

Abstract
Extravascular coagulation leading to fibrin deposition accompanies many immune and inflammatory responses. Although recognized by pathologists for decades, and probably pathologic under certain conditions, the physiologic functions of extravascular coagulation remain to be fully defined. This study demonstrates that thrombin can activate macrophage adhesion and prompt interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1) production in vivo. Peritoneal macrophages were elicited with thioglycollate (TG) and then activated in situ, either by intraperitoneal injection of lipopolysaccharide (LPS) or by injection of antigen into mice bearing antigen-primed T cells. Others previously established that such treatments stimulate macrophage adhesion to the mesothelial lining of the peritoneal cavity. The present study demonstrates that thrombin functions in this process, as macrophage adhesion was suppressed by Refludan, a highly specific thrombin antagonist, and induced by direct peritoneal administration of purified thrombin. Although recent studies established that protease activated receptor 1 (PAR-1) mediates some of thrombin’s proinflammatory activities macrophage adhesion occurred normally in PAR-1–deficient mice. However, adhesion was suppressed in fibrin(ogen)-deficient mice, suggesting that fibrin formation stimulates macrophage adhesion in vivo. This study also suggests that fibrin regulates chemokine/cytokine production in vivo, as direct injection of thrombin stimulated peritoneal accumulation of IL-6 and MCP-1 in a fibrin(ogen)-dependent manner. Given that prior studies have clearly established inflammatory roles for PAR-1, thrombin probably has pleiotropic functions during inflammation, stimulating vasodilation and mast cell degranulation via PAR-1, and activating cytokine/chemokine production and macrophage adhesion via fibrin(ogen).
Introduction
Vasodilation and increased vascular permeability are among the earliest signs of inflammation. These events stimulate the extravasation of inactive coagulant precursors, which become activated upon exposure to extravascular tissues. The ensuing coagulation cascade culminates with the generation of thrombin, a protease that cleaves extravasated fibrinogen, prompting its polymerization and deposition as fibrin. Accordingly, localized extravascular fibrin deposition accompanies many type 1 T helper cell (Th1)–associated responses, including autoimmune neuropathologies,1–4 glomerulonephritis,5,6 rheumatoid arthritis,7–9 Crohn’s disease,10,11 allograft rejection,12,13 delayed-type hypersensitivity,14–19 and viral infections.20,21 For some time, it has been appreciated that such Th1-associated coagulation has physiologic consequences, as the swelling that accompanies delayed-type hypersensitivity responses is suppressed in anticoagulated or fibrinogen-deficient subjects.14–19 However, the full significance of immune-associated extravascular coagulation remains to be defined.
Recent studies suggest that thrombin is a physiologic mediator of inflammatory events. Administration of recombinant hirudin, a highly specific thrombin antagonist, reduces pathology and leukocyte infiltration in a mouse glomerulonephritis model.22 Hirudin analogs also suppress mast cell degranulation and vasodilation in a carrageenin-induced inflammation model,23 and prevent onset and ameliorate established disease in mouse arthritis models.24,25 Together, these studies strongly suggest that thrombin has physiologic functions during immunity/inflammation.
The vasodilatory activities of thrombin likely result from its capacity to stimulate PAR-1, a 7-transmembrane–spanning, G-protein–coupled receptor activated upon cleavage by thrombin.26 Consistent with the aforementioned hirudin studies, PAR-1–deficient mice27 exhibit diminished inflammation in glomerulonephritis and carrageenin models.22,23 In addition, cutaneous injection of a PAR-1–activating peptide stimulates mast cell degranulation and vasodilation in wild-type mice,23 and vascular permeability is suppressed in PAR-1–deficient mice.28 Thus, thrombin-stimulated activation of PAR-1 may constitute one mechanism by which extravascular coagulation influences inflammation.
Thrombin may also influence inflammation through its ability to stimulate fibrin deposition. Indeed, fibrin is a ligand for CD54 (ICAM-1),29,30 CD11b/CD18 (CR3, Mac-1),31–33 and CD11c/CD18 (CR4, p150/95),34,35 adhesion-promoting receptors expressed by endothelial cells, neutrophils, monocytes/macrophages, as well as subsets of dendritic, natural killer, and T cells. Studies using blocking peptides and specific monoclonal antibodies suggest that CD11b/CD18-fibrin interactions regulate leukocyte adherence to vascular clots36 and implanted biomaterials.37,38 Thus, extravascular fibrin may act as a provisional adhesion matrix for leukocyte accumulation at sites of inflammation.
Extravascular fibrin may also directly stimulate leukocyte activities. Fibrin(ogen) reportedly stimulates tumor necrosis factor alpha (TNFα) and IL-1β expression by macrophages39,40 and chemokine secretion by endothelial cells,41,42 fibroblasts,43 and neutrophils.44 We recently demonstrated that fibrinogen also stimulates macrophage chemokine expression, apparently via Toll-like receptor 4 (TLR4).45 As prior studies suggest that TLR4 signals “danger” in response to bacterial lipopolysaccharide (LPS),46–48 we propose that TLR4 may likewise signal danger in response to extravascular coagulation, thereby stimulating the production of chemokines, attracting leukocytes, and enhancing immune surveillance at sites of inflammation.
Until recently, few studies had convincingly evaluated inflammatory roles for fibrin in vivo, in part due to a lack of suitable agents for experimental depletion or antagonism of fibrinogen. Ancrod, a proteolytic enzyme derived from snake venom, proteolyzes fibrinogen, transiently generating a fibrinogen-deficient state. Experimental administration of ancrod produced data consistent with roles for fibrin in experimental encephalomyelitis,1,4 glomerulonephritis,49,50 arthritis,51 transplant rejection,52 and the containment of bacterial infections.53,54 Recently, gene-targeted fibrinogen-deficient mice were generated55 and used to confirm roles for fibrin(ogen) in wound healing56 and the control of bacterial infections.57 Detailed studies of the immune and inflammatory responses in fibrinogen-deficient mice have yet to be reported.
Here, we define and distinguish roles for thrombin, PAR-1, and fibrinogen in a mouse peritonitis model. We demonstrate that thrombin plays an important role in stimulating the adhesion of inflammatory peritoneal macrophages in vivo. Taking advantage of PAR-1–deficient and fibrinogen-deficient mice, we demonstrate that thrombin-stimulated macrophage adhesion is PAR-1 independent, but fibrinogen dependent. We also demonstrate that thrombin stimulates the peritoneal accumulation of cytokines and chemokines in a fibrinogen-dependent manner. Whereas others have clearly established inflammatory roles for PAR-1,22,23,28 our data indicate that extravascular coagulation leading to thrombin production can also regulate inflammation through fibrinogen, presumably via thrombin-stimulated production of fibrin.
