

Abstract
Summary: Methyl-Analyzer is a python package that analyzes genome-wide DNA methylation data produced by the Methyl-MAPS (methylation mapping analysis by paired-end sequencing) method. Methyl-MAPS is an enzymatic-based method that uses both methylation-sensitive and -dependent enzymes covering >80% of CpG dinucleotides within mammalian genomes. It combines enzymatic-based approaches with high-throughput next-generation sequencing technology to provide whole genome DNA methylation profiles. Methyl-Analyzer processes and integrates sequencing reads from methylated and unmethylated compartments and estimates CpG methylation probabilities at single base resolution.
Availability and implementation: Methyl-Analyzer is available at http://github.com/epigenomics/methylmaps. Sample dataset is available for download at http://epigenomicspub.columbia.edu/methylanalyzer_data.html.
Contact: fgh3@columbia.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
DNA methylation plays a critical role in gene silencing and chromatin remodeling (Bird, 2002; Klose and Bird, 2006). DNA methylation is also involved in the transcriptional repression of retrotransposons, genomic imprinting and X-chromosome inactivation in females. In mammals, 5-methylcytosine occurs predominately in CpG dinucleotides. In recent years, a variety of methods, including DNA bisulfite conversion, enzymatic digestion and methylated DNA enrichment, have adapted high-throughput second-generation sequencing to delineate DNA methylation profiles. This presents a bioinformatics challenge in accounting for experimental biases in genome-wide DNA methylation approaches [for a comprehensive review see Robinson et al. (2010)]. Essentially, the concern is the impact of these biases on estimation of CpG methylation probabilities, which is highly dependent on the experimental assay used for DNA methylation profiling (noted above). Methyl-Analyzer was developed to analyze data from the Methyl-MAPS assay (Edwards et al., 2010; Rollins et al., 2006).
Methyl-MAPS utilizes both methylation-sensitive restriction enzymes (RE: AciI, HhaI, HpaII, HpyCH4V, and BstUI) and the methylation-dependent endonuclease (McrBC). Fragments from each digestion are made into mate-pair libraries, and 5′ - and 3′ ends are sequenced to produce forward (F3) and reverse (R3) tags. The challenge in analyzing Methyl-MAPS data is in handling the intraindividual sampling bias, because for each individual sample the methylated and unmethylated compartments from independent digestions by methylation-sensitive RE and -dependent McrBC endonucleases are sequenced separately. To adjust for such biases, Methyl-Analyzer was developed for the analysis of Methyl-MAPS data.
2 METHODS AND ALGORITHMS
Methyl-Analyzer pipeline handles downstream analyses, that is, it takes as input aligned and paired-end reads to a reference genome (Fig. 1). Although the pipeline was initially developed based on Life Technologies SOLiD sequencing platform, it can be easily extended for analysis of sequence data from other next-generation sequencing platforms.
Fig. 1.

Methyl-Analyzer data analysis pipeline.
2.1 Parsing mate-pair reads
Based on the Methyl-MAPS protocol, genomic DNA is digested by RE and McrBC, and used in preparation of two independent mate-pair libraries for next-generation sequencing. F3 and R3 reads of mate-pair libraries are aligned and paired using software specific to the sequencing platform in use. The parsing script screens for properly paired reads that are on the same strand, and having the correct orientation, ordering, and insert size. The fragments formed by properly paired reads are referred to as methylated (from RE digestion) and unmethylated (from McrBC digestion) fragments, respectively.
The parsing script supports both mates format (SOLiD platform) and SAM format. For the SOLiD platform, we have used SOLiD System Analysis Pipeline Tool (Corona Lite) to analyze F3 and R3 reads. Pairing results can be saved in both mates and SAM formats. For other sequencing platforms, the aligned and paired reads are to be saved in SAM format.
2.2 Filtering methylated/unmethylated fragments
Since Methyl-MAPS is an enzymatic-based methylation method, a filtering step is essential to ensure that all correctly paired fragments have an enzyme recognition site in at least one end of the fragment. Fragments that do not contain such recognition sites are removed from further analysis.
