

Abstract
Although human embryonic stem cells (hESC) have been shown to present a stable diploid karyotype 1, many studies have reported that depending on culture conditions they become prone to acquire chromosomal anomalies such as addition of whole or parts of chromosomes. Indeed, during long-term culture, karyotypic alterations are observed when enzymatic or chemical dissociation are used 2,3,4, while manual dissection of colonies for passaging retains a stable karyotype 5. Besides, changes in the environment such as the removal of feeder cells also seem to compromise the genetic integrity of hESC 3,6. Once chromosomal alterations could affect cellular physiology, the characterization of the genetic integrity of hESC invitro is crucial considering hESC as an essential tool in embryogenesis studies and drug testing. Furthermore, for future therapeutic purposes chromosomal changes are a real concern as it is frequently associated to carcinogenesis.
Here we show a simple and useful method to obtain high quality chromosome spreads for subsequent analysis of chromosome set by G-banding, FISH, SKY or CGH techniques 7,8. We recommend checking the chromosomal status routinely with intervals of 5 passages in order to monitor the appearance of translocations and aneuploidies
Priscila Britto and Rafaela Sartore contributed equally to the paper.
Protocol
EQUIPMENT
Tissue culture incubator, 37°C, 5% CO2
Centrifuge
Set of micropipettors (100, 100μL)
Water bath 37°C
Water bath 90°C to be source of steam water
Phase contrast microscope
FIRST STEP
Treating the cells with colcemid
In this procedure we used the hESC line H9 cultured onto mouse embryonic fibroblasts (mEF) inactivated with mitomycin C (Sigma) and maintained in DMEM/F12 (Invitrogen) supplemented with 20% knockout serum replacement (KSR, Gibco) and 8ng/mL of fibroblast growth factor (FGF-2, R&D).
In order to obtain a large number of metaphases spreads, it is recommended to use cultures with high mitotic index in which there are many dividing cells.
Take the culture flask from the incubator and, in the laminar flow, add Colcemid at a final concentration of 0,1μg/mL.
Return the flask to the incubator and wait 3 hours so the inhibitor can cause mitotic arrest on metaphase, when the chromosomes are well condensed and can be easily visualized under the microscope.
Fixing the cells
Take out the medium and add tripsin/EDTA 0.05% sufficient to cover the cells surface. Allow cells to incubate for 5 minutes at room temperature and then inactivate the enzyme either by adding culture medium containing serum or by just adding serum to the cells. NOTE: It is important to make sure that a single cell suspension is achieved to obtain good metaphase spreads.
Transfer the cells to a 15 mL centrifuge tube and centrifuge at 122 g for 5 minutes.
Discard the supernatant, leaving about 200 mL, and resuspend the pellet gently by tapping the tube, as shown in the video. Make sure the cells are well dissociated, so you can't visualize any clumps in the solution.
Next you will need to add 5 mL of hypotonic solution (KCl 75mM) pre-warmed to 37°C, slowly, by the tube wall as in the following description. First add 3 ml, invert the tube to a horizontal position just to mix the cells with the hypotonic solution then add the remaining 2 ml and keep warm at 37°C for 15 minutes.NOTE: At this point the hypotonic solution will cause an increase on the cellular volume and help to untangle the chromosomes. The time of incubation is important to acquire good chromosome spreads. If the timing is too long, the cell membrane may burst too early and chromosomes are lost. If too short, it may be difficult to obtain chromosome spreads because the cell membrane may not disrupt.
Add 3 drops of the fixative solution (methanol/glacial acetic acid 3:1), invert the tube gently to mix the fixative solution with the cells and centrifuge at 122 g for 5 minutes without break. NOTE: The fixative solution should be freshly made. NOTE: It is important to use centrifugation without break to avoid cells attrition, preventing loss of the material.
Discard the supernatant, leaving about 200 mL, and resuspend the pellet gently by tapping the bottom of the tube. NOTE: Again, make sure the cells are well dissociated, so you can't visualize any clumps in the solution.
Add the first 3 mL of fixative solution, slowly by the tube wall, while you rotate the tube. Invert the tube to a horizontal position just to mix the cells, and then add more 2 mL of the fixative solution by the tube wall to take the cells down. Stock at 4°C overnight. NOTE: The fixed cells can be stored in fixative solution for months at 4°C. Besides protecting cells in their swollen state, the fixative solution removes lipids and denatures proteins. These events make the cell membrane fragile and, as a result, make the chromosome spreading easily.
