

Abstract
Cardiac myocytes isolated from adult hearts are widely accepted as a model somewhere half way between embryonic and neonatal muscle cells on one side and a working heart on the other. Thus, cardiomyocytes serve as good models for cardiac cellular physiology and pathophysiology, for pharmaceutical investigations as well as for the exploration of transgenic animal models. Here we describe a method of isolating the cells from the heart. Furthermore we show how a genetic manipulation on cardiac myocytes can be performed without breeding a transgenic animal: This is the combination of long term culture (1 week) and adenoviral gene transfer. The latter one is described from the construction of the virus to the transduction of the cells. It can be used for the expression of genetically encoded biosensors (GEBs), fluorescent fusion proteins, but also for protein over expression and down regulation, e.g. using RNAi. Here we provide an example for the expression of a fusion protein staining a subcellular structure (Golgi). Such protein expression can be visualized by confocal imaging of z-stacks for a 3D-reconstruction of subcellular structures. The protocol comprises state-of-the-art in cell culture, molecular biology and biophysics and thus provides an approach for exploring new horizons in cellular cardiology.
Protocol
The over all design of the protocol containing four major steps is depicted in Figure 1. All steps are described in full detail. Cell isolation, transduction and culture, followed by confocal imaging about 24 hours later is a consecutive and compulsory timescale. The virus construction should be done ahead of the cell isolation and is then used for the genetic transduction.
1. Isolation of adult cardiac myocytes from rat heart
Adult male Wistar rats approximately 250 g in weight (6-12 weeks old) are anesthetised by intraperitoneal injection of 170 μl anaesthesia solution per 100 g bodyweight
Additionally, a citrate solution (2 μl / g body weight) is injected to avoid blood coagulation during the surgery.
Langendorff Perfusion Set-Up should be prepared. All solutions should be saturated with oxygen.
When the rat is anesthetised, the animal is fixed on a surgery board and the upper part of the body is disinfected with 70% ethanol.
The animal is killed by cutting through the carotid.
The Thorax is opened using scissors. The heart and the aorta should now be accessible. The pericard is removed.
Ice-cold solution A+ (5 ml) is injected in both ventricles using a 26-gauge needle to arrest the heart and wash out blood.
The aorta is grabbed with a clamp and truncated at the aortic arch just were the first branching occurs.
The heart is removed from the chest and the aorta is attached to the cannula of a perfusion set-up by forceps and a clamp to allow retrograde Langendorff-perfusion. Be aware not to foraminate the aortic valves.
The heart needs to be fixed by a ligature around the aorta/cannula. Open vessels also need to be ligated.
The heart is retrogradely perfused with oxygen-saturated solution A+ at room temperature (23°C) for 10 min at a pressure of approximately 70 mbar. Make sure no air bubbles enter the aorta.
The perfusion solution is switched to a Liberase solution at a temperature of 37°C for approximately 25 min at a rate of 4 ml/min by means of a peristaltic pump. At the end of the perfusion the heart should look marbled white. The digestion time may need to be adapted to the situation/tissue condition.
The heart is taken from the perfusion set-up and placed in a Petri dish containing solution A at a temperature of 37°C. Remaining vessels and atria are cut off. The vessels are discarded and the atria - if needed - are treated differently (not part of this protocol). The ventricles are devided into approximately 6 pieces.
The pieces are transferred into a 100 ml Erlenmeyer flask containing 20 ml solution A at a temperature of 37°C and slightly agitated in a 37°C water bath for 5 min. Finally the supernatant is discarded.
The procedure described above is repeated once or twice. The supernatant should appear slightly opaque.
The increment of the extracellular calcium concentration is initiated by adding 20 ml of solution B1/2 at 37°C to the cells and tissue remaining a residual of solution A in the Erlenmeyer flask. This is slightly shaken in a 37°C water bath for an additional 5 min. Subsequently the supernatant is discarded.
Add further 20 ml solution B1/2. (The solution/supernatant should now contain viable cells.) A drop of the cells is transferred to a petri dish on an inverted cell culture microscope. If the cells show a high spontaneous activity (contractions) one needs to wait until cells come to rest.
