

Abstract
The fate of infected macrophages has an essential role in protection against Mycobacterium tuberculosis by regulating innate and adaptive immunity. M. tuberculosis exploits cell necrosis to exit from macrophages and spread. In contrast, apoptosis, which is characterized by an intact plasma membrane, is an innate mechanism that results in lower bacterial viability. Virulent M. tuberculosis inhibits apoptosis and promotes necrotic cell death by inhibiting production of prostaglandin E2. Here we show that by activating the 5-lipoxygenase pathway, M. tuberculosis not only inhibited apoptosis but also prevented cross-presentation of its antigens by dendritic cells, which impeded the initiation of T cell immunity. Our results explain why T cell priming in response to M. tuberculosis is delayed and emphasize the importance of early immunity.
Mycobacterium tuberculosis is one of the most important bacterial pathogens1. The ability of M. tuberculosis to persist in people with apparently normal immune systems indicates that M. tuberculosis has developed strategies to avoid, evade and even subvert innate and adaptive immunity2. Although macrophages excel at destroying the biological particles they engulf by phagocytosis, which include dead cells and bacteria, M. tuberculosis has evolved into a parasite of the intracellular milieu of macrophages, where it not only survives but also replicates in the naturally hostile environment of the phagosome3,4. Intracellular pathogens such as M. tuberculosis have developed complex and multifactorial mechanisms by which to evade host defenses. One important strategy used by intracellular pathogens is subversion of the host cell’s death pathways5. Several intracellular pathogens, including Leishmania, Coxiella, Salmonella, Chlamydia and Yersinia, induce host-cell apoptosis as a way of minimizing the inflammatory response and avoid detection6–8. In some of these cases, the induction of host-cell necrosis, sometimes by pyroptosis, leads to lower bacterial viability and is beneficial to the host9,10. Other acute bacterial pathogens, such as Pseudomonas, Neisseria and Streptococcus, are cleared by host-cell apoptosis, and these pathogens evade innate immunity by inhibiting apoptosis11. Virulent strains of M. tuberculosis induce necrosis of both human and mouse macrophages, whereas attenuated M. tuberculosis strains or other nonpathogenic mycobacterial species generally do not12–14. Virulent M. tuberculosis actively inhibits the induction of macrophage apoptosis15, consistent with the identification of M. tuberculosis mutants that induce apoptosis instead of necrosis16,17. In addition, there is accumulating evidence that apoptosis, whether induced by the pathogen itself18,19, pharmacologically20 or by cytotoxic lymphocytes21,22, results in lower viability of M. tuberculosis.
One strategy used by M. tuberculosis to avoid apoptosis is the subversion of host eicosanoid biosynthetic pathways. The attenuated M. tuberculosis strain H37Ra induces the production of prostaglandin E2 (PGE2), which protects the mitochondrial inner membrane and induces the repair of plasma membrane microdisruptions inflicted by the pathogen12,13. These events protect the host macrophages against necrosis and instead promote apoptosis. In contrast, intracellular infection with the virulent H37Rv strain of M. tuberculosis induces the production of lipoxin A4 (LXA4), which inhibits cyclooxygenase 2 production and PGE2 biosynthesis12,13. In a PGE2-poor microenvironment, macrophages cannot prevent the mitochondrial damage or repair the plasma membrane disruptions caused by M. tuberculosis, and this leads to necrosis12,13. Virulent M. tuberculosis in prenecrotic macrophages continues to replicate and to spread to uninfected macrophages after the cells are lysed. Thus, the balance of PGE2 and LXA4 regulates the relative amounts of apoptosis and necrosis after M. tuberculosis infection, with important functional consequences for innate control of the intracellular infection. The essential role of these host lipid pathways in innate immunity has been confirmed by genetic analysis of the susceptibility of zebrafish to Mycobacterium marinum23.
The cross-talk between innate immunity and adaptive immunity in response to intracellular infection is essential, particularly in the case of pathogens that infect macrophages. This is certainly true for immunity to M. tuberculosis, which requires complex synchronization and cooperation between the various arms of the immune system. The regulation of cell death during M. tuberculosis infection should be considered in this context because as well as limiting bacterial replication, apoptotic macrophages seem to provide an important bridge to adaptive immunity. Dendritic cells (DCs) can acquire viral or bacterial antigens through the uptake of apoptotic vesicles derived from infected macrophages24–26. Cross-presentation, initially described in the context of viral infection26, seems to occur during M. tuberculosis infection and to be important for priming of CD8+ T cell responses. For example, the secA2 mutant of M. tuberculosis, which induces more apoptosis of infected macrophages in vitro and in vivo, is associated with enhanced antigen-specific CD8+ T cell immunity, presumably as a consequence of more cross-priming16,27. Furthermore, purified apoptotic vesicles derived from bacillus Calmette-Guérin (BCG)-infected macrophages that are free of any viable bacteria can transfer mycobacterial antigens to human DCs, which can cross-present the antigens to human CD8+ and CD1-restricted T cells28. Similarly, vaccination of mice with purified apoptotic vesicles is sufficient to stimulate T cell immunity against the mycobacterial antigen cargo contained in the vesicles and to elicit protective immunity to challenge with virulent M. tuberculosis29.
The microbiological and immunological data outlined above support the ‘detour’ model30, in which apoptotic blebs derived from cells infected with intracellular pathogens facilitate cross-presentation of microbial antigens to CD8+ T cells. However, it is not known whether apoptosis of infected macrophages occurs in vivo and, if so, whether it has physiological importance. Because the supporting studies used apoptotic vesicles derived from BCG-infected macrophages28,29, it remains unclear whether infection of macrophages with wild-type virulent M. tuberculosis would lead to apoptosis and enhanced T cell immunity or whether virulent M. tuberculosis prevents presentation of its antigens and inhibits initiation of adaptive immunity by other mechanisms. Finally, although immunization with purified apoptotic vesicles leads to cross-priming of antigen-specific T cells by DCs, it is not known whether the same mechanism occurs after infection with M. tuberculosis in vivo. Low-dose aerosol infection in mice is characterized by a delay in the initiation of T cell immunity31,32, but it is unclear whether this delay is due to the inhibition of macrophage apoptosis by virulent M. tuberculosis. Although DCs are required to generate antigen-specific T cell responses33, DCs can be directly infected by M. tuberculosis34,35. M. tuberculosis–infected DCs in the lung traffic to the regional lymph node with kinetics that mirror those of T cell priming, consistent with the idea that M. tuberculosis–infected DCs prime M. tuberculosis–specific T cells directly. This indicates that there might be a priming pathway independent of the ‘detour’ pathway31,32,34. Thus, the role of apoptosis and cross-priming in the generation of adaptive immunity during virulent M. tuberculosis infection remains unclear.