Materials and methods
Animals
Mice aged 6 to 10 weeks old were used for these experiments. C57BL/6 mice were purchased from Taconic (Germantown, NY). Transgenic mice were bred at the Trudeau Institute Animal Breeding Facility. All experimental mice were age- and sex-matched. PAR-1–deficient mice27 (backcrossed 6 generations to C57BL/6 mice) were originally obtained from the Jackson Laboratory (Bar Harbor, ME). Fibrinogen-deficient mice55 (backcrossed 7 generations to C57BL/6 mice) and OT-II T-cell receptor transgenic mice58 were generously supplied by Jay Degen (Children’s Hospital Medical Center, Cincinnati, OH) and Francis Carbone (University of Melbourne, Parkville, Australia)/William Heath (The Walter and Eliza Hall Institute, Parkville, Australia), respectively. Animals were housed in a specific pathogen-free facility and cared for according to the Trudeau Institute Animal Care and Use Committee guidelines.
Generation of antigen-specific Th1 cells
Naïve CD4+ T cells were enriched from spleens and lymph nodes of OT-II T-cell receptor transgenic mice58 using CD4 monoclonal antibody (mAb)–based magnetic cell sorting (Miltenyi Biotec, Auburn, CA) followed by density gradient enrichment of resting naïve cells (interface of 80%/62% percoll). Successful purification was confirmed by flow cytometry (> 85% CD4+Vα2+Vβ5+). We then cultured the OT-II cells (2.5 × 105/mL) with mitomycin C–treated splenic C57BL/6 antigen-presenting cells (1 × 106/mL) in RPMI media containing 7.5% fetal bovine serum, 2 mM glutamine, 50 U/mL penicillin, 50 μg/mL streptomycin, and 50 μM 2-mercaptoethanol. To generate Th1 cells, we supplemented cultures with ovalbumin peptide ISQAVHAAHAEINEAGR (Ova) (10 μM, New England Peptide, Fitchburg, MA), IL-2 (20 U/mL, Roche Molecular Biochemicals, Indianapolis, IN), IL-12 (5 ng/mL, BD Pharmingen, San Diego, CA) and anti–IL-4 (clone 11B11, 10 μg/mL, Trudeau Institute Core Antibody Facility). After 6 days, the differentiated cells were routinely > 90% CD4+Vα2+Vβ5+ by flow cytometry. After washing with phosphate-buffered saline (PBS), we adoptively transferred 10 × 106 of these Th1 effectors to naïve mice by intravenous injection. We also confirmed Th1 differentiation by restimulating cells in vitro on CD3 monoclonal antibody-precoated (10 μg/mL, clone 2C11, BD Pharmingen) plates, collecting supernatants at 48 hours, and measuring interferon gamma production by sandwich enzyme-linked immunosorbent assay (ELISA) using an OptEIA kit (BD Pharmingen).
In vivo assays for peritoneal macrophage adhesion and IL-6/monocyte chemoattractant protein-1 production
To elicit inflammatory macrophages, mice received intraperitoneal injections of 3 mL sterile thioglycollate (TG) broth (Becton Dickinson Microbiology Systems, Cockeysville, MD). Assays were performed 4 days later, when macrophage recruitment was maximal (not shown). For antigen-specific assays, mice received adoptive transfers of OT-II Th1 cells 18 to 24 hours prior to initiation of macrophage activation by intraperitoneal injection of 50 μg Ova in 200 μL sterile PBS (Life Technologies, Rockville, MD). Alternatively, TG-primed mice that had not received Th1 cells were given intraperitoneal injections of Escherichia coli serotype 0111:B4 LPS (Sigma Chemical, St Louis, MO) or human alpha thrombin (Enzyme Research Laboratories, South Bend, IN). The thrombin used in these experiments was found to contain less than 0.1 units/mL endotoxin, as determined by Pyrochrome Limulus Amebocyte Lysate Assay (Associates of Cape Cod, Falmouth, MA). At the indicated times, mice were killed and peritoneal cells and fluid were harvested by washing the cavity with 7 mL PBS. Total cell numbers were determined using a hemocytometer and the percentages of macrophages were assessed by evaluation of Wright-Giemsa–stained cytospin smears (HEMA 3; Fisher Scientific, Pittsburgh, PA). Macrophages were identified as large cells with abundant cytoplasm and a single nucleus containing pale diffuse chromatin. Flow cytometry confirmed similar frequencies of macrophages (forward/side scatterhigh Mac-1+Gr-1−, not shown). IL-6 and monocyte chemoattractant protein-1 (MCP-1) protein levels in the harvested exudate fluid were determined by sandwich ELISA using OptEIA kits (BD Pharmingen).
Anticoagulation with Refludan
Refludan59 (16 000 antithrombin units/mg) was reconstituted as directed by the manufacturer (Hoechst Marion Roussel, Kansas City, MO), diluted in sterile PBS, and injected along with the activating stimuli. In pilot studies, we established that 2 mg/kg Refludan efficiently antagonized coagulation in mice, as reported for recombinant hirudin.22 The short half-life of Refludan led us to evaluate dosages up to 20 mg/kg, which also promoted effective anticoagulation without any apparent toxicity (not shown).
Statistics
Statistical significance was evaluated by Student t test using the program Instat 2.01 (GraphPad Software, San Diego, CA).
Results
Injection of inflammatory stimuli into the peritoneal cavity of mice prompts an initial recruitment of neutrophils, followed by an accumulation of macrophages. Upon subsequent activation in situ, these inflammation-elicited macrophages adhere to the mesothelial lining of the peritoneal cavity,60,61 resulting in a dramatic decrease in the number of macrophages that can be recovered by peritoneal lavage.60,61 This assay provides a quantitative means to monitor the activation of macrophage adhesion in vivo.
Prior studies using this assay established that macrophage adhesion can be triggered by specific antigen in mice bearing antigen-sensitized T cells.60–63 In our version of this model, we inject mice with TG to recruit inflammatory macrophages, adoptively transfer OT-II transgenic T-cell receptor Th1 cells, and then inject Ova, the antigen recognized by OT-II T cells. After 5 hours, we harvest the peritoneal cells and enumerate macrophages by performing differential cell counts. As shown in Figure 1A, administration of TG recruits macrophages to the peritoneal cavity, and adoptive transfer of OT-II TCRtg Th1 cells followed by injection of Ova greatly decreases numbers of recoverable macrophages. This response is T cell and antigen dependent, as neither Th1 cells nor Ova stimulate macrophage adhesion when injected alone (Figure 1A). As previously reported,62 this model can also be used to assay macrophage adhesion stimulated by LPS (Figure 1B).
Figure 1. The activation of inflammation-elicited peritoneal macrophages is thrombin dependent in vivo.

