

Abstract
Most cancer vaccines, thus far, fail to control established tumors. However, their application in preventing tumors is another question that is understudied. In the current study, we investigated the CD8 memory T cell responses of lentivector (lv) immunization and its potential to prevent melanoma using both transplantable B16 tumor and autochthonous melanoma models. We found that lv expressing xenogenic human glycoprotein (hgp100) could induce potent CD8 responses that cross react with mouse gp100. Importantly, the lv primed CD8 response consisted of a high number of memory precursors and could be further increased by recombinant vaccinia virus vector (vv) boost, resulting in enhanced CD8 memory response. These long-lasting CD8 memory T cells played a critical role in immune surveillance and could rapidly respond and expand after sensing B16 tumor cells to prevent tumor establishment. Although CD8 response plays a dominant role after lv immunization, both CD4 and CD8 T cells are responsible for the immune prevention. In addition, we surprisingly found that CD4 help was not only critical for generating primary CD8 responses, but also important for secondary CD8 responses of vv boost. CD4 depletion prior to lv prime or prior to vv boost substantially reduced the magnitude of secondary CD8 effector and memory responses, and severely compromised the effect of cancer immune prevention. More importantly, the CD8 memory response from lv-vv prime-boost immunization could effectively prevent autochthonous melanoma in tumor-prone transgenic (Tg) mice, providing a strong evidence that lv-vv prime-boost strategy is an effective approach for cancer immune prevention.
Keywords: Immune prevention, viral vectors, memory CD8 T cells, genetic immunization
Introduction
Vaccines are the most successful medicines for preventing infectious diseases. Based on the same premise, cancer vaccines have been extensively pursued to stimulate antitumor immunity. However, due to practical and ethical constraints, cancer vaccines, in most cases, have been tested as a therapeutic approach to treat established (and mostly advanced stages of) cancers, which is completely deviated from what vaccine does the best and is intended to do, i.e. to prevent diseases. Therefore, it is no surprise that most cancer vaccines fail to control established cancers (1). It is becoming evident that cancer vaccines alone may not work well for treating tumors due to the immune suppression established in the tumor lesions (1–). But, their efficacy of preventing tumors is totally another question that needs to be addressed. It has been reasoned that cancer vaccines may work effectively in prophylactic setting because the tumor induced immune suppression is not established (4–5). However, cancer immune prevention has been understudied even though it was proposed a decade ago (5–6) possibly due to the difficulty of identifying high risk population, the uncertainty of cancer development, and the risk of developing autoimmune diseases. But, as the advancement of genomic analysis and gene profiling, more accurate prediction of cancer development (or relapse after conventional therapy) and better evaluation of risk-benefit ratio will become possible, which can lead to acceptance of cancer immune prevention in high risk population (4, 7–8).
The researches of tumor immunotherapy in the last two decades yield many different kinds of cancer vaccines including peptide-(9), dendritic cell (DC)-(10), and gene-(11–12) based vaccines that can stimulate potent tumor specific immune responses. Although immunized mice were prevented from tumor cell challenge, the protection was mostly examined when the immune responses were still at the peak (13–14), which has little relevance to the long-term effect of cancer immune prevention. Furthermore, prevention of autochthonous tumor in cancer-prone Tg mice was rarely conducted in a few studies. One report found that DNA immunization against oncoprotein rat Her2/neu could prevent breast cancer in the HER2/NEU Tg mice (15). But, repeated immunization of every 3–4 weeks was required to maintain the prevention effect. Another recent study showed that peptide vaccination against tumor Ag mucin 1 (MUC1) could prevent progression of colitis to colon cancer in the IL-10−/−MUC1 Tg mice (8). But the prevention effect was again examined only 10 weeks after immunization. The long-term prevention effect of cancer vaccines is unknown. In addition, the immune correlates of cancer immune prevention are controversial. For example, while antibodies were found important in preventing autochthonous breast cancer in HER2/NEU Tg mice (15), cytotoxic T cells with (16) or without the help of antibodies (13) were shown to prevent transplantable breast cancer in mice. Thus, there is a need for designing better cancer vaccines and for understanding the mechanisms underpinning the immune prevention.
We and others found that recombinant lentivector (lv) could induce potent CD8 immune responses against self/tumor Ags (17–18) because of their effective transduction of skin DCs in vivo (19–21). Higher percent of CD8 T cells elicited by lv possesses memory phenotype (22), suggesting that the lv primed CD8 T cells will effectively respond to boosting immunization. Heterologous prime-boost immunization strategy has been extensively studied for stimulating potent and long-lasting memory responses to prevent infectious diseases (23–25). It was reported that lv-vv prime-boost could markedly increase melanoma Ag NY-ESO specific effector CD8 responses (26). However, it is not clear if this strategy will generate enhanced memory CD8 T cells and prevent clinically relevant autochthonous cancers. It is also not known if the prime-boost induced memory responses will be qualitatively superior to that from one immunization and better prevent mice from tumor challenge. Therefore, in this study, using self melanoma Ag glycoprotein 100 (gp100), we studied the CD8 memory responses of lv-vv prime-boost immunization and the immune prevention of autochthonous melanoma. We found that much more potent CD8 memory responses could be elicited by lv-vv prime-boost. Critically, we demonstrated that only the CD8 memory T cells from lv-vv prime-boosted mice were able to rapidly respond to tumor cell challenge to prevent tumor establishment. Another important novel finding is that the generation of high level effector and memory CD8 T cells requires the CD4 help during both lv prime and vv boost stage. More importantly, we demonstrated that the lv-vv prime-boost strategy could effectively prevent autochthonous melanoma growth in melanoma-prone metabotropic glutamate receptor 1 (Grm1) Tg mice for at least 12 months.