Macrophages from mice deficient in 5-lipoxygenase (Alox5−/− mice), which cannot synthesize LXA4, undergo more apoptosis after infection with virulent M. tuberculosis12,13. Conversely, macrophages that lack prostaglandin E synthase (Ptges−/− mice), which cannot produce PGE2, undergo more necrosis after infection, even with avirulent M. tuberculosis strains. The greater resistance of Alox5−/− mice36 and the greater susceptibility of Ptges−/− mice13 to infection with virulent M. tuberculosis led us to hypothesize that apoptosis and its regulation by eicosanoids are key determinants of innate and adaptive host resistance to infection. We have developed an adoptive-transfer model using M. tuberculosis–infected macrophages to determine whether inhibition of apoptosis by virulent M. tuberculosis leads to less cross-priming by DCs and to impaired T cell immunity. We find that by inhibiting apoptosis, virulent M. tuberculosis downregulated cross-presentation of M. tuberculosis antigens and impaired T cell responses.
RESULTS
Alox5−/− mice have enhanced T cell responses
After being infected with M. tuberculosis, wild-type mice have lower expression of interleukin 12 (IL-12), interferon-γ (IFN-γ) and inducible NO synthase (iNOS) in their lungs than do Alox5−/− mice, which suggests that lipoxins negatively regulate type 1 immunity to tuberculosis36. To establish that the larger amounts of IL-12, IFN-γ and iNOS in Alox5−/− mice infected with M. tuberculosis correlate with enhanced T cell immunity, we quantified the antigen-specific T cell response in wild-type, Ptges−/− and Alox5−/− mice after aerosol infection with virulent M. tuberculosis. At 2 weeks after infection, the CD8+ T cell response to the immunodominant antigen TB10.4 was greater in the draining lymph nodes of Alox5−/− mice than in those of Ptges−/− or wild-type mice (Fig. 1a). The frequency of TB10.4-specific CD8+ T cells was significantly higher (14%) in the lungs of Alox5−/− mice infected with M. tuberculosis than in those of Ptges−/− mice (4%) or wild-type (5%) mice (Fig. 1b). Moreover, splenic T cells from infected Alox5−/− mice produced more IFN-γ after being stimulated with M. tuberculosis culture-filtrate proteins TB10.4(4–11) (a peptide of amino acids 4–11 of TB10.4, specific for CD8+ T cells) or Ag85B(241–256) (a peptide of amino acids 241–256 of Ag85B, specific for CD4+ T cells) than did cells from infected Ptges−/−or wild-type mice (data not shown). Kinetic analysis of the TB10.4-specific CD8+ T cell response indicated that the response in the Alox5−/− mice was earlier than that of wild-type or Ptges−/− mice (Fig. 1c). At 5 weeks after infection, the M. tuberculosis antigen–specific CD8+ T cell response in the lung and spleen was similar in the various experimental groups, although the lung bacterial burden was significantly higher in Ptges−/− mice and lower in Alox5−/− mice than in wild-type mice (P < 0.05; Supplementary Fig. 1). This observation emphasizes the importance of early T cell immunity in protection against M. tuberculosis infection. Collectively, these data show that after M. tuberculosis infection, the CD4+ and CD8+ T cell response to M. tuberculosis was earlier and stronger in Alox5−/− mice than in either wild-type mice or Ptges−/− mice. Thus, LXA4 negatively regulates T cell immunity to tuberculosis.
Figure 1.

Inhibition by 5-lipoxygenase of the early initiation of an immune response after M. tuberculosis infection. (a,b) Frequency of TB10.4(4–11)-specific CD8+ T cells in the thoracic draining lymph nodes (a) and lungs (b) of Ptges−/−, wild-type (WT) and Alox5−/− mice 2 weeks after M. tuberculosis infection. Numbers adjacent to outlined areas indicate percent CD8+ T cells stained with the TB10.4(4–11)-loaded H-2Kb tetramer (H-2Kb–TB10.4(4–11)). Data are representative of two independent experiments (mean of four to five mice per group). (c) Kinetics of the pulmonary TB10.4-specific CD8+ T cell response in Ptges−/−, wild-type and Alox5−/− mice. *P < 0.05 (one-way analysis of variance (ANOVA)). Data are representative of two experiments (mean ± s.e.m. of four mice per group).
Alox5−/− macrophages enhance CD8+ T cell responses
To determine whether the enhanced T cell response in Alox5−/− mice was due to an intrinsic property of Alox5−/− T cells, we transferred splenic T cells from wild-type, Ptges−/− or Alox5−/− mice infected with M. tuberculosis into sublethally irradiated wild-type mice37,38. At 1 d after T cell transfer, we infected the recipient mice with virulent M. tuberculosis by aerosol. Three weeks later, we detected no difference in colony-forming units (CFU) in the lungs (Supplementary Fig. 2) or spleens (data not shown) of the mice. Thus, the T cells of these mice had similar intrinsic capacity to protect against pulmonary infection with M. tuberculosis.
Because we found a greater T cell response early after infection in Alox5−/− mice, we hypothesized that the initiation of T cell immunity differed in Alox5−/− versus wild-type mice. To study the early events of T cell priming and to determine whether eicosanoids modulate initiation of T cell immunity, we developed an adoptive-transfer model using M. tuberculosis–infected macrophages. The model allows specific analysis of how lipid mediators produced by macrophages infected with M. tuberculosis affect T cell–mediated immunity independently of other eicosanoid-producing cell types.