(A) The activation-induced adhesion of peritoneal macrophages by antigen-specific Th1 cells is thrombin dependent. OT-II transgenic T-cell receptor Th1 effector cells were adoptively transferred to C57Bl/6 mice that had been primed with TG (3 mL intraperitoneally) 3 days earlier. The next day, Ova peptide (50 μg intraperitoneally) or PBS vehicle (200 μL) were administered, and 5 hours later peritoneal exudate cells were harvested and macrophages were enumerated. Where indicated, mice received Refludan (2 mg/kg intraperitoneally) at the time of Ova administration. The data depicts the averages and standard deviations of groups of 3 mice. Th1 cells induced antigen-specific macrophage activation (P < .01), which was suppressed by Refludan (P = .02). This experiment was replicated twice. (B) The activation-induced adhesion of peritoneal macrophages by LPS is thrombin dependent. Peritoneal macrophages were elicited with TG (3 mL intraperitoneally). Four days later, LPS (1 μg intraperitoneally) or vehicle control (200 μL PBS) were administered, and macrophage numbers in peritoneal exudates were determined 5 hours later. Where indicated, mice also received Relfudan (20 mg/kg intraperitoneally) at the time of LPS administration. The data depicts the averages and standard deviations of groups of 5 mice. LPS induced macrophage activation (P < .0001) that was suppressed by Refludan (P < .0001). This experiment has been replicated 4 times.
Roles for thrombin in macrophage adhesion in vivo
As the procoagulant enzyme thrombin has been implicated in a variety of inflammatory responses,22–25,28 and as the nonspecific anticoagulants heparin and warfarin reportedly block macrophage adhesion in peritoneal models,60 we sought to evaluate roles for thrombin in the activation of macrophage adhesion in vivo. To specifically evaluate thrombin, we performed peritoneal macrophage adhesion assays in the presence of Refludan, a commercially available hirudin analog.59 As discussed in the “Introduction,” hirudin analogs were previously used to establish inflammatory roles for thrombin in mouse models.22–25
We found that Refludan significantly inhibited the activation of peritoneal macrophages in vivo. Both OT-II/Ova- and LPS-stimulated macrophage adhesion was suppressed by administration of Refludan (Figure 1A–B), suggesting that thrombin has critical adhesion-promoting functions in these peritoneal models. Notably, further studies confirmed an earlier report64 that intraperitoneal injection of purified thrombin itself can activate macrophage adhesion (Figure 2A).
Figure 2. Purified thrombin activates inflammation-elicited peritoneal macrophages in vivo.
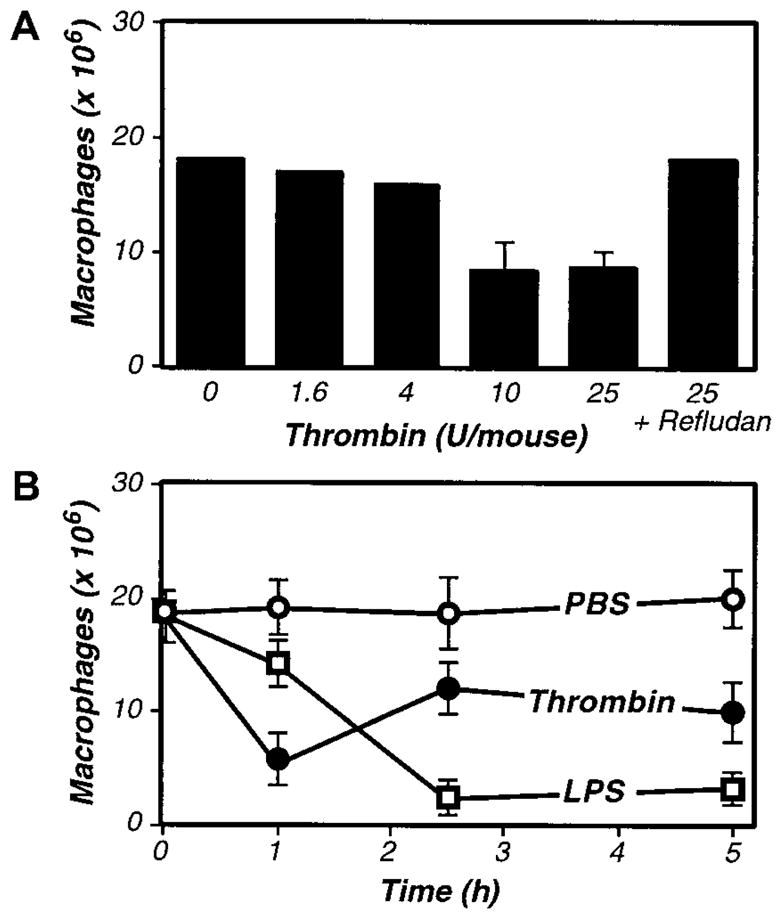
(A) Dose response to thrombin. Four days after TG-stimulated macrophage elicitation (3 mL intraperitoneally), the indicated dosages of purified thrombin were administered, and macrophage numbers in peritoneal exudates were determined after 2.5 hours. (B) Kinetics of macrophage activation in response to thrombin and LPS. Four days after TG-stimulated macrophage elicitation, mice received intraperitoneal injections of thrombin (20 U, ●), LPS (1 μg, □), or vehicle control (200 μL PBS, ○), and macrophage numbers in peritoneal exudates were determined at the indicated times. For A and B, the data represent the averages and standard deviations of 4 animals per group.
As thrombin appeared to be an important mediator of macrophage adhesion, it had to be generated during the course of our peritoneal assays. Indeed, treatment with LPS is well known to up-regulate expression of tissue factor,65,66 an initiator of the coagulation cascade, thereby stimulating thrombin production. Consistent with LPS functioning via the induction of coagulant activities that prompt thrombin production, kinetic analyses revealed that thrombin stimulated macrophage adhesion more rapidly than did LPS (Figure 2B). However, the LPS-stimulated macrophage adhesion was more complete, even at saturating thrombin doses, suggesting that LPS may activate macrophage adhesion via both thrombin-dependent and -independent mechanisms.
Thrombin-stimulated macrophage adhesion is PAR-1 independent and fibrinogen dependent
Having established roles for thrombin in the activation of macrophage adhesion in this model, we next explored its mechanism of action. Recent studies of thrombin’s inflammatory activities have implicated PAR-1 in transmitting thrombin-mediated signals.22,23,28 Thus, we evaluated the activation of macrophage adhesion in PAR-1–deficient mice.27 We first established that TG could stimulate recruitment of inflammatory macrophages to the peritoneal cavity in PAR-1–deficient mice (Figure 3). We then evaluated macrophage adhesion and found no defects in antigen- or LPS-stimulated adhesion in PAR-1–deficient mice (Figure 3). Thus, thrombin stimulates the adhesion of peritoneal macrophages independent of PAR-1.
Figure 3. The activation of inflammation-elicited peritoneal macrophages is not PAR-1 dependent.