2.3 Estimating CpG methylation probability
This is the core module of Methyl-Analyzer that estimates CpG methylation probabilities based on combined coverage of RE (methylated) and McrBC (unmethylated) fragments across CpG dinucleotides. Since for a given sample, these methylated and unmethylated fragments are sequenced in independent sequencing runs, intraindividual sampling bias is a concern that needs to be accounted for in the data analysis. There is no a priori knowledge of the sampling probabilities q1 and q2, respectively, for the sequence fragments from the methylated and unmethylated compartments. However, we can estimate the ratio of sampling probabilities (q1/q2) by,
where the parameters above are defined as: (i) 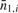 , average number of observed methylated fragments for all CpGs in a 1 kb segment i; (ii)
, average number of observed methylated fragments for all CpGs in a 1 kb segment i; (ii) 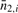 , average of observed unmethylated fragments for all CpGs in a 1 kb segment i; and (iii)
, average of observed unmethylated fragments for all CpGs in a 1 kb segment i; and (iii) 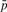 , global CpG methylation level determined via the LUminometric Methylation Assay (LUMA) (Karimi et al., 2006). When extremely biased sampling ratios (
, global CpG methylation level determined via the LUminometric Methylation Assay (LUMA) (Karimi et al., 2006). When extremely biased sampling ratios (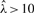 or
or 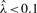 ) of the two libraries are encountered, Methyl-Analyzer generates an error alerting the user to check the data quality. For a CpG site k with observed n1,k, methylated and n2,k unmethylated fragments, the methylation probability is then calculated with,
) of the two libraries are encountered, Methyl-Analyzer generates an error alerting the user to check the data quality. For a CpG site k with observed n1,k, methylated and n2,k unmethylated fragments, the methylation probability is then calculated with,
where 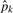 ranges from 0 (unmethylated) to 1 (methylated).
ranges from 0 (unmethylated) to 1 (methylated).
2.4 Visualization of DNA methylation profiles
The Methyl-MAPS assay produces large numbers of sequencing reads that need to be processed and displayed in a user-friendly fashion. Methyl-Analyzer provides scripts to create BED, Wiggle and microarray files that can be uploaded to UCSC Genome Browser as custom tracks. As shown in Supplementary Figure S1, one can visually inspect methylated and unmethylated fragments and CpG methylation probabilities using such custom tracks.
3 RESULTS AND DISCUSSION
Methyl-Analyzer is a python package that processes next-generation sequencing data from the Methyl-MAPS assay. It can be run on any platform with an existing python installation. Methyl-Analyzer has been used for analysis of data from a number of Methyl-MAPS experiments involving human and mouse brain tissue to generate methylomes of these large genomes. Depending on the sequencing depth, the numbers of input reads can vary. However, results from each step of Methyl-Analyzer pipeline are generally consistent. The parsing step passes ~90% of paired F3 and R3 reads on average as properly paired reads, and the filtering step passes an average of 84% of properly paired reads (Supplementary Fig. S2). This demonstrates that the Methyl-MAPS assay can consistently produce reliable DNA methylation profiling data. An example of a regional methylation profile is illustrated in Supplementary Figure S1, where the CpG island is unmethylated with expected low methylation probabilities, and the remaining genic/inter-genic regions showing high methylation. Results for one of our published human DNA methylomes are available at our UCSC Genome Browser mirror site: http://epigenomicspub.columbia.edu/index.html.
We also evaluated the performance of Methyl-Analyzer. Using a dataset with 25 million mate-pair reads, we ran the pipeline in a sequential mode for 29 h on a Mac Pro with dual core and 4 GB of RAM. Supplementary Figure S3 shows runtimes for each chromosome. Alternatively, the scripts specified by chromosomes can be submitted to computer clusters by a job scheduler, which will significantly reduce the total runtime. Methyl-Analyzer is a highly flexible software package suited for processing and running datasets of varying size. In conclusion, Methyl-Analyzer provides a pipeline to build DNA methylation profiles of large genomes. The scripts are easy to use and the pipeline has been automated to a large extent. This package is intended for use in analysis of Methyl-MAPS data to characterize whole-genome DNA methylation patterns in a fast and cost-efficient manner.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Mr. Ramiro Costa for setting up the UCSC Genome Browser Mirror site.
Funding: National Institute of Health (NIH) (HG002915 to F.G.H.); National Institute of Mental Health (NIMH) (MH074118 to F.G.H.).
Conflict of Interest: none declared.
REFERENCES
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Edwards J.R., et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M., et al. LUMA (LUminometric Methylation Assay)–a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res. 2006;312:1989–1995. doi: 10.1016/j.yexcr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., et al. Protocol matters: which methylome are you actually studying? Epigenomics. 2010;2:587–598. doi: 10.2217/epi.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins R.A., et al. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.