Once more the supernatant is discarded and 20 ml of solution B1/2 are added.
The tip of a plastic Pasteur pipette is cut off in such a way that the tissue pieces can pass easily when agitated through the opening. With flame treatment sharp edges are softened (melted).
Now the tissue pieces are slightly and gently triturated. The tissue pieces should "fall apart" into the cells.
The cell containing supernatant is decanted and diluted with solution B to reach the desired cell density, which depends on the type of experiments and should be determined empirically.
2. Construction of an adenovirus for fusion protein transduction
To produce adenoviruses the Transpose-Ad Adenoviral Vector System from MP Biomedicals is used1. This protocol starts with the available and tested entry vector (pCR259) for a fluorescent fusion protein or GEB s2.
An amount of 10 ng of the above mentioned initial pCR259 vector containing the gene of interest is transformed with 100 μl HighQ-1 Transpose-AD™ 294 competent cells.
This mixture is incubated for 20 min on ice followed by a heat shock for 45 s at 42°C and stored on ice for an additional 2 min.
Add 900 μl of unselective LB-medium and grow at 37°C with gentle agitation for 4 hours.
Plate 50 μl, 100 μl and 200 μl on plates containing LB-medium with Chloramphenicol (Cm, 20 μg/ml), Ampicillin (100 μg/ml), tetracycline (15 μg/ml), X-Gal (280 μg/ml) and IPTG (40 μg/ml). (A more intense and faster blue staining is possible by using the more expensive dye Bluo-Gal with 300μg/ml. This should be used in particular when you are not so familiar in recognizing and distinguishing blue/white colonies)
Grow for 24 hours at 37°C. Leave at 4°C until good identification of blue colonies is possible. When there are problems to decide which colony is truly white pick some of the colonies and replicate on another plate containing the three antibiotics, X-Gal and IPTG and let them grow over night at 37°C. Remark) The HighQ-1 Transpose-AD™ 294 cells contain two important plasmids: The adenovirus backbone (Transpose-Ad™ 294) and a helper plasmid expressing a transposase, which mediates the insertion of the gene of interest into its target sites, located in the LacZ gene of the Transpose-AD 294 plasmid.
To identify positive colonies containing the gene of interest in the virus backbone perform colony-PCR as described in 2.7-2.9. Use vector primers flanking the multiple cloning site (forward 5'CCCAGTCACGACGTTGTAAAACG3', reverse 5'GCGGATAACAATTTCACACAGG3') and make a colony PCR with 100% white colonies and use one blue colony as a control for Transpose-AD 294 plasmids without inserted gene of interest. Blue colonies will give you a short PCR fragment (~200bp), colonies containing your gene of interest show a large PCR product (depending on the length of your gene of interest) and colonies with two fragments are multiple cell colonies and have to be separated if further used.
According to the table below a master mix needs to be prepared.
Pick a single 100% white colony with an autoclaved toothpick and first make a replicate of this colony on a LB-plate with Cm.
Transfer the colony into the PCR-mix. Twirl it in the PCR-mix 3-4 times than remove the toothpick before it could soak a lot of the PCR-mix. The PCR-protocol depends on the kb of the insert and the used polymerase. The annealing temperature of the primers is 57°C and a use of 35 cycles is recommended.
Grow over night cultures (LB-medium with Cm) of the positive clones and isolate the plasmids with a commercial available kit without spin columns, because the centrifugal force disrupts the huge virus vector.
Transform 0.5 μg of the isolated plasmids in 100 μl DH5α competent cells. Incubate DNA with cells on ice for 30 min. Heat shock 45 s at 42°C and set on ice for 2 min.
Add 900 μl unselective LB-medium and grow at 37°C with gentle agitation for 1 hour.
Plate 50 μl on a plate containing LB-medium with Cm, X-Gal and IPTG. Incubate at 37°C for 24 hours.
Pick and replicate up to 20 100% white colonies on separate Ampicillin, Tetracycline and Chloramphenicol plates containing IPTG and X-Gal. Let them grow over night at 37°C. Select colonies which are 100% white, chloramphenicol resistant, ampicillin and tetracyclin sensitive.