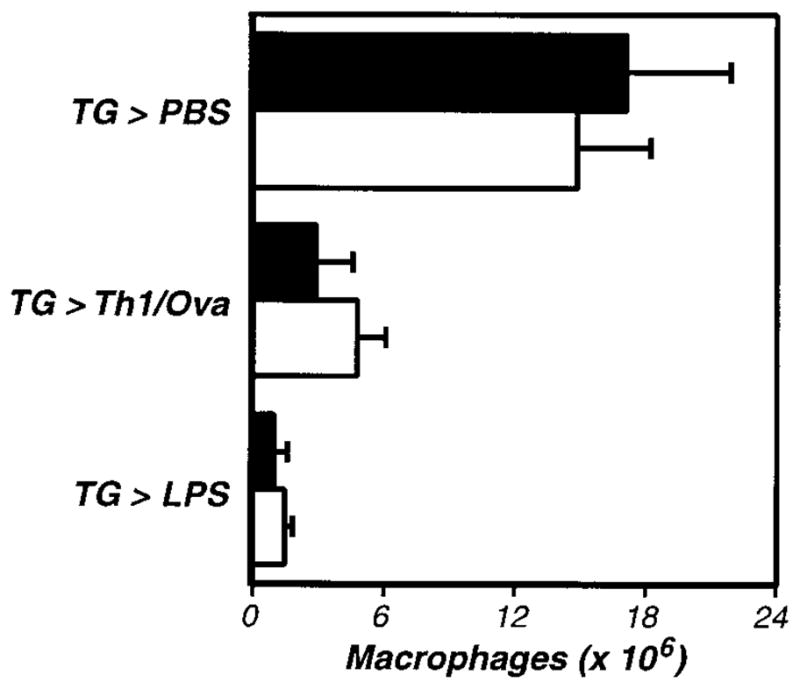
PAR-1–deficient (−/−, □) and littermate control (+/−, ■) mice were compared, using the assays described in Figure 1. The data represent the averages and standard deviations of 5 animals per group. Neither TG-induced macrophage recruitment nor macrophage activation were significantly impaired in PAR-1–deficient mice.
Having ruled out PAR-1, we next examined whether thrombin-stimulated fibrin formation accounts for thrombin’s role in macrophage adhesion. Indeed, prior studies had established that peritoneal macrophages harvested soon after antigen stimulation are coated with fibrin(ogen).67 To explore functional roles for fibrin, we evaluated macrophage adhesion in fibrinogen-deficient mice.55 Again, we began by demonstrating that fibrinogen deficiency does not suppress the TG-stimulated elicitation of macrophages to the peritoneal cavity (Figure 4). Subsequent analyses revealed that antigen-, LPS-, and thrombin-stimulated macrophage adhesion were all fibrinogen dependent, each being significantly suppressed in fibrinogen-deficient mice (Figure 4 and Figure 5). As antigen and LPS are known to stimulate macrophage procoagulant activity leading to thrombin production, and as thrombin stimulates the conversion of fibrinogen to fibrin, the simplest interpretation of our data is that thrombin-mediated fibrin formation functions in the adhesion of inflammatory macrophages.
Figure 4. The activation of inflammation-elicited peritoneal macrophages is fibrinogen dependent.
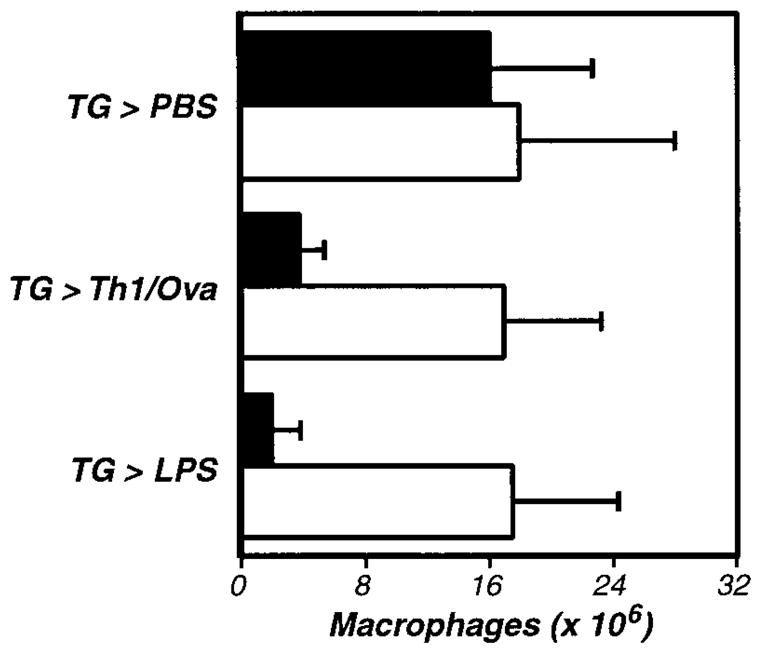
Fibrinogen-deficient (−/−, □) and littermate control (+/−, ■) mice were compared, using the assays described in Figure 1. The data represent the averages and standard deviations of 5 animals per group. TG-induced recruitment of inflammatory macrophages was not significantly impaired in fibrinogen-deficient mice, but macrophage activation was significantly diminished in the absence of fibrinogen (Th1/Ova, P < .002; LPS, P < .002). This experiment was repeated 3 times.
Figure 5. Thrombin stimulates fibrinogen-dependent cytokine and chemokine production in vivo.
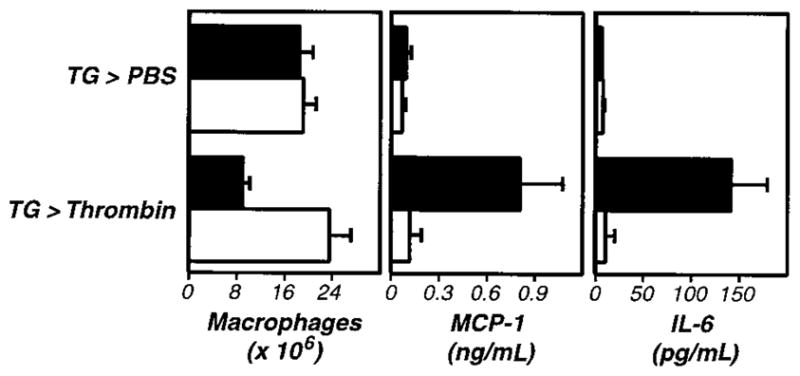
Four days after TG administration, wild-type (+/+,■) or fibrinogen-deficient (−/−, □) mice received intraperitoneal injections of thrombin (20 U). Macrophage numbers in peritoneal exudates were determined after 5 hours (left panel). Thrombin-stimulated macrophage activation was fibrinogen dependent (P < .001). Examination of the exudate fluid by ELISA revealed that thrombin also stimulated increases in peritoneal levels of MCP-1 (middle panel, P < .005) and IL-6 (right panel, P < .001) that were fibrinogen dependent (P < .005 and P < .001, respectively). The data represent averages and standard deviations of 4 animals per group. We repeated this experiment twice.