We measured the migration of uninfected CD45.2+ macrophages in the airways and lung tissues at various time points after intratracheal adoptive transfer of these cells into CD45.1+ congenic mice. We detected the transferred macrophages for at least 7 d (Supplementary Fig. 3), which indicated that these cells can migrate into the lung tissue and persist. We next infected wild-type, Ptges−/− and Alox5−/− macrophages in vitro with M. tuberculosis using a low multiplicity of infection. We transferred the infected macrophages by the intratracheal route into wild-type recipient mice. We found that 17 d later, there were significantly more TB10.4-specific CD8+ T cells in both the thoracic draining lymph node and the lungs of mice that received M. tuberculosis–infected Alox5−/− macrophages than in those of mice that received M. tuberculosis–infected wild-type or Ptges−/− macrophages (Fig. 2a). Lymphocytes from the thoracic lymph nodes and lungs of mice that received M. tuberculosis-infected Alox5−/− macrophages also secreted more IFN-γ after stimulation with M. tuberculosis antigens recognized by CD8+ T cells (TB10.4(4–11) and 32c(309–318)) or CD4+ T cells (Ag85B(241–256) and ESAT6(3–15); Fig. 2b). The enhanced T cell response after adoptive transfer of Alox5−/− macrophages seemed to be the result of earlier initiation of the immune response, just as observed for intact Alox5−/− mice (Fig. 1c). We detected the TB10.4-specific CD8+ T cell response earlier in the pulmonary draining lymph nodes and the lungs of the recipient mice after adoptive transfer of M. tuberculosis-infected Alox5−/− macrophages than after wild-type macrophage transfer (Fig. 2c). Moreover, 4 weeks after adoptive transfer, the pulmonary bacterial burden was lower in wild-type mice that had received infected Alox5−/− macrophages than in mice that had received infected wild-type or Ptges−/− macrophages (Fig. 2d). At this time point, the number of TB10.4-specific CD8+ T cells in the lung and spleen was similar in the groups (Supplementary Fig. 4), which suggested that the early initiation of T cell immunity contributes substantially to protection against M. tuberculosis. The ability to recapitulate the early T cell response and lower pulmonary bacterial burden of Alox5−/− mice produced solely by transfer of M. tuberculosis–infected Alox5−/− macrophages into unmanipulated mice shows the important influence of eicosanoid production by infected macrophages on the regulation of T cell immunity and host resistance to pulmonary M. tuberculosis infection. Thus, the predominant production of PGE2 by infected Alox5−/− macrophages, which in vitro is the cause of more apoptosis, correlates with a greater T cell response during the early phase of infection in vivo.
Figure 2.

Transfer of M. tuberculosis–infected Alox5−/− macrophages into wild-type mice recapitulates the phenotype of intact Alox5−/− mice. (a) Frequency of TB10.4(4–11)-specific CD8+ T cells in the thoracic draining lymph nodes (top) and lungs (bottom) 17 d after intratracheal transfer of M. tuberculosis H37Rv–infected Alox5−/−, Ptges−/− or wild-type macrophages into wild-type mice. Numbers adjacent to outlined areas indicate percent CD8+ T cells stained with H-2Kb–TB10.4(4–11). Data are representative of four independent experiments (mean and s.e.m. of 16 total mice per group). (b) Enzyme-linked immunospot assay of IFN-γ production showing the frequency of M. tuberculosis–specific CD4+ and CD8+ T cells 17 d after intratracheal transfer of M. tuberculosis H37Rv–infected Ptges−/−, wild-type or Alox5−/− macrophages into wild-type mice; splenic T cells from the wild-type recipient mice were cultured with APCs and Ag85B(241–256), ESAT6(3–15), TB10.4(4–11) or 32c(309–318). Data are representative of three experiments. (c) Kinetics of PLN and lung TB10.4-specific CD8+ T cell responses in recipient mice after transfer of infected Ptges−/−, wild-type or Alox5−/− macrophages. SFC, spot-forming cells. Data are representative of two experiments (mean ± s.e.m. of four mice per group). (d) Bacterial burden in the lung 28 d after intratracheal transfer of M. tuberculosis H37Rv–infected Alox5−/−, Ptges−/− or wild-type macrophages into wild-type mice. Each symbol represents an individual mouse; small horizontal bars indicate the mean. Data are from two independent experiments (n = 4 mice per group). NS, not significant; *P < 0.05, compared with wild-type (one-way ANOVA).
Alox5−/− alveolar macrophages enhance T cell responses
We next investigated whether transfer of infected alveolar Alox5−/− macrophages elicited an increased pulmonary T cell response. We obtained minimally manipulated infected alveolar macrophages by bronchoalveolar lavage 3 h after infection of wild-type or Alox5−/− mice. We transferred the lavaged cells into naive wild-type mice via the intratracheal route. On day 17 after transfer of alveolar macrophages, we detected no TB10.4-specific CD8+ T cells in the lungs, pulmonary lymph nodes (PLNs) or spleens of mice that received infected wild-type alveolar macrophages (Fig. 3a). In contrast, we identified TB10.4-specific CD8+ T cells in all three tissues in mice that received infected Alox5−/− alveolar macrophages. The percentage of CD8+ T cells specific for TB10.4(4–11) was significantly higher in the lungs and PLNs of mice that received infected Alox5−/− alveolar macrophages than that in those of recipients of wild-type alveolar macrophages (Fig. 3b).
Figure 3.

Transfer of infected Alox5−/− alveolar macrophages generates pulmonary and systemic CD4+ and CD8+ T cell responses. (a) Frequency of TB10.4(4–11)-specific CD8+ T cells in the lungs, PLN and spleen 17 d after intratracheal transfer of alveolar macrophages (MΦ) from wild-type mice (left) or Alox5−/− mice (right) infected by aerosol with M. tuberculosis (virulent Erdman strain). Numbers adjacent to outlined areas indicate percent CD8+ T cells stained with H-2Kb–TB10.4(4–11) or streptavidin-phycoerythrin alone (Control; bottom row). Data are from one experiment. (b) Frequency of H-2Kb–TB10.4(4–11)–stained CD8+ T cells in the lungs, PLN and spleens of mice treated as described in a (n = 5 per group). Staining with isotype-matched control antibody (Isotype) is shown for the wild-type group only. *P < 0.05 (one-way ANOVA with the Bonferroni post-test). Data are from one experiment (mean and s.e.m.).
We measured the frequency of IFN-γ-secreting T cells in lymphocytes from lungs, PLNs and spleens of recipient mice stimulated with TB10.4(4–11) or 32c(309–318) (CD8+ epitopes) or with Ag85B(241–256) or ESAT6(3–15) (CD4+ epitopes). As noted after transfer of peritoneal macrophages (Fig. 2b), we detected a greater T cell response encompassing both CD4+ and CD8+ T cells after the transfer of infected Alox5−/− alveolar macrophages than after the transfer of wild-type alveolar macrophages (Supplementary Fig. 5). Thus, similar to results obtained with thioglycollate-elicited peritoneal macrophages, the dysregulated production of prostanoids by alveolar macrophages is associated with the initiation of an earlier CD4+ and CD8+ T cell response.
