligation, the animal is placed in a sealed chamber infused with nitrogen until a level of 6.0% O2 is reached. The animal is then exposed to 35 min of hypoxia.
After hypoxia exposure, the mouse recovers on a heating pad (33°C) for 30 min and then is returned to the dam. For creation of brain injury with combined hypoxia/ischemia and infection/inflammation, the animal is then injected intraperitoneally with 0.015 ml of lipopolysaccharide (LPS, 1 mg/kg).
Four days post-hypoxia/ischemia, mice are anesthetized with indirect cooling on ice to the point of unresponsiveness to noxious stimuli, then perfused with saline and then 4% paraformaldehyde. Brains are removed and cryoprotected.
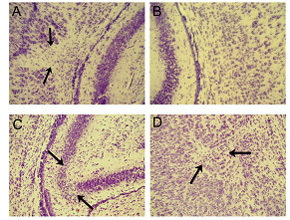 Figure 1. Characterization of brain injury in the hypoxic/ischemic mouse model with H & E staining. There is focal necrosis (arrows) in the cerebral white matter ipsilateral (A) to the carotid ligation, compared to the contralateral cerebral white matter (B). Ipsilateral necrosis and focal microinfarcts are observed in the hippocampus (C) and thalamus (D) in the brains with ipsilateral cerebral white matter injury.
Figure 1. Characterization of brain injury in the hypoxic/ischemic mouse model with H & E staining. There is focal necrosis (arrows) in the cerebral white matter ipsilateral (A) to the carotid ligation, compared to the contralateral cerebral white matter (B). Ipsilateral necrosis and focal microinfarcts are observed in the hippocampus (C) and thalamus (D) in the brains with ipsilateral cerebral white matter injury.
Representative Results and Figures:
Mouse models of PVL with hypoxia/ischemia and/or infection/inflammation are achieved by unilateral carotid ligation followed by exposure to hypoxia and injection of LPS. We are defining the new mouse models as suitable PVL models. Neuropathological examination of coronal sections stained with hematoxylin-and-eosin (H & E) through the entire forebrain reveals focal necrosis in the central white matter of the cerebral hemisphere ipsilateral to the carotid ligation (Fig. 1A), with relative sparing of neurons in the overlying cerebral cortex compared to the contralateral cerebral white matter (Fig. 1B). In the focal necrotic white matter, there is marked increase in cellularity consisting of macrophages and reactive astrocytes, as is characteristic of focal human PVL. In the contralateral central white matter, the oligodendroglial nuclei are arranged in fascicular-like bundles; this architecture is completely disrupted by the focal necrosis on the ipsilateral side. Of note, we also observe ipsilateral necrosis in the hippocampus (Fig. 1C) and thalamus (focal microinfarcts) (Fig. 1D) in the brains with ipsilateral cerebral white matter injury. Human PVL also does not occur in complete isolation from accompanying gray matter lesions. The hallmark of PVL is the relative, although not complete, sparing of the cerebral cortex overlying the injured white matter - a feature that we believe is present in our mouse models.
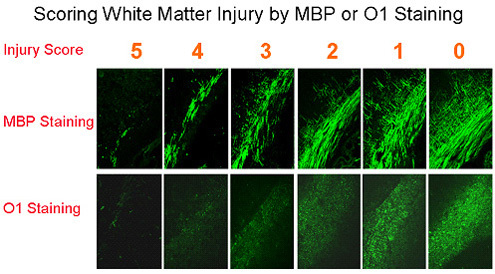 Figure 2. Scoring scale of 0-5 for evaluating white matter injury by MBP or O1 in mouse models of periventricular leukomalacia.
Figure 2. Scoring scale of 0-5 for evaluating white matter injury by MBP or O1 in mouse models of periventricular leukomalacia.
Our mouse models show characteristics of PVL at both the phenotypic and molecular levels. We have used immunocytochemical detection of myelin basic protein (MBP) or O1-antigen to evaluate injury, have established a standard scale from 0 to 5 to score the white matter injury (Fig. 2), and have characterized the white matter pathology (Fig. 3). More recently, a non-biased stereological assay has been implemented to substitute for the initial semi-quantitative method. In addition, to test for correlative effects of white matter injury on functional outcomes, we have performed behavioral tests for motor function. We have demonstrated that the mice exhibit striking motor deficits at P10-21 following hypoxia/ischemia plus LPS at P6. We have established scoring criteria with a scale of 0-4 to define contralateral limb dysfunction in injured mice at P10-21. Typically, normal mice can scamper up a 30° incline (score of 4) and climb up a 45° incline without sliding (score of 3). Injured mice have lower scores in correlation with the degrees of white matter injury.