Materials and Methods
Cell lines and Mice
Cell lines CV-1, TK-143, 293T and B16F10 were acquired from ATCC and maintained in complete DMEM media. C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD). Hgp100 specific TCR Tg mice (pmel-1 mice) were purchased from Jackson Laboratory (Bar Harbor, ME). The melanoma prone Grm1 Tg mice were kindly provided by Dr. Suzie Chen of Rutgers University (27). All the mice were housed under SPF conditions in Laboratory Animal Services of Georgia Health Sciences University. Animal care protocols were approved by the IACUC of Georgia Health Sciences University.
Construction of viral vectors and immunization
Plasmid hgp100 DNA was kindly provided by Dr. Walter Storkus, University of Pittsburgh Cancer Institute and Department of Dermatology, University of Pittsburgh. To construct lv expressing xenogenic hgp100, gene fragment containing the N-terminal 340aa of hgp100 was obtained using high fidelity PCR and cloned into the lv plasmid. The recombinant lv was designated as hgp100-lv. Hgp100-lv was prepared and titered as previously described (20). For immunization, 1.5 × 107 transduction units (TUs) of hgp100-lv were injected into the footpad.
To construct recombinant vv expressing the hgp100, a shuttle plasmid vector pG10 was used (Fig.1A). The hgp100 340aa fragment was cloned behind the p7.5 early gene promoter to obtain pG10-hgp100. CV-1 cells in 6-well plates were infected with a wild type vaccinia virus of WR strain at MOI of 0.1, and then transfected with pG10-hgp100 by SuperFect reagent (Qiagen, Inc.). The recombinant vv was selected in human TK- 143 cells with addition of BrdU in the medium. After three rounds of plaque purification, the purity of the virus was verified by PCR assays for presence of the transgene and deletion of the viral thymidine kinase gene, and by fluorescence of DsRED in the infected cells. The virus, designated as hgp100-vv, was amplified in HeLa cells and purified by a standard procedure (28). For immunization, 1.5 × 107 pfu of hgp100-vv was injected i.p. or via footpad.
Fig.1. Hgp100-lv immunization stimulates potent CD8 responses that can cross-react with mgp100 epitope.

A. The schematic diagram of hgp100-lv and hgp100-vv was shown. Hgp100-N indicated the N-terminal 340aa of hgp100. B. Pmel-1 cells in the popliteal LN were induced to proliferate by hgp100-lv or hgp100-vv footpad immunization. C57BL/6 mice were immunized with hgp100-lv or hgp100-vv and then adoptively transferred with CFSE labeled pmel-1 T cells. Three days later, pmel-1 T cells were analyzed for proliferation by gating on Thy1.1+ CD8 T cells. A representative of 3 mice in each group was shown. C. Two weeks after immunization, ICS of IFNγ of the peripheral blood cells was performed following 3hr ex vivo stimulation with hgp100 peptide. No IFNγ + CD8 T cells were detected in control mice (not shown). The number in the parenthesis of upper right quadrant represents the percentage of IFNγ+ T cells among total CD8 cells. A summary of 3 mice was shown on the right. D. Cross reactivity of hgp100-lv induced CD8 responses to mgp100 epitope in peripheral blood was demonstrated by ICS of IFNγ after ex vivo stimulation with indicated peptides. Summary data of three mice was also shown. The experiment was repeated twice with similar data. Statistical analysis was done with unpaired t-test.
In vivo proliferation of pmel-1 by hgp100-lv or hgp100-vv immunization
To examine the induction of Ag specific T cell proliferation by recombinant lv and vv in vivo, mice were immunized with either hgp100-lv or hgp100-vv and then injected with 1×106 pmel-1 cells labeled with carboxyfluorescein succinimidyl ester (CFSE) dye as previously described (19). Three days later, vaccine draining LN was collected to determine the proliferation of pmel-1 cells by analyzing the progressive dilution of CFSE dye.
Flow cytometry analysis of immune responses
To detect gp100 specific CD8 T cells, single cells from peripheral blood or spleen were stained with hgp10025–33/Db tetramer (kindly provided by the NIH tetramer core facility at Emory University) together with anti-CD127, anti-CD8, and anti-CD62L (Biolegend, San Diego, CA). In addition, to measure the cytokine production, single cell suspensions of peripheral blood or spleen were stimulated for 3hrs with 1μg/ml of hgp100 peptide (GeneScript, New Jersey) and then intracellularly stained (ICS) for IFN-γ as described (20). Cells were collected using a FACScanto system (BD Bioscience, San Jose, CA). Data were analyzed using FCS Express V3 software (De Novo Software, Ontario, Canada).
Immune prevention of cancer development
To study the cancer immune prevention effect, B16F10 (3 × 105) cells were inoculated subcutaneously into the shaved flank of the immunized C57BL/6 mice. Tumor growth was monitored. To investigate the prevention effect in autochthonous melanoma model, lv prime with or without vv boost was conducted on 1.5 or 3 months old melanoma prone Grm1 Tg mice. Melanoma growth on the tail and ear were monitored.
Depletion of T cell subsets in vivo
To study the immune correlates of cancer prevention, immunized mice were depleted of CD4, CD8, or both by in vivo injection of 500μg of functional grade of monoclonal Abs (GK1.5 for CD4 depletion, 2.43 for CD8 depletion, Bio X Cell, Inc, West Lebanon, NH) before tumor challenge. The effect of T cell subset depletion was monitored by examining the CD4 and CD8 T cells in the peripheral blood.
Statistical analysis
Data were analyzed using student’s unpaired t-test, ANOVA, or Logrank test of the Prism software (GraphPad Prism, La Jolla, CA).