CLEANING THE GLASS SLIDES
Before starting making your chromosome spreads make sure that your slides are properly cleaned or you may lose some nuclei and also metaphases. This step is as well important if you want to try any hybridization technique 9.
Place slides in a beaker containing 6M HCl for at least 3 h at room temperature.
After this time, wash slides in running tap water for 10 minutes.
Rinse the slides in distilled water and let they dry at room temperature.
Now the slides can be stored into 96% ethanol and dried with a lint-free immediately prior to use.
NOTE: For in situ hybridization methods, the slides cannot be stored for more than 2 days into 96% ethanol
SECOND STEP
Washing the cells
On the next day, centrifuge the cells at 122 g for 5 minutes without break.
Discard the supernatant leaving about 200 ml. Resuspend the pellet by gently flicking the tube then add 5 ml of fresh fixative solution (methanol/glacial acetic acid 3:1), slowly by the tube wall while rotating the tube. Repeat this wash step 2 more times for a total of three washes. NOTE: The fixative solution should be freshly made.
Preparing the slides
At the last centrifugation step leave sufficient amount of solution to homogenize the cell pellet and obtain an adequate density of cells in the slides (see figure 1).
Add 20-30 μL of cell suspension onto a clean dry slide by moving the pipette very close and parallel to the surface. As the fixative solution evaporates, the surface of slide becomes grainy.
Immediately, expose the slide, face up, into the steam of hot water (90°) for 30 seconds to cause the cells blow up. NOTE: At this moment, the methanol from the fixative solution evaporates, resulting in an increased acetic acid concentration, which together with the water stimulates the chromosome spreading.
Look at a phase contrast microscope just to check if the concentration as well as the cell distribution is good (see figure 1).
Now the slides are ready to be used to check the set of chromosomes by cytogenetics approaches as G-banding (Giemsa), FISH, SKY or CGH.
IMPORTANT HINTS
How choose an adequate metaphase to be analyzed
Choosing round and isolated metaphases (figure 2D), you may avoid considering false aneuploidies as the gain of chromosomes generated by two or more spreads mixed together or the loss of chromosomes generated by a launched chromosome. So, avoid choosing metaphases near nuclei because some chromosomes may be hidden by them (figure 2A and 2B). Besides, look for round metaphases and be sure that any chromosome is separated from the metaphase (figure 2C).
Feeder cells and the hESC karyotype
For G-banding and SKY analysis, the presence of feeder cells in hESC culture does not interfere with the results, because feeder cells are not under a dividing state (inactivated by mitomycin C or irradiation) and will not be part of the metaphase population. However, for the application of FISH technique in interphase nuclei, it is preferred to separate the hESC population from feeder cells by harvesting the colonies manually before trypsinization for FISH analysis.
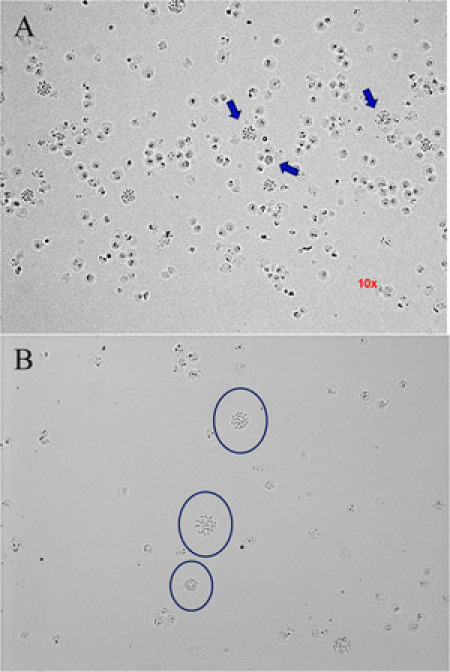 Figure 1: How know the appropriate cell density in the slides. Images of phase contrast microscope in 10x objective. (A) The cell density is too high. The arrows point to metaphase spreads too close of nuclei. (B) A slide with good cell density. The circles show metaphases spreads isolated from nuclei or other metaphases. Please click here to see a larger version of figure 1.