Thrombin stimulates fibrinogen-dependent IL-6 and MCP-1 production in vivo
During the course of these studies, we discovered that levels of the cytokine IL-6 (Figure 5, right panel) and the chemokine MCP-1 (Figure 5, middle panel) were significantly elevated in peritoneal fluid harvested after administration of thrombin. This thrombin-stimulated cytokine/chemokine production was suppressed by Refludan (not shown) and failed to occur in fibrinogen-deficient mice (Figure 5). As with macrophage adhesion, thrombin-stimulated cytokine/chemokine production proceeded normally in PAR-1–deficient mice (not shown). Stimulation by OT-II Th1 cells/Ova or LPS also prompted cytokine/chemokine production, but only the thrombin-stimulated cytokine/chemokine production was suppressed by Refludan and failed to occur in fibrinogen-deficient mice (not shown). Thus, although thrombin is not absolutely required for cytokine/chemokine production in response to antigen or LPS, thrombin clearly has the capacity to stimulate secretion of inflammatory mediators in vivo in a fibrin(ogen)-dependent manner.
Discussion
In vitro, thrombin reportedly stimulates leukocyte chemotaxis68–70 and proliferation,71–73 and activates mast cell degranulation.74 In theory, gene-targeting techniques could provide an unambiguous means to study inflammatory roles for thrombin in vivo. However, targeted deletion of thrombin or earlier components of the coagulation cascade (ie, tissue factor, factor VII, factor V, or factor X)75–81 results in embryonic or perinatal lethality. Thus, it has not yet been possible to produce adult animals genetically lacking the capacity to generate thrombin.
Recent studies using analogs of recombinant hirudin suggest that thrombin is a physiologic mediator of inflammatory events. These highly specific thrombin antagonists reduce pathology in murine glomerulonephritis,22 arthritis,24,25 and carrageenin-induced inflammation models.23 Mechanistically, some proinflammatory activities of thrombin apparently result from its capacity to stimulate PAR-1, as PAR-1–deficient mice exhibit diminished inflammation in the glomerulonephritis and carrageenin models.22,23
In this report we demonstrated additional inflammatory roles for thrombin in vivo using a mouse peritonitis model. Specifically, we found that Refludan, a hirudin-based pharmacologic thrombin antagonist, suppressed antigen- or LPS-stimulated activation of macrophage adhesion. We also demonstrated that intraperitoneal injection of purified thrombin activates macrophage adhesion, and simultaneously stimulates the peritoneal accumulation of IL-6 and MCP-1. Despite the aforementioned studies implicating PAR-1 in thrombin-stimulated inflammation, we found that the proinflammatory activities of thrombin in these peritoneal models were PAR-1 independent. Rather, both thrombin-stimulated cytokine/chemokine production and macrophage adhesion required fibrinogen, as each was suppressed in fibrinogen-deficient mice.
Mechanistically, the simplest interpretation of our data are that (1) antigen-specific T cells and LPS elicit expression of procoagulant activity stimulating thrombin production, (2) thrombin cleaves fibrinogen, prompting fibrin formation, and (3) fibrin then functions in macrophage adhesion.
Although others have clearly shown that activation of inflammatory peritoneal macrophages stimulates their adhesion to the mesothelial lining of the peritoneal cavity,61 we are presently unable to distinguish between several potential models of fibrin-stimulated macrophage adhesion. One possibility is that fibrin directly mediates macrophage adhesion by simultaneously binding CD11b/CD18 and ICAM-1, fibrin-binding receptors expressed by macrophages and mesothelial cells, respectively. Indeed, an analogous fibrin-mediated bridging model probably accounts for leukocyte adhesion to endothelial cells.29,30 However, as neutrophils also express high levels of CD11b/CD18, but are not depleted from the peritoneal exudates upon macrophage activation62,64 (not shown), we consider a simple bridging model to be an unlikely explanation for thrombin/fibrin(ogen)-stimulated macrophage adhesion.
Alternatively, fibrin could stimulate macrophage adhesion by activating mesothelial cell expression of ligands for macrophage adhesion molecules. Numerous recent studies have established that fibrin(ogen) can activate expression of molecules by endothelial cells, fibroblasts, and leukocytes.39–45 However, if fibrin stimulates mesothelial cell expression of adhesion-promoting ligands, those ligands would need to be macrophage-specific, since neutrophils were not depleted during our assays.
We favor a third model, in which fibrin directly binds and cross-links receptors on macrophages, thereby transmitting signals that stimulate adhesion. Indeed, peritoneal macrophages harvested shortly after stimulation are coated with fibrin(ogen),67 and fibrin-(ogen) can directly stimulate macrophage secretion of cytokines and chemokines in vitro.40,45 Here, we demonstrated that injection of thrombin activates fibrin(ogen)-dependent cytokine/chemokine secretion in vivo. Thus, we believe that thrombin-stimulated fibrin formation directly stimulates peritoneal macrophages, prompting adhesion and secretion of inflammatory mediators. Although we cannot exclude contributions by other cell types, our preliminary data strongly suggest that macrophages are the source of cytokine/chemokine production in our model, as plastic adherent peritoneal cells harvested shortly after the injection of thrombin secreted elevated levels of IL-6 and MCP-1 in vitro without any further stimulation (not shown).
Notably, our studies cannot distinguish between activities of fibrin(ogen) and those of fibrin(ogen)-degradation products (FDPs). FDPs reportedly stimulate vascular permeability,82 endothelial cell retraction,83,84 monocyte/macrophage IL-1 and IL-6 production,85,86 and leukocyte chemotaxis.87–89 FDPs can be generated via plasmin-mediated proteolysis of fibrin, and mice with reduced or no plasmin have been generated by gene-targeted deletion of plasminogen activators or plasminogen, respectively.90,91 Such plasmin-deficient mice display increased pathology in glomerulonephritis92 and arthritis51,93 models, suggesting that plasmin-generated FDPs may well function in inflammation.
Regardless of the precise mechanism, our data are relevant to a number of human pathologies. Extravascular coagulation accompanies many Th1-associated diseases, including autoimmune neuropathologies,1–4 glomerulonephritis,5,6 rheumatoid arthritis,7–9 Crohn’s disease,10,11 and allograft rejection.12,13 As our studies indicate that thrombin and fibrin(ogen) function to stimulate cytokine/chemokine production and macrophage adhesion in vivo, extravascular coagulation likely exacerbates Th1-associated chronic inflammation. Thus, treatment modalities that specifically block inflammation-associated thrombin formation, fibrin deposition, and/or fibrin degradation may constitute novel approaches for controlling pathologic Th1 responses associated with autoimmunity and transplantation. They may likewise provide novel means to attenuate acute inflammation resulting from trauma, burns, or infections.
Our finding that thrombin/fibrin(ogen) can regulate cytokine/chemokine production and macrophage adhesion may also be relevant to septic shock. Bacterial endotoxins prompt expression of procoagulant activities,65,66 and recent studies indicate that therapeutic administration of physiologic vascular anticoagulants reduces septic mortality,94–97 suggesting that procoagulants and/or their products (eg, fibrin, FDPs) play pathologic roles in sepsis. Future studies will be required to clarify roles for thrombin/fibrin(ogen)-stimulated cytokine/chemokine production and/or macrophage adhesion during septic shock.