Results
Lv expressing xenogenic hgp100 stimulates potent CD8 responses that cross react with mouse gp100 epitope
To characterize the lv induced immune responses and to study the potential of lv immunization for immune prevention, we constructed recombinant lv expressing melanoma Ag human gp100 (hgp100) because it was previously demonstrated that xenogenic hgp100 DNA could better protect mice from B16 tumor cell challenge than mouse gp100 (mgp100) (29). As a comparison, recombinant vv was also constructed (Fig.1A). We found that, compared to hgp100-vv, hgp100-lv was more potent at inducing in vivo proliferation of hgp10025–33 specific CD8 T cells, pmel-1 (Fig.1B). In addition, by measuring the IFNγ+ T cells, we found that activation of endogenous CD8 T cells by lv was substantially higher than recombinant vv (Fig.1C), which was in agreement with our previous findings that lv was superior to other viral vectors (19). Approximately 7% of CD8 T cells in the peripheral blood from hgp100-lv immunized mice produced IFNγ in responding to a 3hr ex vivo hgp10025–33 peptide stimulation, whereas the hgp100 CD8 response was undetectable in hgp100-vv immunized mice. Critically, the hgp100-lv activated CD8 T cells could effectively cross recognize the mgp100 epitope (mgp10025–33) and produce IFNγ (Fig.1D). These data indicate that lv expressing xenogenic hgp100 elicits potent CD8 T cell responses that can cross react with mgp100 peptide. We also showed that although recombinant vv is a proven safe vaccine vector and has been widely utilized for immunization, it is not effective for stimulating immune responses against the transgene encoded Ag.
The gp100 specific CD8 T cells elicited by hgp100-lv consist of effector memory precursor cells with the phenotype of CD127hi CD62Llo
The ultimate purpose of vaccine administration is to generate long-lasting memory immune cells in the immunized host. To further characterize the CD8 immune responses elicited by hgp100-lv immunization, we determined the CD127 (IL-7Rα) expression on the hgp10025–33 tetramer positive cells because it was previously shown that CD127 was selectively expressed on the precursors of memory CD8 T cells (30–31). Using tetramer staining, we found that while the ratio of hgp100 specific CD8 T cells were low following hgp100-vv immunization, hgp100-lv immunization dramatically increased the hgp10025–33 tetramer positive CD8 T cells (Fig.2A), consistent with the data of cytokine staining (Fig.1C). Approximately 30–40% of CD8 T cells in the peripheral blood were hgp10025–33 tetramer positive two weeks after immunization. Five weeks after immunization, the proportion of hgp10025–33 tetramer positive cells remained at 10% of total CD8 T cells. More importantly, ~20% of tetramer positive CD8 T cells were also CD127+at the peak of immune responses (day 15) (Fig.2B). Most of the hgp10025–33 tetramer positive cells were CD62L-. The CD127+CD62L- phenotype of activated CD8 T cells is consistent with the markers of effector memory T cells (31), suggesting lv activated T cells contain a high level of precursors of effector memory CD8 T cells in the peripheral at early stage of immune responses. The ratio of CD127+ cells increased with time (Fig.2C), in agreement with the notion that memory T cells are gradually accumulated as the effector cells waned (32).
Fig.2. Lv stimulated hgp100 specific CD8 T cells consist of high percent of CD127+ memory precursors.

Two weeks after immunization, peripheral blood cells from immunized C57BL/6 mice were stained with hgp100/Db tetramer and CD127 or CD62L. A. In agreement with data of Fig.1, hgp100-lv induced more potent CD8 response than hgp100-vv. A representative of five mice was presented. The kinetics of tetramer positive cells was also shown on the right. B. The tetramer positive CD8 T cells were gated and analyzed for CD127 expression. Approximately 20–30% of hgp100 specific CD8 T cells were CD127+ and most of hgp100/Db tetramer positive CD8 T cells were CD62L-. C. The percent of memory CD127+ hgp100 specific CD8 T cells increased with time. Five mice were in each group and the experiment was repeated twice with similar observation.
Lv-vv prime-boost markedly increases the magnitude of effector and memory CD8 responses
The presence of CD127+ CD8 memory precursor T cells suggests that lv primed immune responses have the potential to be substantially increased with boosting immunization. To examine this hypothesis, lv primed mice were then boosted with hgp100-vv. The kinetics of immune responses was determined by repeatedly examining the IFNγ+ CD8 T cells in the peripheral blood. Consistent with the data in Fig.1 and Fig.2, hgp100-vv alone did not induce measurable immune responses. However, hgp100-vv boost immunization rapidly and markedly increased the magnitude of CD8 responses in the hgp100-lv primed mice (Fig.3A). Five days after boost, the CD8 responses approximately reached the peak with ~15% of CD8 T cells in the blood were IFNγ+. Reversing the order of prime-boost to vv prime and lv boost only induced the same magnitude of immune response as that of the lv elicited primary responses. The CD8 responses induced by lv-vv prime-boost were long-lasting. More than two months (70 days) after boost, the ratio of IFNγ+ CD8 T cells in the PBL was still at the level of 5% (Fig.3A). And at this stage, approximately 20% of CD8 T cells in the blood were hgp10025–33 tetramer positive cells (Fig.3B). Importantly, more than 90% of the hgp100 specific CD8 T cells in the PBL were CD127+, suggesting most of hgp100 specific CD8 T cells become memory cells (lower panel, Fig.3B). Although the hgp10025–33 tetramer positive cells were markedly higher in the prime-boosted mice compared to lv immunization alone, the ratio of CD127+ remained the same (lower panel, Fig.3B). Thus, vv boost does not preferentially increase memory cells, but simply increases the overall Ag specific responses that give rise to a higher number of memory CD8 T cells.