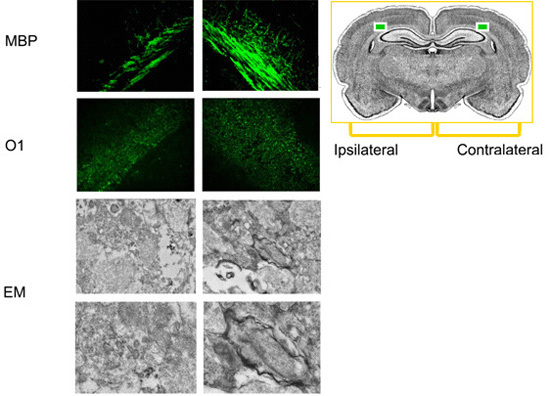 Figure 3. Characterization of white matter injury by MBP or O1 staining and myelin structure by electron microscope (EM). MBP and O1 staining show myelin loss in cerebral white matter ipsilateral to the ligation, compared to contralateral white matter. EM examination shows myelinated axons in cerebral white matter contralateral to the ligation and degenerated tissue in ipsilateral white matter.
Figure 3. Characterization of white matter injury by MBP or O1 staining and myelin structure by electron microscope (EM). MBP and O1 staining show myelin loss in cerebral white matter ipsilateral to the ligation, compared to contralateral white matter. EM examination shows myelinated axons in cerebral white matter contralateral to the ligation and degenerated tissue in ipsilateral white matter.
We anticipate that the establishment of the new mouse models of PVL will allow us to study specific mechanisms of PVL-like injury to the immature brain. We will address pathogenesis using transgenic mouse strains to examine the role of individual molecules of interest and evaluate targets of neuroprotection.
Discussion
Injury to the developing brain leads to devastating neurological consequences. Strikingly, the pattern of perinatal brain injury is highly age-dependent. In term infants, it predominantly affects cerebral cortex with characteristic neuronal loss. However, in premature infants, it selectively affects cerebral white matter with prominent injury to developing oligodendrocytes, a disorder termed periventricular leukomalacia (PVL). The magnitude of the problem of brain injury in the premature infant is extraordinary. Premature delivery and improved neonatal intensive care have led to nearly 90% of survival of the 13 million premature infants worldwide (approximately 56,000 in the United States) born yearly with a birthweight under 1500 grams, i.e., very low-birth-weight infants. Approximately 10% of the survivors subsequently exhibit cerebral palsy and approximately 50% have cognitive and behavioral deficits. PVL is the most important brain pathology underlying cerebral palsy in premature infants. No specific therapy for PVL presently exists, in part because the pathogenesis has not been well understood1,2.
Although the etiology of PVL is multifactorial, perinatal hypoxia/ischemia with maternal intrauterine infection is thought to be a primary cause of PVL. A hypoxic/ischemic model of PVL in the rat at P7 has been previously established3. This model is unique in that it resembles PVL in premature infants. The hallmark of this model is selective white matter pathology, in contrast to the majority of the stroke models that are characterized by gray matter infarction. However, the limitations of the hypoxic-ischemic model in the rat should not be overlooked. First, the etiology of PVL is often multifactorial. In addition to hypoxia/ischemia, maternal-fetal infection/inflammation is strongly associated with PVL. Thus, combined hypoxia/ischemia and infection/inflammation would likely create more PVL-like models. The rodent age that best correlates with the human developmental period of greatest risk for PVL lesions (24-32 weeks of gestation) is postnatal because oligodendroglial differentiation is predominantly a postnatal event in rodents. As it is impossible to produce a maternal-fetal infection/inflammation in postnatal ages, it would be reasonable to simulate an infection at a relevant postnatal developmental stage to create a rodent model of PVL. We have sought to combine LPS injection with the hypoxic/ischemic model in mice at P6, to produce more clinically relevant PVL lesions. Second, the optimal proof-of-concept experimental approach for a causative role of a molecule necessitates the use of transgenic mice; genetic manipulations are much harder in the rat than in the mouse. Thus, we have sought to convert our experimental model of PVL from the rat to the mouse.
Mouse models of PVL will greatly facilitate 1) the study of disease biology and pathogenesis using available transgenic mouse strains; 2) conduction of drug trials to discover candidate agents with therapeutic potential; 3) testing of the utility of stem cells using immunodeficient mouse strains.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work is in part supported by grants from National Institutes of Health (RO1 NS059043 and RO1 ES015988), National Multiple Sclerosis Society, Roche Foundation for Anemia Research, Feldstein Medical Foundation, and Shriners Hospitals for Children.
We would like to thank Dr. David Pleasure for his suggestions. This work was in part supported by grants to W.D. from National Institutes of Health (RO1 NS059043 and RO1 ES015988), National Multiple Sclerosis Society, and Shriners Hospitals for Children. We declare no competing interests related to this article.
References
- Volpe JJ. Neurology of newborn. Philadelphia: Saunders; 2001. pp. 217–276. [Google Scholar]
- Deng W, Pleasure J, Pleasure D. Progress in periventricular leukomalacia. Arch. Neurol. 2008;65:1291–1295. doi: 10.1001/archneur.65.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J. Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]