Increased vascular permeability leading to plasma exudation is among the earliest signs of inflammation. As plasma contains coagulant precursors that become activated upon exposure to extravascular cells, inflammation prompts extravascular thrombin and fibrin formation. Accumulating evidence suggests that this extravascular coagulation has pleiotropic immune and inflammatory functions. Thrombin clearly plays multiple roles, evidenced by the diminished inflammatory responses of both PAR-1– and fibrinogen-deficient mice. Notably, PAR-3 and PAR-4 are also activated by thrombin,98 though we have yet to assess inflammatory functions of those thrombin receptors. Our data suggest that extravascular fibrin also has multiple inflammatory roles, including the regulation of macrophage adhesion and the stimulation of cytokine/chemokine production. Given these pleiotropic activities, we hypothesize that extravascular coagulation may function as nature’s adjuvant, both signaling “danger” at sites of inflammation and providing a provisional fibrin matrix that spatially localizes the ensuing host response.
Acknowledgments
We are indebted to the employees of the Trudeau Institute Animal Breeding and Experimental Animal Maintenance Facilities for dedicated care of the mice used for these studies. We also wish to thank Gail Huston for sharing expertise with the OT-II model, Jean Brennan for assistance with differential cell analyses, and Sachin Mani for technical assistance.
Supported by funds from Trudeau Institute.
References
- 1.Paterson PY. Experimental allergic encephalomyelitis: role of fibrin deposition in immunopathogenesis of inflammation in rats. Fed Proc. 1976;35:2428–2434. [PubMed] [Google Scholar]
- 2.Paterson PY, Koh CS, Kwaan HC. Role of the clotting system in the pathogenesis of neuroimmunologic disease. Fed Proc. 1987;46:91–96. [PubMed] [Google Scholar]
- 3.Claudio L, Raine CS, Brosnan CF. Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol. 1995;90:228–238. doi: 10.1007/BF00296505. [DOI] [PubMed] [Google Scholar]
- 4.Inoue A, Koh CS, Shimada K, Yanagisawa N, Yoshimura K. Suppression of cell-transferred experimental autoimmune encephalomyelitis in defibrinated Lewis rats. J Neuroimmunol. 1996;71:131–137. doi: 10.1016/s0165-5728(96)00150-6. [DOI] [PubMed] [Google Scholar]
- 5.Holdsworth SR, Tipping PG. Macrophage-induced glomerular fibrin deposition in experimental glomerulonephritis in the rabbit. J Clin Invest. 1985;76:1367–1374. doi: 10.1172/JCI112112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neale TJ, Tipping PG, Carson SD, Holdsworth SR. Participation of cell-mediated immunity in deposition of fibrin in glomerulonephritis. Lancet. 1988;2:421–424. doi: 10.1016/s0140-6736(88)90413-8. [DOI] [PubMed] [Google Scholar]
- 7.Zvaifler NJ. The immunopathology of joint inflammation in rheumatoid arthritis. Adv Immunol. 1973;16:265–336. doi: 10.1016/s0065-2776(08)60299-0. [DOI] [PubMed] [Google Scholar]
- 8.Zacharski LR, Brown FE, Memoli VA, et al. Pathways of coagulation activation in situ in rheumatoid synovial tissue. Clin Immunol Immunopathol. 1992;63:155–162. doi: 10.1016/0090-1229(92)90008-c. [DOI] [PubMed] [Google Scholar]
- 9.Gabazza EC, Osamu T, Yamakami T, et al. Correlation between clotting and collagen metabolism markers in rheumatoid arthritis. Thromb Haemost. 1994;71:199–202. [PubMed] [Google Scholar]
- 10.Wakefield AJ, Sawyerr AM, Dhillon AP, et al. Pathogenesis of Crohn’s disease: multifocal gastrointestinal infarction. Lancet. 1989;2:1057–1062. doi: 10.1016/s0140-6736(89)91078-7. [DOI] [PubMed] [Google Scholar]
- 11.Hudson M, Hutton RA, Wakefield AJ, Sawyerr AM, Pounder RE. Evidence for activation of coagulation in Crohn’s disease. Blood Coagul Fibrinolysis. 1992;3:773–778. doi: 10.1097/00001721-199212000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Dvorak HF, Mihm MC, Jr, Dvorak AM, Barnes BA, Manseau EJ, Galli SJ. Rejection of first-set skin allografts in man: the microvasculature is the critical target of the immune response. J Exp Med. 1979;150:322–337. doi: 10.1084/jem.150.2.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labarrere CA, Nelson DR, Faulk WP. Myocardial fibrin deposits in the first month after transplantation predict subsequent coronary artery disease and graft failure in cardiac allograft recipients. Am J Med. 1998;105:207–213. doi: 10.1016/s0002-9343(98)00246-0. [DOI] [PubMed] [Google Scholar]
- 14.Nelson DS. The effects of anticoagulants and other drugs on cellular and cutaneous reactions to antigen in guinea pigs with delayed-type hypersensitivity. Immunology. 1965;9:219–234. [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen S, Benacerraf B, McCluskey RT, Ovary Z. Effect of anticoagulants on delayed hypersensitivity reactions. J Immunol. 1967;98:351–358. [PubMed] [Google Scholar]
- 16.Colvin RB, Johnson RA, Mihm MC, Jr, Dvorak HF. Role of the clotting system in cell-mediated hypersensitivity, I: fibrin deposition in delayed skin reactions in man. J Exp Med. 1973;138:686–698. doi: 10.1084/jem.138.3.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colvin RB, Dvorak HF. Role of the clotting system in cell-mediated hypersensitivity, II: kinetics of fibrinogen/fibrin accumulation and vascular permeability changes in tuberculin and cutaneous basophil hypersensitivity reactions. J Immunol. 1975;114:377–387. [PubMed] [Google Scholar]
- 18.Edwards RL, Rickles FR. Delayed hypersensitivity in man: effects of systemic anticoagulation. Science. 1978;200:541–543. doi: 10.1126/science.644314. [DOI] [PubMed] [Google Scholar]
- 19.Colvin RB, Mosesson MW, Dvorak HF. Delayed-type hypersensitivity skin reactions in congenital afibrinogenemia lack fibrin deposition and induration. J Clin Invest. 1979;63:1302–1306. doi: 10.1172/JCI109425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy GA, Leibowitz JL, Edgington TS. Induction of monocyte procoagulant activity by murine hepatitis virus type 3 parallels disease susceptibility in mice. J Exp Med. 1981;154:1150–1163. doi: 10.1084/jem.154.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Fung LS, Chung S, et al. Monoclonal anti-prothrombinase (3D4.3) prevents mortality from murine hepatitis virus (MHV-3) infection. J Exp Med. 1992;176:689–697. doi: 10.1084/jem.176.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med. 2000;191:455–462. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cirino G, Cicala C, Bucci MR, Sorrentino L, Maraganore JM, Stone SR. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varisco PA, Peclat V, van Ness K, Bischof-Delaloye A, So A, Busso N. Effect of thrombin inhibition on synovial inflammation in antigen induced arthritis. Ann Rheum Dis. 2000;59:781–787. doi: 10.1136/ard.59.10.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marty I, Peclat V, Kirdaite G, Salvi R, So A, Busso N. Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest. 2001;107:631–640. doi: 10.1172/JCI11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 27.Connolly AJ, Ishihara H, Kahn ML, Farese RV, Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- 28.Vogel SM, Gao X, Mehta D, et al. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics. 2000;4:137–145. doi: 10.1152/physiolgenomics.2000.4.2.137. [DOI] [PubMed] [Google Scholar]