Fig.3. Lv primed CD8 responses can be markedly increased by vv boost, generating long-lasting memory CD8 responses.

A. The kinetics of CD8 response after lv or vv immunization was monitored by ICS of IFNγ. Forty-five days later, some of the lv primed mice were boosted with vv or vice versa. Five mice were in each group. B. Seventy days after boost, the hgp100 specific CD8 T cells and their memory marker of CD127 were measured. Only the CD8 T cells were shown. Summaries of the tetramer and CD127 staining (5 mice) were presented on the right. The experiment was repeated twice with similar observation. Unpaired t-test or ANOVA was used for statistical analysis.
The CD8 memory T cells generated by lv-vv prime-boost have superior quality
In order to prevent tumor cells from becoming established tumor, it is critical that the vaccine induced tumor specific memory T cells can immediately sense the emergence of tumor cells and be rapidly reactivated and expanded to kill the malignant or premalignant cells in the incipient stage. It is known that tumor cells, such as the low immunogenic B16 tumor cells are rarely detected by the immune system because of the low level or absence of MHC molecules. Inoculation as low as a thousand B16F10 cells is sufficient to form tumor in C57BL/6 mice (33). To investigate if the memory CD8 T cells activated by lv-vv prime-boost can protect mice from tumor cell challenge, we first examined whether the memory CD8 T cells can sense the inoculation of B16F10 tumor cells and be re-activated. Approximately 2.5 months after vv boost and prior to tumor cell challenge, the lv-vv prime-boosted mice maintained a significantly higher IFNγ+ CD8 T cells compared to other mice (Fig.4A). This data suggests that memory CD8 T cells can rapidly respond to ex vivo Ag stimulation to execute effector function. But, the most significant finding was that the memory CD8 T cells in the lv-vv prime-boosted mice could sense the B16 tumor cell challenge and immediately expanded to a significantly higher level (Fig.4B). The hgp10025-33 specific CD8 T cells in lv-vv prime-boosted mice were increased by 2-3 folds after B16 tumor cell challenge. In contrast, there was no measurable increase of CD8 T cells from the lv primed, vv primed, or the vv-lv prime-boosted mice in responses to the inoculation of B16 tumor cells (Fig.4C). Our data strongly suggest that the hgp100 specific memory CD8 T cells generated from lv-vv prime-boost may be qualitatively different from those generated from one immunization. The memory CD8 T cells generated by lv-vv prime-boost are better prepared and ready to be re-activated when cognate Ag is sensed.
Fig.4. Memory CD8 T cells from lv-vv prime-boosted mice immediately respond to tumor cell challenge.

A–C: Two and half months after boost, CD8 memory responses in the lv, vv, lv-vv, or vv-lv immunized C57BL/6 mice were examined by ICS of IFNγ following 3hr ex vivo hgp100 peptide re-stimulation in the peripheral blood of mice pre (A) and post (B) B16 tumor cell challenge. One representative of 5 mice was shown. A summary of CD8 memory responses before and after tumor challenge was shown (mean ± SEM) in C. The experiment was repeated twice with similar data. Unpaired t-test was used for analysis. D. The mice were monitored for tumor growth for 2 months after B16 tumor challenge. Prevention of B16 tumor challenge was significantly more effective in the lv-vv immunized mice. Logrank test was used for statistical analysis.
The higher magnitude of memory CD8 T cells and their rapid responsiveness to tumor cell challenge in the lv-vv prime-boosted mice were translated into better protection. Eighty percent of the mice from lv-vv prime-boosted group were protected (Fig.4D). Only a small fraction of vv-lv prime-boosted mice were protected from B16 tumor challenge. On the other hand, although lv or vv alone immunization could delay tumor growth, but all mice eventually succumbed to tumor growth.
Both CD8 and CD4 effector T cells are required for cancer immune prevention
To study the role of CD8 and CD4 effector T cells in cancer immune prevention, CD8, CD4, or both were depleted 2 weeks after lv immunization at the peak of immune responses. Depletion of CD8 or both CD4 and CD8 effector T cells completely deprived of the immune protection of mice (Fig.5). On the other hand, mice with CD4 depletion retained some of the immune protection effect. These data suggest that both CD4 and CD8 effector cells are required for immune prevention even though CD8 T cells may be the dominant effector cells.
Fig.5. Both CD4 and CD8 effector T cells are required for immune protection of mice from tumor cell challenge.
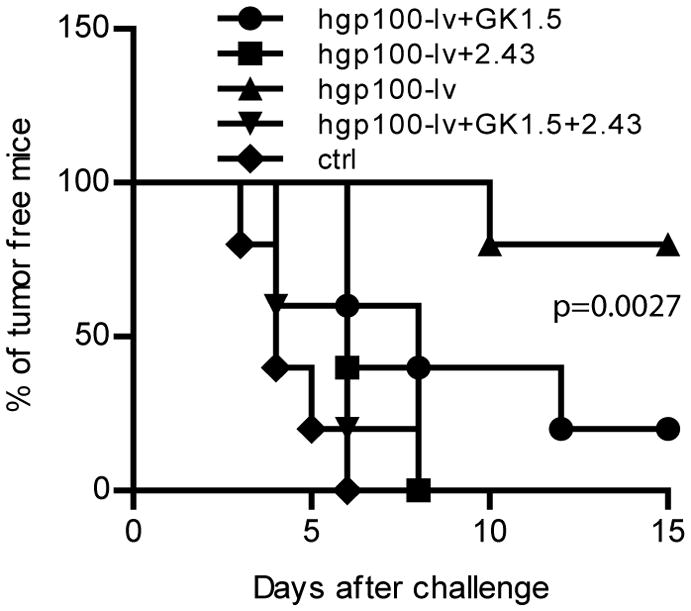
Two weeks after lv immunization, CD4, CD8, or both were depleted with monoclonal antibodies in vivo. Two days later, mice were challenged with B16F10 tumor cells. Ctrl group indicated mice without immunization, while hgp100-lv represented the hgp100-lv immunized mice without depletion. A cohort data from two experiments (10 mice in each group) was presented. Logrank test was used for analysis.