Alox5−/− macrophages enhance CD8+ T cell cross-priming
We considered two potential mechanisms by which infected macrophages might affect T cell immunity: first, Alox5−/− macrophages could directly prime naive T cells; and second, macrophage cell death could affect the cross-presentation of M. tuberculosis antigens by DCs. We first evaluated whether Alox5−/− macrophages have a greater ability than other macrophages to stimulate M. tuberculosis–specific T cells in vitro. We pulsed wild-type, Ptges−/− and Alox5−/− macrophages with total M. tuberculosis antigens (M. tuberculosis sonicate), intact Ag85 protein or Ag85B(241–256) and found no difference in their ability to stimulate IL-2 production by the Ag85B-specific T cell hybridoma BB739 (Supplementary Fig. 6); this indicated that the intrinsic ability of these macrophages to process and present antigen was unaffected by PGE2 synthase or 5-lipoxygenase.
To determine how infected Alox5−/− macrophages elicit an enhanced T cell response, we evaluated the ability of M. tuberculosis–infected macrophages to prime CD8+ T cells. We used OT-I T cell antigen receptor–transgenic CD8+ T cells that were specific for the ovalbumin (OVA) epitope SIINFEKL and expressed the CD90.2 congenic T cell marker. We labeled these cells with the cytosolic dye CFSE (carboxyfluorescein diacetate succinimidylester) and transferred them into CD90.1+ congenic recipient mice. Then, 1 d later, we injected wild-type, Ptges−/− or Alox5−/− macrophages pulsed with SIINFEKL peptide and infected with M. tuberculosis into the footpads of wild-type mice. We analyzed T cells in the draining popliteal lymph nodes 4 d later. The proliferation and population expansion of OT-I T cells were significantly greater after the administration of M. tuberculosis-infected SIINFEKL-pulsed Alox5−/− macrophages than after administration of similarly treated wild-type macrophages (Fig. 4a,b). After transfer of wild-type macrophages that had been pulsed with peptide and infected with M. tuberculosis, we detected only a small population of SIINFEKL-specific CD8+ T cells (~500 cells per lymph node). This was not substntially different from the number found in mice that received only OT-I cells, without macrophages. In contrast, we found an expanded population of OVA-specific T cells after transfer of Alox5−/− macrophages that had been pulsed with SIINFEKL and infected with M. tuberculosis (~1 × 104 cells per lymph node; 20-fold more than after transfer of wild-type macrophages treated similarly). We found no difference in OT-I T cell proliferation after the administration of wild-type or Ptges−/− macrophages (data not shown).
Figure 4.

CD8+ T cell activation induced by M. tuberculosis–infected Alox5−/− macrophages requires cross-presentation by CD11c+ DCs. (a,b) CD8+ T cell proliferation (a) and population expansion of cells positive for the H-2Kb–SIINFEKL pentamer (b) 5 d after intravenous transfer of CFSE-labeled Thy-1.2+ splenic OT-I CD8+ T cells into Thy-1.1+ B6.PL mice, followed within 24 h by M. tuberculosis H37Rv–infected wild-type or Alox5−/− macrophages pulsed with SIINFEKL. Numbers above bracketed lines (a) indicate percent CFSE− cells (left) or CFSE+ cells (right); numbers adjacent to outlined areas (b) indicate percent CD8+ T cells stained with H-2Kb–SIINFEKL. (c) Frequency of SIINFEKL-specific CD8+ T cells in the draining lymph nodes of CD11c-DTR–transgenic mice (CD11c-DTR) and nontransgenic littermate control mice (Non-TG) given intravenous injection of OT-I CD8+ T cells, followed by subcutaneous transfer of M. tuberculosis H37Rv–infected, SIINFEKL-pulsed Alox5−/− macrophages and treatment with diphtheria toxin. No Mϕ (right), control mice that did not receive macrophages. Numbers adjacent to outlined areas indicate percent CD8+ T cells stained with H-2Kb–SIINFEKL after 5 d. Data are representative of three experiments (mean and s.e.m. of four to five mice per group). (d) OVA-specific OT-I CD8+ T cells in draining lymph nodes of β2-microglobulin-deficient (B2m−/−), TAP-1-deficient (Tap1−/−) or Thy-1.1+ recipient mice 5 d after the transfer of CFSE-labeled Thy-1.2+ splenic OT-I CD8+ T cells and M. tuberculosis H37Rv–infected, SIINFEKL-pulsed wild-type or Alox5−/− macrophages. Data are representative of two to three independent experiments (mean ± s.e.m. of three to five mice per group). *P < 0.05 (one-way ANOVA).
Although the transfer of infected Alox5−/− macrophages elicited a greater OT-I response than did the transfer of wild-type cells, we found no difference in OT-I T cell proliferation after the transfer of uninfected, SIINFEKL-pulsed Alox5−/− or wild-type macrophages, with or without lipopolysaccharide (Supplementary Fig. 7). In addition, the transfer of uninfected, SIINFEKL-pulsed Alox5−/− or wild-type macrophages that had been treated with the apoptosis-inducing agent staurosporine poorly stimulated OT-I proliferation (Supplementary Fig. 7). These data confirm that there is no intrinsic difference between Alox5−/− and wild-type macrophages in their ability to stimulate T cell responses and instead support the idea that the greater apoptosis of M. tuberculosis-infected Alox5−/− macrophages leads to more T cell priming than do wild-type or Ptges−/− macrophages.
We next investigated whether the enhanced capacity of Alox5−/− macrophages to prime antigen-specific T cells was due to direct presentation by the M. tuberculosis–infected Alox5−/− macrophages or required cross-priming by DCs. We transferred infected macrophages and OT-I T cells into transgenic mice that expressed the diphtheria toxin receptor (DTR) fused to green fluorescent protein (GFP) under the control of the CD11c promoter (B6.FVB-Tg-Itgax-DTR/EGFP-57Lan/J, CD11c-DTR–transgenic mice). Administration of diphtheria toxin results in transient deletion of CD11c+ antigen-presenting cells (APCs; Supplementary Fig. 8). We adoptively transferred CFSE-labeled OT-I T cells into CD11c-DTR–transgenic mice and their non-transgenic littermates. After 24 h, we treated the mice with diphtheria toxin and transferred SIINFEKL-pulsed, M. tuberculosis–infected Alox5−/− macrophages into them. The transfer of SIINFEKL-pulsed, infected Alox5−/− macrophages into nontransgenic littermate controls treated with diphtheria toxin induced significant T cell population expansion in the popliteal lymph nodes 4 d after transfer, as described above (Fig. 4c). However, when we transferred SIINFEKL-pulsed, M. tuberculosis–infected Alox5−/− macrophages into CD11c-DTR–transgenic mice treated with diphtheria toxin, we found only a small population of OT-I cells. This population was not significantly different from that in mice that had not received Alox5−/− macrophages (Fig. 4c). Both the percentage and the total number of CD8+ T cells specific for SIINFEKL were much smaller after deletion of CD11c+ APCs (Fig. 4c), which indicated that the transferred Alox5−/− macrophages were not sufficient to prime CD8+ T cells and that endogenous CD11c+ DCs were required.