- 29.Languino LR, Plescia J, Duperray A, et al. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73:1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- 30.Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. Regulation of leukocyte-endothelium interaction and leukocyte trans-endothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci U S A. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altieri DC, Bader R, Mannucci PM, Edgington TS. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diamond MS, Springer TA. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol. 1993;120:545–556. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loike JD, Sodeik B, Cao L, et al. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the Aα chain of fibrinogen. Proc Natl Acad Sci U S A. 1991;88:1044–1048. doi: 10.1073/pnas.88.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nham SU. Characteristics of fibrinogen binding to the domain of CD11c, an alpha subunit of p150,95. Biochem Biophys Res Commun. 1999;264:630–634. doi: 10.1006/bbrc.1999.1564. [DOI] [PubMed] [Google Scholar]
- 36.Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to αIIbβ3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100:2085–2093. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang L, Eaton JW. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J Exp Med. 1993;178:2147–2156. doi: 10.1084/jem.178.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang L, Ugarova TP, Plow EF, Eaton JW. Molecular determinants of acute inflammatory responses to biomaterials. J Clin Invest. 1996;97:1329–1334. doi: 10.1172/JCI118549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan ST, Edgington TS. Integrin regulation of leukocyte inflammatory functions: CD11b/CD18 enhancement of the tumor necrosis factor-α responses of monocytes. J Immunol. 1993;150:2972–2980. [PubMed] [Google Scholar]
- 40.Perez RL, Roman J. Fibrin enhances the expression of IL-1β by human peripheral blood mononuclear cells: implications in pulmonary inflammation. J Immunol. 1995;154:1879–1887. [PubMed] [Google Scholar]
- 41.Qi J, Kreutzer DL. Fibrin activation of vascular endothelial cells: induction of IL-8 expression. J Immunol. 1995;155:867–876. [PubMed] [Google Scholar]
- 42.Harley SL, Powell JT. Fibrinogen up-regulates the expression of monocyte chemoattractant protein 1 in human saphenous vein endothelial cells. Biochem J. 1999;341:739–744. [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Piela-Smith TH. Fibrin(ogen)-induced expression of ICAM-1 and chemokines in human synovial fibroblasts. J Immunol. 2000;165:5255–5261. doi: 10.4049/jimmunol.165.9.5255. [DOI] [PubMed] [Google Scholar]
- 44.Walzog B, Weinmann P, Jeblonski F, Scharffetter-Kochanek K, Bommert K, Gaehtgens P. A role for β2 integrins (CD11/CD18) in the regulation of cytokine gene expression of polymorphonuclear neutrophils during the inflammatory response. Faseb J. 1999;13:1855–1865. doi: 10.1096/fasebj.13.13.1855. [DOI] [PubMed] [Google Scholar]
- 45.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 46.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 47.Vogel SN, Johnson D, Perera PY, et al. Functional characterization of the effect of the C3H/HeJ defect in mice that lack an Lpsn gene: in vivo evidence for a dominant negative mutation. J Immunol. 1999;162:5666–5670. [PubMed] [Google Scholar]
- 48.Hoshino K, Takeuchi O, Kawai T, et al. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 49.Thomson NM, Moran J, Simpson IJ, Peters DK. Defibrination with ancrod in nephrotoxic nephritis in rabbits. Kidney Int. 1976;10:343–347. doi: 10.1038/ki.1976.120. [DOI] [PubMed] [Google Scholar]
- 50.Cole EH, Glynn MF, Laskin CA, Sweet J, Mason N, Levy GA. Ancrod improves survival in murine systemic lupus erythematosus. Kidney Int. 1990;37:29–35. doi: 10.1038/ki.1990.4. [DOI] [PubMed] [Google Scholar]
- 51.Busso N, Peclat V, Van Ness K, et al. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J Clin Invest. 1998;102:41–50. doi: 10.1172/JCI2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Munda R, Glas-Greenwalt P, Weiss MA, Pollak VE, Alexander JW. Prolongation of survival of a heart xenograft by defibrination with ancrod. Transplantation. 1983;35:620–622. [PubMed] [Google Scholar]
- 53.Koch T, Duncker HP, Axt R, Schiefer HG, Van Ackern K, Neuhof H. Effects of hemorrhage, hypoxia, and intravascular coagulation on bacterial clearance and translocation. Crit Care Med. 1993;21:1758–1764. doi: 10.1097/00003246-199311000-00027. [DOI] [PubMed] [Google Scholar]
- 54.Koch T, Annuss C, Schiefer HG, van Ackern K, Neuhof H. Impaired bacterial clearance after activation of the complement and coagulation systems. Shock. 1997;7:42–48. doi: 10.1097/00024382-199701000-00005. [DOI] [PubMed] [Google Scholar]
- 55.Suh TT, Holmback K, Jensen NJ, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 56.Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound-healing defects in mice lacking fibrinogen. Blood. 2001;97:3691–3698. doi: 10.1182/blood.v97.12.3691. [DOI] [PubMed] [Google Scholar]
- 57.Goguen JD, Bugge T, Degen JL. Role of the pleiotropic effects of plasminogen deficiency in infection experiments with plasminogen-deficient mice. Methods. 2000;21:179–183. doi: 10.1006/meth.2000.0989. [DOI] [PubMed] [Google Scholar]
- 58.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 59.Greinacher A, Volpel H, Janssens U, et al. Recombinant hirudin (lepirudin) provides safe and effective anticoagulation in patients with heparin-induced thrombocytopenia: a prospective study. Circulation. 1999;99:73–80. doi: 10.1161/01.cir.99.1.73. [DOI] [PubMed] [Google Scholar]
- 60.Nelson DS. Reaction to antigen in vivo of the peritoneal macrophages of guinea pigs with delayed-type hypersensitivity: effects of anticoagulants and other drugs. Lancet. 1963;2:175–176. doi: 10.1016/s0140-6736(63)92808-3. [DOI] [PubMed] [Google Scholar]
- 61.Nelson DS, North RJ. The fate of peritoneal macrophages after the injection of antigen into guinea pigs with delayed-type hypersensitivity. Lab Invest. 1965;14:89–101. [PubMed] [Google Scholar]