CD4 help is required for both primary and secondary CD8 immune response
In addition to the direct role at effector phase to prevent tumor cell challenge (Fig.5), CD4 cell has been widely recognized for helping CD8 responses (34). To study the role of CD4 help in the induction of CD8 response by lv-vv prime-boost strategy, we immunized the mice that were pretreated with GK1.5 or isotype control Ab as depicted in Fig.6A. Following GK1.5 administration, CD4 T cells were completely deleted two days later and were undetectable for 3 weeks in the peripheral blood (data not shown). We found that in the absence of CD4 T cells, the primary CD8 response by lv immunization were markedly reduced (Fig.6B), suggesting that the induction of primary CD8 response by lv immunization is CD4 dependent.
Fig.6. CD4 help is required for generating both primary and secondary CD8 responses.

A. The experimental strategy was shown. Three experimental groups of C57BL/6 mice were included: Non-depl (no CD4 depletion), Pre-prime (CD4 depletion prior to hgp100-lv prime), and Pre-boost (CD4 depletion prior to hgp100-vv boost). Isotype Ab was rat IgG2b. Immune responses were monitored by collecting peripheral blood and ICS staining of IFNγ at the time points indicated in D. B. CD4 help was required for primary CD8 response. Mice were immunized with hgp100-lv with or without prior CD4 depletion. CD8 response was examined 2 weeks later by ICS of IFNγ after 3hr ex vivo stimulation with hgp100 peptide. Summary of data from two experiments (8 mice) was shown on the right. C. Five days after vv boost, the secondary CD8 response was examined by ICS of IFNγ after 3hr ex vivo stimulation with hgp100 peptide. A summary of 4 mice was shown on the right. CD4 help at both prime and at boost stage was needed for generating secondary CD8 responses. D. The kinetics of CD8 responses also demonstrated that CD4 depletion prior to lv prime or vv boost could markedly reduce the secondary CD8 response. E. Forty-five days after vv boosting immunization, mice were challenged with B16 tumor cells. CD4 depletion abolished the cancer immune prevention effect of lv-vv prime-boost. Two experiments were repeated with similar data. Unpaired t-test and Logrank test were used for analysis.
To examine the CD4 help on the secondary CD8 immune responses, CD4 T cells were depleted either prior to lv prime or prior to vv boost. We found that the secondary CD8 responses boosted by vv were severely damaged, especially when CD4 depletion happened prior to lv prime (Fig. 6C). This conclusion was supported by the kinetics of the primary and secondary CD8 responses (Fig.6D). In addition, in the absence of CD4, the secondary CD8 response was sharply lower 5 weeks after vv boost, suggesting that the magnitude of CD8 memory T cells was also dependent on CD4 help (Fig.6D). Not surprisingly, CD4 depletion, irrespective of time, severely compromised the cancer immune prevention effect. All the mice with CD4 depletion succumbed to tumor growth (Fig.6E). These data suggest that CD4 help is not only important for primary CD8 responses, but also critical for generating potent secondary effector and memory CD8 responses to protect mice from tumor cell challenge.
To further examine the effect of CD4 help on the magnitude of secondary CD8 responses and on the formation of memory CD8 T cells, we also analyzed the hgp10025–33 tetramer specific CD8 T cells and the memory phenotype of CD127 after vv boost immunization with or without prior CD4 depletion. The data showed that, in agreement with the result of cytokine staining presented in Fig.6C, the magnitude of secondary CD8 responses was markedly reduced in the absence of CD4 help, especially when the CD4 depletion happened prior to hgp100-lv prime (Fig. 7A). These data suggest that CD4 help at both prime and boost stage is required to generate effective secondary CD8 responses. However, although the magnitude of secondary CD8 responses was markedly reduced without CD4 help, the ratio of CD127+ memory CD8 T cells was not markedly changed when the CD4 depletion occurred before prime (Fig.7B). In contrast, CD4 depletion prior to vv boost also significantly reduced the ratio of CD127+ memory CD8 T cells (Fig.7B). It is not clear why CD4 depletion prior to vv boost reduces the memory CD8 T cell ratio. In summary, CD4 help at both prime and boost stages is important for the magnitude of primary and secondary CD8 effector and memory responses.
Fig.7. CD4 help is required for secondary CD8 responses.

The experimental strategy was the same as Fig.6. CD4 depletion was conducted two days prior to lv prime or vv boost. Two weeks after vv boost, the hgp100 specific CD8 T cells were analyzed by tetramer (A) and CD127 (B) staining. CD4 depletion markedly reduced the secondary hgp100-Db tetramer positive CD8 T cells in the blood. In addition, CD4 depletion prior to hgp100-vv boost also reduced the ratio of CD127+ T cells. The summary of data from 4 mice was shown. Statistical analysis was done with unpaired t-test.