To further investigate whether cross-priming was required for the response described above, we transferred CFSE-labeled OT-I T cells into mice deficient in TAP1, the transporter associated with antigen processing, or into mice deficient in β2-microglobulin, which lack mature major histocompatibility complex (MHC) class I molecules. In the absence of TAP1 or β2-microglobulin in recipient mice, OT-I T cell populations did not expand after transfer of SIINFEKL-pulsed, M. tuberculosis–infected Alox5−/− macrophages. This observation shows that the population expansion of OT-I T cells after transfer of infected Alox5−/− macrophages depended on the processing and presentation of SIINFEKL by the host-derived MHC class I pathway (Fig. 4d). Collectively, these data indicate that M. tuberculosis–infected Alox5−/− macrophages enhance CD8+ T cell responses through DC-dependent cross-priming.
Macrophage apoptosis enhances T cell immunity
We next tested whether inhibition of apoptosis would reverse the greater T cell immunity seen after transfer of M. tuberculosis–infected Alox5−/− macrophages. Infection of Alox5−/− macrophages with the virulent H37Rv strain of M. tuberculosis led to apoptosis, as reported before12. The apoptosis of M. tuberculosis–infected Alox5−/− macrophages was significantly inhibited in vitro by an inhibitor of caspase 8 and the extrinsic apoptotic pathway, by an inhibitor of caspase-9 and the intrinsic apoptotic pathway, and by a combination of both inhibitors (Fig. 5a).
Figure 5.
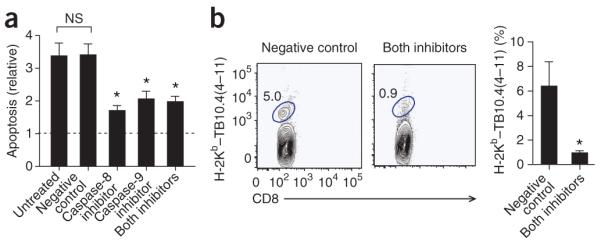
Caspase-dependent apoptosis of M. tuberculosis-infected Alox5−/− macrophages is required for the early initiation of T cell immunity. (a) Apoptosis of Alox5−/− macrophages infected with M. tuberculosis H37Rv (multiplicity of infection, ~2), assessed 3 d after treatment for 2 h with a negative control peptide (Negative control) or with an inhibitor of caspase-8 or caspase-9 or both (0.5 μM each), presented relative to that of uninfected Alox5−/− macrophages (Untreated). *P < 0.05, compared with control group (one-way ANOVA with Dunnett’s multiple-comparison test). Data are representative of three experiments (mean and s.e.m.). (b) Frequency of TB10.4(4–11)-specific CD8+ T cells in the lungs of mice 17 d after the intratracheal transfer of M. tuberculosis H37Rv–infected Alox5−/− macrophages treated with inhibitors of caspase-8 and caspase-9 (Both inhibitors) or a negative control peptide (Negative control); results (right) are normalized by division of the absorbance by the absorbance of cultures of uninfected macrophages. Numbers adjacent to outlined areas indicate percent CD8+ T cells stained with H-2Kb–TB10.4(4–11). *P < 0.05 (Mann-Whitney test). Data are representative of two independent experiments (mean and s.e.m. of four mice per group).
Supplementary Figure 1. The immune response is similar in Ptges−/−, wild type, and Alox5−/− mice five weeks after Mtb infection. The frequency of TB10.4(4-11)-specific CD8+ T cells was determined five weeks after aerosol Mtb infection (~100 CFU) in the lungs (a) and the spleens (b) of Ptges−/−, wild type, and Alox5−/− mice. The percentage of CD8+ T cells that stained with the H-2 Kb–TB10.4(4-11) tetramer is indicated in each representative FACS plot. Each bar is the mean ± SE, of 5 individual mice. (c) The pulmonary bacterial burden in Ptges−/−, wild type, and Alox5−/− mice (n=5/group) five weeks after infection with aerosolized Mtb. Results are representative of two independent experiments. *, p<0.05 by a one-way ANOVA compared to wild type group, or †, p<0.05 Alox5−/− vs. Ptges−/−.
Supplementary Figure 2. The intrinsic ability of T cells from Ptges−/−, wild type, and Alox5−/− mice to provide protection against pulmonary Mtb infection is similar. (a) Sublethally irradiated wild type mice were used as recipients for splenic T cells (CD3+) from Mtb infected Ptges−/−, wild type, or Alox5−/− mice. Recipient mice were infected with Mtb by the aerosol route within 24 hrs after transfer of purified T cells. Three weeks after infection, the bacterial burden in the lung and spleen were determined. Each point represents data from an individual mouse, and the bars represent the mean (n=4-5 mice per group). The differences in lung CFU between Ptges−/− and wild type, or wild type and Alox5−/− mice were not significant (as determined by a one-way ANOVA). (b) Control experiment done under the same conditions as (a). Recipient mice were irradiated and purified splenic T cells from Mtb infected syngeneic mice were transferred intravenously (WT). These were compared to control mice that did not receive T cells (No Tx). These two groups were infected as above and analyzed after three weeks. The difference in lung CFU was very significant as determined using a t-test (p = 0.0002).
Supplementary Figure 3. Adoptively transferred macrophages traffic into the lung. Uninfected CD45.2+ macrophages were transferred into CD45.1 recipient mice via the intratracheal route. The presence of donor CD45.2+ macrophages was evaluated in the air space (by bronchoalveolar lavage) and lung tissue (after collagenase digestion) 6h, 24h, 48h, and 7 days after adoptive transfer.
Supplementary Figure 4. The frequency of TB10.4(4-11) specific CD8+ T cells in the lungs and spleens of wild type mice is similar 28 days after the intratracheal transfer of H37Rv-infected Ptges−/−, wild type, or Alox5−/− macrophages. Representative of FACS plots showing the H-2 Kb–TB10.4(4-11) tetramer staining of CD8+ T cells from the lung (top row) or spleen (bottom row) of recipient mice 4 weeks after infected macrophages transfer. The frequency of TB10.4(4-11)-specific CD8+ T cells is indicated in each plot and in the bar graph. Bars, mean ± SE bar graph of 4 mice per group.