Figure 1: How know the appropriate cell density in the slides. Images of phase contrast microscope in 10x objective. (A) The cell density is too high. The arrows point to metaphase spreads too close of nuclei. (B) A slide with good cell density. The circles show metaphases spreads isolated from nuclei or other metaphases. Please click here to see a larger version of figure 1.
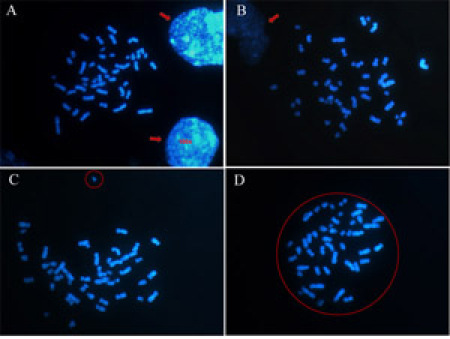 Figure 2: How to choose an adequate metaphase to be analysed. Chromosomes are stained with DAPI and the images obtained by a fluorescent microscope in 100x objective. (A) (B) Avoid metaphases near nuclei (indicated by the arrows) (C) and metaphases not round that present chromosomes separated (circle). (D) A round metaphase is a good metaphase to be analysed. Please click here to see a larger version of figure 2.
Figure 2: How to choose an adequate metaphase to be analysed. Chromosomes are stained with DAPI and the images obtained by a fluorescent microscope in 100x objective. (A) (B) Avoid metaphases near nuclei (indicated by the arrows) (C) and metaphases not round that present chromosomes separated (circle). (D) A round metaphase is a good metaphase to be analysed. Please click here to see a larger version of figure 2.
Discussion
The preparation of chromosome spreads is a critical step for a successful analysis of the genetic status of embryonic stem cells by routinely techniques as G-banding, and more sophisticated techniques such as FISH, SKY and CGH.
This procedure can be applied for many cell types by varying the period of colcemid incubation, which depends on the cell cycle length. For colonies of embryonic stem cells, we wait 3 hours while for embryoid bodies (EB) this time can be up to 6 hours.
In the case of working with cell aggregates (like embryoid bodies) you will need to dissociate very well in order to obtain a single cell solution. In this case, after trypsin and mechanical dissociation, you can make use of a 40μm nylon cell strainer (BD Biosciences) and then give sequence to the procedure (from step 4 of the procedures topic).
In the procedure here described it is worth noting that the cell suspension is dropped "closely" onto the slides in order to avoid excessive spreading of the chromosomes and consequently generation of artifacts.
We believe that this protocol is an useful and simple tool to be routinely applied not only by laboratories dealing with embryonic stem cells but by other investigators interested in studying chromosomal instability in other stem cells.
References
- Thomson JA. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1145. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Draper JS. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22(1):53–53. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Maitra A. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37(10):1099–1099. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- Mitalipova MM. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23(1):19–19. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- Buzzard JJ, Gough NM, Crook JM, Colman A. Karyotype of human ES cells during extended culture. Nat Biotechnol. 2004;22(4):381–381. doi: 10.1038/nbt0404-381. [DOI] [PubMed] [Google Scholar]
- Caisander G. Chromosomal integrity maintained in five human embryonic stem cell lines after prolonged in vitro culture. Chromosome Res. 2006;14(2):131–131. doi: 10.1007/s10577-006-1019-8. [DOI] [PubMed] [Google Scholar]
- Imreh MP. In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. J Cell Biochem. 2006;99(2):508–508. doi: 10.1002/jcb.20897. [DOI] [PubMed] [Google Scholar]
- Robin-Wesselschmidt JL. In: Human Stem Cell Manual. Loring JF, Robin-Wesselschmidt JL, Robin-Wesselschmidt Schwartz, editors. California: Elsevier's Science & Technology Rights; 2007. pp. 59–59. [Google Scholar]
- Rooney DE. Human Cytogenetics. Third Edition. Oxford University Press; 2001. [Google Scholar]
- Heslop-Harrison P, Schwarzacher T. In: Practical in situ hybridization. Heslop-Harrison P, Schwarzacher T, editors. Oxford: BIOS Scientific Publishers; 2000. pp. 51–51. [Google Scholar]