Lv-vv prime-boost induces potent CD8 responses in the melanoma prone Grm1 Tg mice and prevents autochthonous melanoma growth
The ultimate goal of immune prevention is to prevent spontaneous tumor growth in high risk population. To examine if the lv-vv prime-boost immunization strategy can prevent tumor growth in autochthonous tumor model, we utilized the Grm1 Tg mice. These mice contain transgene Grm1 under the control of tyrosinase related protein 2 promoter, which allows transformation of melanocytes. Pigmentation begins to emerge at 3 months old and eventually all mice will develop melanoma even though malignant metastasis is rare (27). Another advantage of using the Tg mice is that melanoma appearance can be easily visible on the tail and ear. The Grm1 Tg mice were immunized with hgp100-lv at age of 1.5 or 3 months old with or without boost with hgp100-vv one month later. Lv immunization stimulated potent CD8 responses in the immunized mice. Vv boost increased the secondary and memory CD8 T cell responses in Grm1 Tg mice (Fig.8A). However, compared to the CD8 responses in C57BL/6 mice, the primary CD8 responses elicited by hgp100-lv in Grm1 Tg mice is higher. On the other hand, the boosting effect by vv in Grm1 Tg mice is not as impressive as those in C57BL/6 mice. At present, the mechanisms underpinning these differences are not clear. But nevertheless, approximately 90% of the mice immunized by lv-vv prime-boost at early age could be fully protected from melanoma for at least 12 months. Lv immunization alone can protect up to 50% of the mice from melanoma growth (Fig.8B). However, if preventive immunization started at 3 months old when skin pigmentation was developing, the protection rate declined (Fig.8C), a phenomenon was more obvious in the lv alone group. The reduced prevention effect in elder mice was also observed previously (35–36), and is consistent with the notion that immune prevention effect of cancer vaccines decline when tumors begin to grow. While the lv-vv prime-boosted mice were protected from melanoma growth, a mild hair depigmentation (patched vitiligo) was visible in the tumor free mice. But the overall condition of the immunized mice was very healthy. No weight loss and no wasting signs were observed. These data suggest that effective tumor prevention can be induced by lv-vv prime-boost immunization strategy in cancer prone mice without stimulating severe autoimmune diseases.
Fig.8. Lv-vv prime-boost prevents tumor growth in melanoma-prone Tg mice.

Melanoma prone Grm1 Tg mice were immunized at age of 1.5 months old (B) or 3 months old (C) with or without vv boost a month later. The immune responses were monitored by examining the IFNγ+ CD8 T cells in the peripheral blood (A). Tumor appearance (heavy pigmentation and small tumor nodules) was monitored. A cohort of 10 mice in each group from 3 experiments was presented. Pictures were taken at age of 12 months (B: immunization started at 1.5 months old) or 6 months (C: immunization started at 3 months old). Statistical analysis was done with either ANOVA or Logrank test.
Discussion
In the current study, we found that lv expressing xenogenic hgp100 melanoma Ag could induce potent CD8 T cell responses that can cross react with mgp100 Ag. A significant portion of the gp100 specific CD8 T cells expressed the memory marker of CD127 at the peak of immune responses, suggesting that they were CD8 memory precursor cells. The memory CD8 T cells were markedly increased by vv boost. Importantly, CD8 memory T cells from the lv-vv prime-boosted mice could be rapidly reactivated and expanded after sensing tumor cells, which was required for prevention of B16 tumor cell challenge and for prevention of autochthonous melanoma in melanoma-prone Tg mice. The cancer prevention effect is mediated by both CD4 and CD8 T cells. In addition, both the lv elicited primary and vv boosted secondary gp100 specific CD8 responses were CD4 dependent. Depletion of CD4 prior to lv prime or vv boost markedly reduced the primary and secondary CD8 effector responses and the memory CD8 T cells and compromised the cancer prevention effect of lv-vv prime-boost immunization. It was also found that immunization at early stage could protect mice better than immunization at later time from development of autochthonous melanoma. The cancer prevention is long-lasting. Lv-vv prime-boost immunization at 1.5 months old could prevent Tg mice from autochthonous melanoma for at least 12 months.
Like vaccines for infectious diseases, the main purpose of cancer vaccines is to activate and expand the usually rare tumor Ag specific immune cells to a higher level and equip them with memory characteristics so that they can be rapidly reactivated to execute their effector function once they sense the tumor cells. Compared to peptide vaccines, lv immunization was shown to induce higher number of memory CD8 T cells (22). Using CD127 staining, we not only confirmed this finding, but also found that the CD8 memory cells could be markedly increased by vv boost. It is not known why lv immunization can stimulate high level of CD8 memory cells. One explanation can be the immunization associated inflammation. It was reported that the formation of memory CD8 T cells was negatively affected by the presence of inflammation during the priming phase (37–38). Lv injection induces no obvious inflammation, thus can facilitate generation of memory CD8 T cells at early stage. Another reason for the high level of CD127+ memory precursor cells after lv immunization was speculated that the CD4 helper epitopes in the lv particles may promote development of CD127+ cells (22) because CD4 T cell help was demonstrated to be required for the emergence of CD127+ CD8 T cells after acute lymphocytic choriomeningitis virus infection (39).