Supplementary Figure 5. The frequency of Mtb-specific CD4+ and CD8+ T cells following adoptive transfer of Mtb infected alveolar macrophages from wild-type or Alox5−/− mice into naïve wild type mice. Seventeen days after the adoptive transfer of Mtb infected alveolar macrophages from wild type or Alox5−/− mice into wild type recipient mice, the frequency of Mtb-specific CD4+ and CD8+ T cells was enumerated using an IFN-γ elispot to measure the response to Ag85B(247-256), ESAT6(3-15), TB10.4(4-11), or 32c(309-318) synthetic peptides in the lungs, pulmonary lymph nodes (PLN), and spleens. Mice that received Alox5−/− alveolar macrophages had larger IFN-γ responses to ESAT6 (CD4+ T cell epitope) and TB10.4 (CD8+ T cell epitope) in the PLN and spleen, respectively, at this early time point (p<0.05, t-test). Statistical analysis of the other elispot responses is difficult because many of the mice that received wild type alveolar macrophages had no detectable response, and the Alox5−/− alveolar macrophages group showed significant variability in their responses. To compare the magnitude of the response, recipient mice were categorized as responders or non-responders based on detection of IFN-γ secreting T cells. When all the possible responses were analyzed in aggregate, it was clear that transfer of infected Alox5−/− alveolar macrophages induced a better response than elicited by infected wild type alveolar macrophages (p < 0.0001 by Fischer exact test).
Supplementary Figure 6. Ptges−/−, wild type, and Alox5−/− macrophages have a similar ability to process and present antigen. Ptges−/−, wild type, and Alox5−/− macrophages were pulsed with different concentration of sonicated Mtb, Mtb-culture filtrate proteins, Ag85 protein, or Ag85B(241-256) peptide, for 6 hrs. BB7 hybidoma cells were added to macrophages and IL-2 production was measured in the supernatants 24 hrs later. The ratio of the macrophages to T cells was 1:1. Values represent mean ± SE of triplicate samples.
Supplementary Figure 7. Wild type and Alox5−/− macrophages have a similar capacity to enhance OVA-antigen specific T cell response in the absence of Mtb infection. (a) CD8+ T cell proliferation 5 days after IV transfer of CFSE-labeled Thy1.2+ splenic OT-I CD8+ T cells into Thy1.1+ B6.PL mice followed within 24 hrs by uninfected wild type or Alox5−/− macrophages pulsed with SIINFEKL. (b) Purified CD45.1+ OT-1 CD8+ T cells were injected IV into CD45.2+ C57BL/6 mice (n=2-5/group). Within 24 hours, uninfected wild type or Alox5−/− macrophages cultured with SIINFEKL (pOVA) and treated with LPS (100 ng/ml) or staurosporine (1 μM) for 6 hrs. OT-1 cells were identified as CD45.1+CD8+Vβ5+Vα2+ cells. The frequency of OT-1 cells in each draining LN is shown. Bars represent mean ± SE.
Supplementary Figure 8. In vivo depletion of CD11c+ cells. Three different doses of diphtheria toxin (DT) (25, 50, 100 ng/mouse) were administered by the IP route to CD11c–DTR TG and non-TG littermate mice. The frequency of CD11c+ cells were measured in the spleens 24 hours later. The majority of CD11c+ cells were depleted following 100 ng of DT compared to their littermate non-tg controls.
We postulated that DC uptake and cross-presentation of the antigen cargo contained in apoptotic vesicles derived from M. tuberculosis–infected Alox5−/− macrophages augments the T cell response in vivo. To test this possibility, we treated M. tuberculosis–infected Alox5−/− macrophages with the inhibitors of caspase-8 and caspase-9 described above or with an inactive caspase-inhibitor control and subsequently transferred the cells by the intratracheal route into the lungs of uninfected wild-type mice. As expected, the transfer of M. tuberculosis–infected Alox5−/− macrophages treated with the negative control peptide augmented the TB10.4-specific CD8+ T cell response, as described above (Fig. 5b). In contrast, the transfer of M. tuberculosis–infected Alox5−/− macrophages treated with inhibitors of caspase-8 and caspase-9 did not induce early population expansion of TB10.4-specific CD8+ T cells in the lung (Fig. 5b). Thus, apoptosis of M. tuberculosis-infected macrophages is required for the early initiation of T cell immunity after pulmonary infection with M. tuberculosis.
DISCUSSION
Pulmonary macrophages are the primary hosts for the replication of M. tuberculosis. The ability of M. tuberculosis to inhibit macrophage antimicrobial defenses makes it a successful pathogen. The induction of apoptosis, including formation of the apoptotic envelope, is inhibited in macrophages infected with virulent M. tuberculosis14,40,41. PGE2 can prevent necrosis of M. tuberculosis–infected macrophages by protecting against mitochondrial inner membrane instability and promoting the repair of plasma membrane damage, two independent insults induced by virulent M. tuberculosis12,13. By preventing necrosis and increasing apoptosis of M. tuberculosis–infected macrophages, PGE2 enhances innate immune control of the initial infection both in vitro and in vivo12. Here we have used an adoptive-transfer model in which macrophages were infected in vitro with M. tuberculosis and then transferred into recipient mice to address whether apoptotic macrophages contribute to the initiation of adaptive immunity. We found that the host lipid mediator PGE2 was required for the induction of apoptosis in M. tuberculosis–infected macrophages, which in turn promoted T cell priming in vivo. An enhanced antigen-specific CD8+ T cell response required presentation of apoptotic macrophage-derived antigens by the ‘detour’ pathway, as defined by the TAP-1-dependent and MHC class I–restricted cross-presentation by DCs. In addition to CD8+ T cell responses, CD4+ T cell responses were also considerably enhanced after the transfer of proapoptotic M. tuberculosis–infected macrophages. We have shown that the adoptive transfer of proapoptotic M. tuberculosis–infected macrophages led to an early and robust antigen-specific T cell response that correlated with considerable pulmonary protection. Thus, virulent M. tuberculosis inhibits PGE2 production and prevents macrophage apoptosis, which impairs both innate and adaptive immunity to M. tuberculosis.