Colony-PCR (optional): positive clones can be tested again with colony-PCR to check that they contain the insert.
Pick a single colony of a positive recombinant and grow in 600 ml LB-medium with Cm.
Isolate the plasmid using a plasmid maxi kit.
Digest 5 μg of plasmid with Pac I for two hours at 37°C and stop the reaction at 65°C for 20 min.
Concentrate the whole digest in a SpeedVac at room temperature for 30 min to a final volume of 10 μl or use a phenol-chloroform extraction method.
Thaw a vial of frozen QBI-HEK 293 cells with gentle agitation in a 37°C water bath. Rinse the outside of the vial with 70% ethanol and proceed aseptically in a sterile hood.
Transfer cells to a sterile 15 ml tube, add dropwise 10 ml DMEM (+10% FCS) while gentle agitating and centrifuge 10 min at 250xg to pellet the cells.
Discard supernatant. Resuspend the pellet in 10 ml DMEM (+10% FCS) by gently pipetting up and down.
Seed in a 75 cm2 flask and add 10 ml DMEM with 10% FCS to the cells. Incubate over night at 37°C in a 5% CO2 incubator.
The next day verify the cell density. QBI-HEK 293 cells should be split at a 1:10 to 1:20 ratio.
Remove the medium from the cells and trypsinize with 2 ml Trypsin for 1-3 min. Give 10 ml DMEM to the cells and centrifuge at 250xg for 10 min.
Remove carefully the medium and give 10 ml fresh medium to the cells, resuspend and count the cells.
Plate in a six-well one well with 2x105, 3x105, 4x105, 5x105 and 6x105 cells. Incubate the dish at 37°C in a CO2-incubator.
At the following day use a well with 50% confluence to transfect with the concentrated Pac I digest. We have good experience with Lipofectamine 2000 transfection, but also other methods (such as calcium phosphate transfection) should be suitable.
Change the medium in the well 3-4 h before transfection to get the cells into exponential growth during the procedure.
Mix 5 μl Lipofectamine with 250 μl DMEM and incubate at room temperature for 5 min.
Give the Lipofectamine-Medium Mixture to the DNA (Pac I digest) and mix by tapping against the reaction tube, do not mixing by pipetting. Incubate the mixture 20 min at room temperature.
Drop the transfection mixture to the cells and rock the plate to mix. Incubate at 37°C with 5% CO2. Change medium after 4-5 hours.
Two days after transfection take the medium from the cells and give it into a fresh culture flask (75 cm2).
Trypsinize the cells with 500 μl trypsin for up to 5 min. Give 1 ml DMEM to the cells, take the whole mixture and put it, together with 17 ml fresh medium (DMEM with FCS) into the culture flask. Cultivate the cells at 37°C with 5%CO2 until cytopathic effects (CPE) are visible. See Figure 2 to learn how CPE should look like.
Collect the cells with the medium in a 50 ml tube and freeze the cells at -80°C. Break the cells with three freeze/thaw cycles (-80°C/37°C water bath).
Centrifuge 20 min at 8000xg and 4°C. Decant the supernatant into a 100mm-diameter dish and discard the pellet. Use a sterile filter with 0.45μm pore size to filtrate the supernatant. This is the first virus stock (passage 1).
Use 1/3 of this stock to transduce a 75cm2 flask with QBI-HEK 293 cells with 70% confluence. Incubate at 37°C and 5%CO2 until CPE s are visible.
If the cells show CPE s collect them and extract the viruses as done before. This is passage 2 of the virus.
Repeat steps 2.37 and 2.38 with virus passage 2 and so on until the amplification of the viruses reaches a sufficient virus stock. The virus can be concentrated with specific centrifugation tubes (Amicon Ultra-15 Centrifugal filter unit with ultracel-100 membrane from Millipore) and stored for several months at -80°C. To count the infectious particles use a plaque assay.
Use virus passage 2 or higher to transduce cells and for further amplification of the virus.
3. Primary cell culture and adenoviral transduction
Sterile round cover slips of 20 mm diameter are placed in standard 12-well plates.