Although it is widely believed that CD4 help may be dispensable for primary CD8 responses induced by viral infections (40–42), exceptions do exist (43). Heath and colleagues demonstrated that in both herpes simplex virus infection model and OVA coated splenocyte immunization model, the primary CD8 response was markedly reduced in the absence of CD4 help (43). Another study also reported that the magnitude of primary CD8 effector responses against H-Y Ag reduces by 2–3 folds in the absence of CD4 help (44). In the current study, we found that CD4 help was needed for inducing effective primary CD8 T cell responses. CD4 depletion prior to lv immunization markedly reduced the magnitude of primary CD8 responses (Fig.6B). Regarding to the issue whether the secondary CD8 response requires CD4 help, Shedlock et al demonstrated that memory CD8 T cells formed in the presence of CD4 could effectively respond to boost immunization regardless the presence of CD4 during the Ag boost (41). However, our data demonstrated that the secondary CD8 response was also dependent on CD4 help. CD4 depletion prior to vv boost substantially reduced the magnitude of CD8 responses even if the primary CD8 effector and memory responses were primed by lv in the presence of CD4 help (Fig. 6C and 6D). Apparently, in our system, the CD8 memory T cells generated in the presence of CD4 help from the primary immunization were not responding well without CD4 help during the secondary stimulation, suggesting the secondary CD8 response by boost immunization was also CD4 dependent. The reasons for such divergence on CD4 dependence may include the nature of Ag, the inflammation status during priming, and the DC subsets involved.
In our opinion, the stringent and appropriate criteria to measure the quality and usefulness of memory CD8 T cells are to determine their responsiveness to tumor cell challenge. On this aspect, we found that only the CD8 memory T cells from the lv-vv prime-boosted mice were able to sense and respond to the B16 tumor cell challenge (Fig. 4B and 4C), resulting in effective cancer prevention in both transplantable B16 tumor and autochthonous melanoma models. It remains to be studied why only the memory CD8 T cells from lv-vv prime-boosted mice are able to recognize and respond to B16 tumor cell challenge. It was reported that the MHC I and II molecules on B16 tumor cells are not detectable in vitro and remain low in vivo (33), which allows B16 tumor cells easily evade the surveillance of immune system. It is possible that memory CD8 T cells generated from multiple stimulations (such as the prime-boost) are qualitatively different from that generated by one immunization. The CD8 memory T cells from lv-vv prime-boosted mice may be more prepared and ready to respond, which allows them to recognize the B16 tumor cells and expand even when the MHC molecules are low (45). An alternative explanation is that the absolute number of gp100 specific CD8 T cells in the lv-vv prime-boosted mice is higher and contributes to more IFNγ production upon encountering the tumor cells, which then increases the MHC expression to a higher level, making the B16 tumor cells become visible by the memory T cells. A third explanation is that the high number of effector memory CD8 T cells can immediately execute their effector function including tumor lyses, which release sufficient tumor Ag to be cross-presented by APCs to further expand the memory T cells and to generate Ag spreading to prevent tumor growth. All these explanations suggest that for an effective cancer immune prevention, a high level and qualitatively fit memory CD8 T cells may be required in order for them to recognize, respond, and kill the tumor cells to prevent tumor establishment.
The perspective clinical applications of cancer immune prevention depend on accurate evaluation of risk-benefit ratio and the safety of both the immunization vector and the vaccine induced immune responses. With technology development such as gene profiling and genomic analysis, the risk of developing certain malignancy can be better measured and the risk-benefit can be more accurately estimated. Under such conditions, high risk population will be more likely to accept the procedure of cancer immune prevention if the risks associated with immune prevention can be minimized. Recombinant lv has been demonstrated to be less likely to induce tumorigenesis compared to conventional oncoretroviral vector in mice (46). The data from recent clinical trials using lv modified cells (including stem cells) demonstrated no severe side effects (47–49). In addition, lv immunization with the purpose of targeting non-stem cells (such as skin DCs and epidermal cells) should be safer. Thus, based on safety profile of lv and vv and the potency of lv induced Ag specific immune responses against desired tumor Ag, we reason that lv-vv prime-boost immunization strategy has a great potential for cancer immune prevention in human.
Acknowledgments
Research in this study was supported by NIH grant R01 CA16444 and Distinguished Investigator Fund from Georgia Research Alliance to YH.
We would like to thank the NIH tetramer core facility at Emory University for synthesizing the hgp10025–33/Db tetramer and Dr. Suzie Chen of Rutgers University for kindly providing the melanoma prone Grm1 Tg mice. The authors also greatly appreciate the stimulating discussion on this project with Drs. Andrew Mellor and David Munn, Immunotherapy Center, Georgia Health Sciences University.