One way that M. tuberculosis evades host defense mechanisms is by delaying the initiation of T cell priming31,32. There is a longer delay between M. tuberculosis infection and the initiation of adaptive immunity than for other intracellular pathogens, including Salmonella enteritica42, Listeria monocytogenes43, Leishmania major44 and the influenza virus45. In people infected with tuberculosis, the development of adaptive immunity requires 5–6 weeks (ref. 46); in mice, the earliest T cell response is detected 8–10 d after aerosol infection31. Virulent M. tuberculosis can disrupt the crosstalk between innate and adaptive immunity by altering macrophage and DC functions that are central for the initiation and instruction of the immune response. The delayed dissemination of M. tuberculosis into the draining lymph nodes correlates with delayed initiation of T cell immunity in susceptible C3H mice31. Delayed onset of immunity has consequences for the control of bacterial replication. Transient depletion of DCs at the time of infection delays the initiation of T cell immunity by 7–10 d (ref. 32). Although the T cell response reaches normal levels within 3 weeks of infection, the mycobacterial burden remains higher than that in control, undepleted mice for at least 2 months32. Thus, even a transient delay in the initiation of adaptive immunity leads to long-term impairment of bacterial control. Given the important role of DCs in T cell priming and transport of M. tuberculosis from the lungs to the lymph nodes, it is likely that M. tuberculosis interrupts this process, either by delaying the acquisition of antigen by DCs (apoptosis inhibition) or by impairing DC trafficking (prevention of DC activation). The links between apoptotic macrophages, DC activation and early T cell immunity in the control of virulent M. tuberculosis infection require further elucidation.
Understanding how cellular death affects innate and adaptive immunity is important for elucidating disease pathogenesis. In non-infectious diseases such as autoimmunity, cell death through necrosis enhances T cell–mediated immunity to specific antigens because loss of plasma-membrane integrity and release of the cellular contents acts as an adjuvant. In contrast, apoptosis might prevent autoimmunity because apoptotic cells maintain the integrity of their cell membranes and are rapidly removed by phagocytes47. During infection with M. tuberculosis, cellular necrosis and loss of plasma membrane integrity allow bacteria to infect other cells12, whereas apoptosis of M. tuberculosis–infected cells enhances the innate control of intracellular bacterial replication as well as T cell–mediated immunity. Published studies have emphasized the importance of apoptosis in the cross-presentation of M. tuberculosis antigens by human DCs in vitro and the role of apoptotic vesicles purified from BCG-infected macrophages in vivo16,29. However, the response of host macrophages to live M. tuberculosis in vivo has not been addressed.
We developed an infected macrophage adoptive-transfer model that allows study of how altering macrophage function in an otherwise normal host affects immunity to M. tuberculosis. Just as infection with proapoptotic bacterial mutants generates greater CD8+ T cell responses16, a propensity for the host macrophage to undergo apoptosis also enhanced T cell immunity and led to better protection. Enhanced T cell–mediated immunity was not restricted to CD8+ T cells. M. tuberculosis–specific CD4+ responses were also greater after the transfer of infected proapoptotic Alox5−/− macrophages. Thus, whereas apoptosis directly increased CD8+ T cell responses by cross-presentation, it also enhanced MHC class II–restricted antigen presentation. Presumably, phagocytosis of apoptotic vesicles by DCs delivers their M. tuberculosis antigen cargo to the endocytic system, which intersects with the MHC class II processing pathway, ultimately increasing the priming of CD4+ T cells.
Whereas DCs are required for priming mycobacterial specific T cell immunity33, the demonstration that DCs are directly infected by M. tuberculosis and facilitate the local dissemination of M. tuberculosis to regional lymph nodes34 suggests that M. tuberculosis–infected DCs might be crucial for T cell priming. Our finding that intratracheal transfer of alveolar or peritoneal M. tuberculosis–infected Alox5−/− macrophages led to earlier initiation of CD4+ and CD8+ T cell immunity suggests that the transfer of antigen to uninfected DCs is sufficient for T cell priming. The thioglycollate-elicited peritoneal macrophages used in our study here were activated and recruited into the peritoneal space, just as most macrophages found in the lungs of infected mice were recruited from the periphery. Because peritoneal macrophages can directly prime CD8+ T cells in vivo48, we determined whether M. tuberculosis–infected macrophages directly prime T cells or whether their propensity to undergo apoptosis leads to the entry of mycobacterial antigens into the detour pathway. We found that infected Alox5−/− macrophages could not directly prime T cells and instead that T cell priming required endogenous DCs. A limitation of the CD11c-DTR model is macrophage toxicity owing to ingestion of diphtheria toxin from dying DCs, which can mask macrophage priming of T cells49. However, by using mice deficient in TAP-1 or β2-microglobulin as recipients, we have demonstrated the importance of the endogenous MHC class I processing and presentation pathway. Similar to results obtained with DC depletion, we did not observe OT-I T cell population expansion when we administered proapoptotic M. tuberculosis–infected Alox5−/− macrophages to mice deficient in TAP-1 or β2-microglobulin. These experiments showed that CD8+ T cell priming required cross-presentation of antigens acquired by DCs from apoptotic, M. tuberculosis–infected macrophages through the detour pathway.
Finally, we speculate that the proinflammatory function of apoptosis during infection, in contrast to its anti-inflammatory propensity during physiological cell death, might be a consequence of Toll-like receptor ligands and other microbe-derived signals that are contained in the apoptotic vesicles. Our data indicate that Alox5−/− macrophages are not intrinsically more stimulatory than are wild-type macrophages, as cells of both genotypes processed and presented antigen to T cells in vitro and in vivo with similar efficiency. Instead, we found that apoptosis under proinflammatory conditions led to more efficient cross-priming. These results are in keeping with the idea that apoptotic cells do not stimulate primary immune responses unless there are additional danger signals, such as infection, that lead to inflammation by triggering Toll-like receptor or other pattern-recognition receptor signaling.