20 μl ECM solution is equally distributed on each cover slip. Remains are discarded. For drying allow a minimum of 1 h at 37° C. Alternatively the coating can by dried over night (12 h) at room temperature.
Each well is filled with 1 ml Medium M199 with supplements comprising antibiotics and ITS.
A cell suspension (from step 1.21) of 200 to 400 μl is added to each well. Culturing of the cells occurs at 37°C in a 5% CO2 incubator.
Cells are allowed to sediment and after 1 h medium is changed.
Immediately after changing the medium an adequate amount of virus containing solution (from passage 2 or higher) is mixed by gently rocking the plate.
In case of a long term culture medium is changed every 2 days.
24 hours after transfection GEB expression is detectable.
4. Confocal imaging for 3D subcellular structures and Ca2+ Signaling
Confocal imaging can be performed on any confocal microscope. However, calcium-imaging experiments require an acquisition speed of at least video rate (25-30 Hz). This restricts usable technologies to either multi beam scanners (e.g. Nipkow disc, swept field or kilo-beam array scanner) or the very fast single beam scanner (e.g. resonant scanner or acousto optical deflector (AOD)-scanner). For an overview see Ref. 3. The 3D-subcellular imaging requires a z-drive. Here a kilo-beam array scanner (VTinfinity, VisiTech Int., Sunderland, UK) is used.
The microscope and peripheral equipment needs to be set into an operation mode. Especially for the laser this may require a warming up period until a stable output power is reached.
Cells need to be transferred to an experimental chamber (cover-slip transfer). The coverslips with cells are transferred to an experimental chamber. 1mL Tyrode solution (containing 1.8 mM extracellular calcium) is then added.
The experimental chamber needs to be placed on the microscope stage. Stimulation electrodes, perfusion set-up and further peripheral devices, e.g. temperature control, need to be arranged accordingly.
Cells will be focused at and selected using white light and/or epifluorescent illumination.
Acquisition parameters such as laser power, exposure time etc. or an entire experiment protocol needs to be set-up.
Time series and/or Z-section images can be acquired.
On the set-up further advanced multimode measurements can be done using electrophysiology (patch-clamp) or/and optical manipulation like fluorescence redistribution after photobleach (FRAP) or uncaging of substances including two photon photolysis.
Image analysis, especially 3D-reconstruction is performed on dedicated analysis software such as Imaris (Bitplane AG, Zürich, Switzerland), VeloCity (Improvision, Coventry, UK) or Arivis Browser (arivis GmbH, Rostock, Germany).
5. Representative Results:
The final outcomes of the protocol are image sequences that can be used to extract further data.
An example is the investigation of subcellular structures and organelles. Figure 3 depicts the example of the 3-dimensional arrangement of the Golgi apparatus. In contrast to electron microscopy all investigations can be performed on living cells.
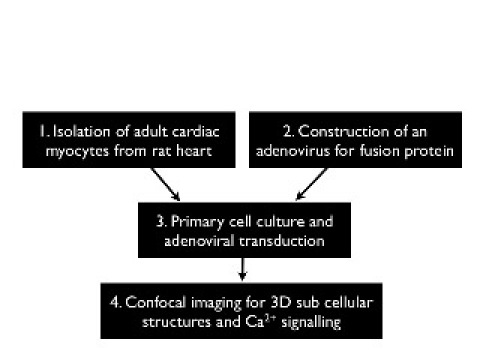 Figure 1: General flow of the major experimental steps.
Figure 1: General flow of the major experimental steps.
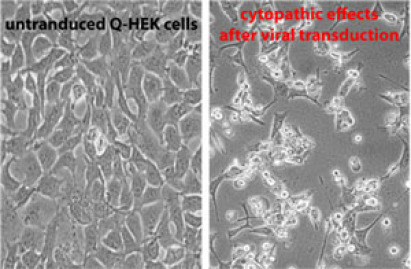 Figure 2: The left image depicts the Q-HEK cells in culture with 90-95% confluency which are not transduced. Cytopatic effects after viral transduction are shown in the right part. Cells round up and detach from the bottom of the plate. In these cells the nucleus occupies the major part of the cells as a result of active virus production.