- 62.Nelson DS, Boyden SV. The loss of macrophages from peritoneal exudates following the injection of antigens into guinea-pigs with delayed-type hypersensitivity. Immunology. 1963;6:264–275. [PMC free article] [PubMed] [Google Scholar]
- 63.Sonozaki H, Cohen S. The effect of sensitized lymphocytes on peritoneal exudate macrophages in the guinea pig. J Immunol. 1971;106:1404–1406. [PubMed] [Google Scholar]
- 64.Jokay I, Karczag E. Thrombin-induced “macrophage disappearance reaction” in mice. Experientia. 1973;29:334–335. doi: 10.1007/BF01926512. [DOI] [PubMed] [Google Scholar]
- 65.Colucci MR, Balconi R, Lorenzet A, et al. Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest. 1983;71:1893–1896. doi: 10.1172/JCI110945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drake TA, Cheng J, Chang A, Taylor FB., Jr Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993;142:1458–1470. [PMC free article] [PubMed] [Google Scholar]
- 67.Colvin RB, Dvorak HF. Fibrinogen/fibrin on the surface of macrophages: detection, distribution, binding requirements, and possible role in macrophage adherence phenomena. J Exp Med. 1975;142:1377–1390. doi: 10.1084/jem.142.6.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bar-Shavit R, Kahn A, Wilner GD, Fenton JW. Monocyte chemotaxis: stimulation by specific exosite region in thrombin. Science. 1983;220:728–731. doi: 10.1126/science.6836310. [DOI] [PubMed] [Google Scholar]
- 69.Bizios R, Lai L, Fenton JW, Malik AB. Thrombin-induced chemotaxis and aggregation of neutrophils. J Cell Physiol. 1986;128:485–490. doi: 10.1002/jcp.1041280318. [DOI] [PubMed] [Google Scholar]
- 70.Grandaliano G, Valente AJ, Abboud HE. A novel biologic activity of thrombin: stimulation of monocyte chemotactic protein production. J Exp Med. 1994;179:1737–1741. doi: 10.1084/jem.179.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen LB, Teng NN, Buchanan JM. Mitogenicity of thrombin and surface alterations on mouse splenocytes. Exp Cell Res. 1976;101:41–46. doi: 10.1016/0014-4827(76)90409-2. [DOI] [PubMed] [Google Scholar]
- 72.Bar-Shavit R, Kahn AJ, Mann KG, Wilner GD. Identification of a thrombin sequence with growth factor activity on macrophages. Proc Natl Acad Sci U S A. 1986;83:976–980. doi: 10.1073/pnas.83.4.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naldini A, Sower L, Bocci V, Meyers B, Carney DH. Thrombin receptor expression and responsiveness of human monocytic cells to thrombin is linked to interferon-induced cellular differentiation. J Cell Physiol. 1998;177:76–84. doi: 10.1002/(SICI)1097-4652(199810)177:1<76::AID-JCP8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 74.Razin E, Marx G. Thrombin-induced degranulation of cultured bone marrow-derived mast cells. J Immunol. 1984;133:3282–3285. [PubMed] [Google Scholar]
- 75.Bugge TH, Xiao Q, Kombrinck KW, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc Natl Acad Sci U S A. 1996;93:6258–6263. doi: 10.1073/pnas.93.13.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carmeliet P, Mackman N, Moons L, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383:73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 77.Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384:66–68. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 78.Rosen ED, Chan JC, Idusogie E, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390:290–294. doi: 10.1038/36862. [DOI] [PubMed] [Google Scholar]
- 79.Xue J, Wu Q, Westfield LA, et al. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci U S A. 1998;95:7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun WY, Witte DP, Degen JL, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci U S A. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dewerchin M, Liang Z, Moons L, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost. 2000;83:185–190. [PubMed] [Google Scholar]
- 82.Sueishi K, Nanno S, Tanaka K. Permeability enhancing and chemotactic activities of lower molecular weight degradation products of human fibrinogen. Thromb Haemost. 1981;45:90–94. [PubMed] [Google Scholar]
- 83.Rowland FN, Donovan MJ, Picciano PT, Wilner GD, Kreutzer DL. Fibrin-mediated vascular injury: identification of fibrin peptides that mediate endothelial cell retraction. Am J Pathol. 1984;117:418–428. [PMC free article] [PubMed] [Google Scholar]
- 84.Dang CV, Bell WR, Kaiser D, Wong A. Disorganization of cultured vascular endothelial cell monolayers by fibrinogen fragment D. Science. 1985;227:1487–1490. doi: 10.1126/science.4038818. [DOI] [PubMed] [Google Scholar]
- 85.Robson SC, Shephard EG, Kirsch RE. Fibrin degradation product D-dimer induces the synthesis and release of biologically active IL-1 beta, IL-6 and plasminogen activator inhibitors from monocytes in vitro. Br J Haematol. 1994;86:322–326. doi: 10.1111/j.1365-2141.1994.tb04733.x. [DOI] [PubMed] [Google Scholar]
- 86.Lee ME, Rhee KJ, Nham SU. Fragment E derived from both fibrin and fibrinogen stimulates interleukin-6 production in rat peritoneal macrophages. Mol Cells. 1999;9:7–13. [PubMed] [Google Scholar]
- 87.Kay AB, Pepper DS, McKenzie R. The identification of fibrinopeptide B as a chemotactic agent derived from human fibrinogen. Br J Haematol. 1974;27:669–677. doi: 10.1111/j.1365-2141.1974.tb06633.x. [DOI] [PubMed] [Google Scholar]
- 88.Saldeen K, Christie N, Nelson WR, Movat HZ. Effect of a fibrin(ogen)-derived vasoactive peptide on polymorphonuclear leukocyte emigration. Thromb Res. 1985;37:85–89. doi: 10.1016/0049-3848(85)90035-0. [DOI] [PubMed] [Google Scholar]
- 89.Gross TJ, Leavell KJ, Peterson MW. CD11b/CD18 mediates the neutrophil chemotactic activity of fibrin degradation product D domain. Thromb Haemost. 1997;77:894–900. [PubMed] [Google Scholar]
- 90.Carmeliet P, Schoonjans L, Kieckens L, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 91.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. doi: 10.1101/gad.9.7.794. [DOI] [PubMed] [Google Scholar]
- 92.Kitching AR, Holdsworth SR, Ploplis VA, et al. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang YH, Carmeliet P, Hamilton JA. Tissue-type plasminogen activator deficiency exacerbates arthritis. J Immunol. 2001;167:1047–1052. doi: 10.4049/jimmunol.167.2.1047. [DOI] [PubMed] [Google Scholar]
- 94.Taylor FB, Jr, Chang A, Esmon CT, D’Angelo A, Vigano-D’Angelo S, Blick KE. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79:918–925. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Creasey AA, Chang AC, Feigen L, Wun TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2856. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Minnema MC, Chang AC, Jansen PM, et al. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood. 2000;95:1117–1123. [PubMed] [Google Scholar]
- 97.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 98.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]