Despite the worldwide application of BCG vaccination and other anti–M. tuberculosis interventions, M. tuberculosis remains one of the most successful human pathogens. Eight to ten million new cases of active tuberculosis occur each year due in large part to the large reservoir of asymptomatic people chronically infected with M. tuberculosis1. Estimates suggest that up to a third of the world’s population is latently infected with M. tuberculosis, which indicates that M. tuberculosis can persist over the long term in humans1. An understanding of the strategies that M. tuberculosis uses to evade host immunity is essential if an effective vaccine is to be developed50. The work reported here has demonstrated that by inhibiting PGE2 production and apoptosis, M. tuberculosis impairs T cell immunity. Moreover, we have established a direct link between apoptosis of M. tuberculosis–infected macrophages and T cell cross-priming by DCs in vivo. This improved mechanistic understanding might help in optimizing vaccination.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/natureimmunology/.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Koller (University of North Carolina) for Alox5−/− and Ptges−/− mice. Supported by the US National Institutes of Health (AI 067731 to S.M.B. and AI072143 to H.G.R.), the Fonds de la Recherche en Santé du Québec (M.D.) and Fundação para a Ciência e Tecnologia of Portugal (C.N.-A.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Barry CE, III, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007;27:347–362. doi: 10.1007/s10875-007-9084-0. [DOI] [PubMed] [Google Scholar]
- 3.Russell DG, Mwandumba HC, Rhoades EE. Mycobacterium and the coat of many lipids. J. Cell Biol. 2002;158:421–426. doi: 10.1083/jcb.200205034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturgill-Koszycki S, Schaible UE, Russell DG. Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO J. 1996;15:6960–6968. [PMC free article] [PubMed] [Google Scholar]
- 5.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters NC, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.John B, Hunter CA. Immunology. Neutrophil soldiers or Trojan horses? Science. 2008;321:917–918. doi: 10.1126/science.1162914. [DOI] [PubMed] [Google Scholar]
- 8.Bergsbaken T, Cookson BT. Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007;3:e161. doi: 10.1371/journal.ppat.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haimovich B, Venkatesan MM. Shigella and Salmonella: death as a means of survival. Microbes Infect. 2006;8:568–577. doi: 10.1016/j.micinf.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tunbridge AJ, et al. Inhibition of macrophage apoptosis by Neisseria meningitidis requires nitric oxide detoxification mechanisms. Infect. Immun. 2006;74:729–733. doi: 10.1128/IAI.74.1.729-733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Divangahi M, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 2006;176:3707–3716. doi: 10.4049/jimmunol.176.6.3707. [DOI] [PubMed] [Google Scholar]
- 15.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 16.Hinchey J, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J. Clin. Invest. 2007;117:2279–2288. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velmurugan K, et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Remold HG, Ieong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J. Immunol. 2006;176:4267–4274. doi: 10.4049/jimmunol.176.7.4267. [DOI] [PubMed] [Google Scholar]
- 19.Duan L, Gan H, Arm J, Remold HG. Cytosolic phospholipase A2 participates with TNF-α in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J. Immunol. 2001;166:7469–7476. doi: 10.4049/jimmunol.166.12.7469. [DOI] [PubMed] [Google Scholar]
- 20.Gan H, et al. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat. Immunol. 2008;9:1189–1197. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oddo M, et al. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 1998;160:5448–5454. [PubMed] [Google Scholar]
- 22.Brookes RH, et al. CD8+ T cell-mediated suppression of intracellular Mycobacterium tuberculosis growth in activated human macrophages. Eur. J. Immunol. 2003;33:3293–3302. doi: 10.1002/eji.200324109. [DOI] [PubMed] [Google Scholar]
- 23.Tobin DM, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yrlid U, Wick MJ. Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J. Exp. Med. 2000;191:613–624. doi: 10.1084/jem.191.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inaba K, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 27.Sadagopal S, et al. Reducing the activity and secretion of microbial antioxidants enhances the immunogenicity of BCG. PLoS. One. 2009;4:e5531. doi: 10.1371/journal.pone.0005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaible UE, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat. Med. 2003;9:1039–1046. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 29.Winau F, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–117. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Winau F, Kaufmann SH, Schaible UE. Apoptosis paves the detour path for CD8 T cell activation against intracellular bacteria. Cell. Microbiol. 2004;6:599–607. doi: 10.1111/j.1462-5822.2004.00408.x. [DOI] [PubMed] [Google Scholar]
- 31.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf AJ, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2007;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J. Immunol. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- 34.Wolf AJ, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 35.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J. Immunol. 1997;159:635–643. [PubMed] [Google Scholar]
- 36.Bafica A, et al. Host control of Mycobacterium tuberculosis is regulated by 5- lipoxygenase-dependent lipoxin production. J. Clin. Invest. 2005;115:1601–1606. doi: 10.1172/JCI23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-γ, and kill intracellular bacteria. PLoS Pathog. 2008;4:e1000239. doi: 10.1371/journal.ppat.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodworth JS, Wu Y, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells require perforin to kill target cells and provide protection in vivo. J. Immunol. 2008;181:8595–8603. doi: 10.4049/jimmunol.181.12.8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramachandra L, Noss E, Boom WH, Harding CV. Processing of Mycobacterium tuberculosis antigen 85B involves intraphagosomal formation of peptide-major histocompatibility complex II complexes and is inhibited by live bacilli that decrease phagosome maturation. J. Exp. Med. 2001;194:1421–1432. doi: 10.1084/jem.194.10.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan L, Gan H, Golan DE, Remold HG. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J. Immunol. 2002;169:5181–5187. doi: 10.4049/jimmunol.169.9.5181. [DOI] [PubMed] [Google Scholar]
- 41.Gan H, et al. Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J. Infect. Dis. 2005;191:1292–1300. doi: 10.1086/428906. [DOI] [PubMed] [Google Scholar]
- 42.Srinivasan A, Foley J, Ravindran R, McSorley SJ. Low-dose Salmonella infection evades activation of flagellin-specific CD4 T cells. J. Immunol. 2004;173:4091–4099. doi: 10.4049/jimmunol.173.6.4091. [DOI] [PubMed] [Google Scholar]
- 43.Kursar M, et al. Organ-specific CD4+ T cell response during Listeria monocytogenes infection. J. Immunol. 2002;168:6382–6387. doi: 10.4049/jimmunol.168.12.6382. [DOI] [PubMed] [Google Scholar]
- 44.Lira R, Doherty M, Modi G, Sacks D. Evolution of lesion formation, parasitic load, immune response, and reservoir potential in C57BL/6 mice following high- and low-dose challenge with Leishmania major. Infect. Immun. 2000;68:5176–5182. doi: 10.1128/iai.68.9.5176-5182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moskophidis D, Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 1998;188:223–232. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallgren A. The time-table of tuberculosis. Tubercle. 1948;29:245–251. doi: 10.1016/s0041-3879(48)80033-4. [DOI] [PubMed] [Google Scholar]
- 47.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J. Immunol. 2005;175:2071–2081. doi: 10.4049/jimmunol.175.4.2071. [DOI] [PubMed] [Google Scholar]
- 49.Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock KL. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.