Figure 2: The left image depicts the Q-HEK cells in culture with 90-95% confluency which are not transduced. Cytopatic effects after viral transduction are shown in the right part. Cells round up and detach from the bottom of the plate. In these cells the nucleus occupies the major part of the cells as a result of active virus production.
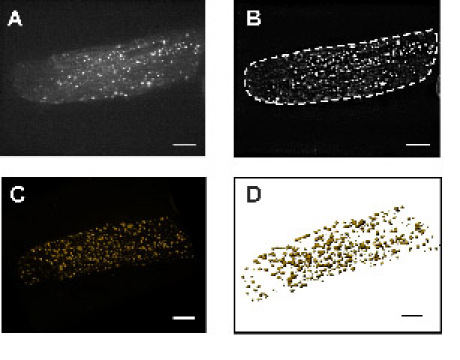 Figure 3: Adult rat cardiac myocyte transfected with an YFP-fusion protein targeting the Golgi apparatus. Cardiac myocytes after adenoviral gene transfer (one day in culture) express the fluorescent fusion protein. Panel A depicts a single confocal slice taken from a z-stack. In panel B the same cell is depicted after deconvolution using a theoretical point spread function (PSF). This de-blurred image stack is subsequently used to render the cell and get a 3D volume reconstruction as shown in panel C or a 3D surface reconstruction (D). The bar represents 10 μm.
Figure 3: Adult rat cardiac myocyte transfected with an YFP-fusion protein targeting the Golgi apparatus. Cardiac myocytes after adenoviral gene transfer (one day in culture) express the fluorescent fusion protein. Panel A depicts a single confocal slice taken from a z-stack. In panel B the same cell is depicted after deconvolution using a theoretical point spread function (PSF). This de-blurred image stack is subsequently used to render the cell and get a 3D volume reconstruction as shown in panel C or a 3D surface reconstruction (D). The bar represents 10 μm.
Discussion
The procedure described for the isolation of rat cardiomyocytes can be adapted to other species, e.g. mouse4, if necessary. A parameter that needs adaptation is the enzyme mix for digestion (see below) and also the duration of digestion. Be aware that the cells of some species (e.g. mouse) might be more fragile than others (e.g. rat).
The choice of collagenase is probably the most crucial step in the isolation procedure. We choose Liberase Blendzyme because it is a synthetic enzymatic mix with virtually no batch-to-batch variations. The conventional method is the use of so called crude collagenase extracted from Clostridium histolyticum, which is also a valid method. However, batch-to-batch variations require new optimizing for each new batch.
Apart from the fluorescent fusion proteins and the GEB transduction the adenoviral gene transfer can also be used for over expression of functional proteins or their down regulation (e.g. by RNAi5). This holds true because transduction efficiency is high (> 95%) and reproducible.
Acknowledgments
This work was supported by the German National Science Foundation (DFG) and by the Federal Institute for Risk Assessment (BfR, Germany).
References
- Kaestner L, Ruppenthal S, S . In: Molecular Imaging II. Licha K, Lin CP, editors. Vol. 7370. Bellingham: SPIE; 2009. pp. 737008–737008. [Google Scholar]
- Viero C, Kraushaar U, Ruppenthal S. In vivo imaging of Drosophila melanogaster pupae with mesoscopic fluorescence tomography. Cell Calcium. 2008;43(1):59–59. [Google Scholar]
- Kaestner L, Lipp P. Shorte SL, Frischknecht F. Imaging Cellular and Molecular Biological Functions. Berlin Heidelberg: Springer; 2007. pp. 289–289. [Google Scholar]
- Kaestner L, Lipp P. In: Optics in Life Science. Popp J, von Bally G, editors. Vol. 6633. Munich: SPIE; 2007. pp. 66330K–66330K. [Google Scholar]
- Rinne A, Littwitz C, Bender K. Adenovirus-mediated delivery of short hairpin RNA (shRNA) mediates efficient gene silencing in terminally differentiated cardiac myocytes. Methods Mol Biol. 2009;515:107–107. doi: 10.1007/978-1-59745-559-6_7. [DOI] [PubMed] [Google Scholar]