Abbreviations used in the paper
- lv
lentivector
- vv
vaccinia virus vector
- gp100
glycoprotein 100
- Tg
transgenic
- ICS
intracellular staining
- Grm1
Metabotropic glutamate receptor 1
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, Chandler P, Munn DH, Mellor A, Fu N, He Y. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol. 2010;185:5082–5092. doi: 10.4049/jimmunol.1001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13:5256–5261. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]
- 4.Disis ML. The ultimate in cancer chemoprevention: cancer vaccines. Cancer Prev Res (Phila) 2010;3:406–409. doi: 10.1158/1940-6207.CAPR-10-0043. [DOI] [PubMed] [Google Scholar]
- 5.Finn OJ, Forni G. Prophylactic cancer vaccines. Curr Opin Immunol. 2002;14:172–177. doi: 10.1016/s0952-7915(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 6.Forni G, Lollini PL, Musiani P, Colombo MP. Immunoprevention of cancer: is the time ripe? Cancer Res. 2000;60:2571–2575. [PubMed] [Google Scholar]
- 7.Weiner LM, Surana R, Murray J. Vaccine prevention of cancer: can endogenous antigens be targeted? Cancer Prev Res (Phila) 2010;3:410–415. doi: 10.1158/1940-6207.CAPR-10-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beatty PL, Narayanan S, Gariepy J, Ranganathan S, Finn OJ. Vaccine against MUC1 antigen expressed in inflammatory bowel disease and cancer lessens colonic inflammation and prevents progression to colitis-associated colon cancer. Cancer Prev Res (Phila) 2010;3:438–446. doi: 10.1158/1940-6207.CAPR-09-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elsawa SF, Rodeberg DA, Celis E. T-cell epitope peptide vaccines. Expert review of vaccines. 2004;3:563–575. doi: 10.1586/14760584.3.5.563. [DOI] [PubMed] [Google Scholar]
- 10.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Anderson RJ, Schneider J. Plasmid DNA and viral vector-based vaccines for the treatment of cancer. Vaccine. 2007;25(Suppl 2):B24–34. doi: 10.1016/j.vaccine.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 12.Harrop R, John J, Carroll MW. Recombinant viral vectors: cancer vaccines. Advanced drug delivery reviews. 2006;58:931–947. doi: 10.1016/j.addr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Pilon SA, Piechocki MP, Wei WZ. Vaccination with cytoplasmic ErbB-2 DNA protects mice from mammary tumor growth without anti-ErbB-2 antibody. J Immunol. 2001;167:3201–3206. doi: 10.4049/jimmunol.167.6.3201. [DOI] [PubMed] [Google Scholar]
- 14.Vollmer CM, Jr, Eilber FC, Butterfield LH, Ribas A, Dissette VB, Koh A, Montejo LD, Lee MC, Andrews KJ, McBride WH, Glaspy JA, Economou JS. Alpha-fetoprotein-specific genetic immunotherapy for hepatocellular carcinoma. Cancer Res. 1999;59:3064–3067. [PubMed] [Google Scholar]
- 15.Rovero S, Amici A, Di Carlo E, Bei R, Nanni P, Quaglino E, Porcedda P, Boggio K, Smorlesi A, Lollini PL, Landuzzi L, Colombo MP, Giovarelli M, Musiani P, Forni G. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–5142. doi: 10.4049/jimmunol.165.9.5133. [DOI] [PubMed] [Google Scholar]
- 16.Reilly RT, Machiels JP, Emens LA, Ercolini AM, Okoye FI, Lei RY, Weintraub D, Jaffee EM. The collaboration of both humoral and cellular HER-2/neu-targeted immune responses is required for the complete eradication of HER-2/neu-expressing tumors. Cancer Res. 2001;61:880–883. [PubMed] [Google Scholar]
- 17.Liu Y, Peng Y, Mi M, Guevara-Patino J, Munn DH, Fu N, He Y. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J Immunol. 2009;182:5960–5969. doi: 10.4049/jimmunol.0900008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esslinger C, Chapatte L, Finke D, Miconnet I, Guillaume P, Levy F, MacDonald HR. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest. 2003;111:1673–1681. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Y, Zhang J, Donahue C, Falo LD., Jr Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24:643–656. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y, Zhang J, Mi Z, Robbins P, Falo LD., Jr Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol. 2005;174:3808–3817. doi: 10.4049/jimmunol.174.6.3808. [DOI] [PubMed] [Google Scholar]
- 21.Furmanov K, Elnekave M, Lehmann D, Clausen BE, Kotton DN, Hovav AH. The role of skin-derived dendritic cells in CD8+ T cell priming following immunization with lentivectors. J Immunol. 2010;184:4889–4897. doi: 10.4049/jimmunol.0903062. [DOI] [PubMed] [Google Scholar]
- 22.Chapatte L, Colombetti S, Cerottini JC, Levy F. Efficient induction of tumor antigen-specific CD8+ memory T cells by recombinant lentivectors. Cancer Res. 2006;66:1155–1160. doi: 10.1158/0008-5472.CAN-05-2597. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Ouyang K, Fang J, Zhang H, Wu G, Ma Y, Zhang Y, Hu X, Jin L, Cao R, Fan H, Li T, Liu J. Improved efficacy of DNA vaccination against prostate carcinoma by boosting with recombinant protein vaccine and by introduction of a novel adjuvant epitope. Vaccine. 2009;27:5411–5418. doi: 10.1016/j.vaccine.2009.06.089. [DOI] [PubMed] [Google Scholar]
- 24.Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 25.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, Vuola JM, Blanchard TJ, Gothard P, Watkins K, Hannan CM, Everaere S, Brown K, Kester KE, Cummings J, Williams J, Heppner DG, Pathan A, Flanagan K, Arulanantham N, Roberts MT, Roy M, Smith GL, Schneider J, Peto T, Sinden RE, Gilbert SC, Hill AV. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–735. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 26.Palmowski MJ, Lopes L, Ikeda Y, Salio M, Cerundolo V, Collins MK. Intravenous injection of a lentiviral vector encoding NY-ESO-1 induces an effective CTL response. J Immunol. 2004;172:1582–1587. doi: 10.4049/jimmunol.172.3.1582. [DOI] [PubMed] [Google Scholar]
- 27.Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, Marin Y, Roberts KG, Yudt LM, Chen A, Cheng J, Incao A, Pinkett HW, Graham CL, Dunn K, Crespo-Carbone SM, Mackason KR, Ryan KB, Sinsimer D, Goydos J, Reuhl KR, Eckhaus M, Meltzer PS, Pavan WJ, Trent JM, Chen S. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 28.Guo ZS, Naik A, O’Malley ME, Popovic P, Demarco R, Hu Y, Yin X, Yang S, Zeh HJ, Moss B, Lotze MT, Bartlett DL. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–9998. doi: 10.1158/0008-5472.CAN-05-1630. [DOI] [PubMed] [Google Scholar]
- 29.Gold JS, Ferrone CR, Guevara-Patino JA, Hawkins WG, Dyall R, Engelhorn ME, Wolchok JD, Lewis JJ, Houghton AN. A single heteroclitic epitope determines cancer immunity after xenogeneic DNA immunization against a tumor differentiation antigen. J Immunol. 2003;170:5188–5194. doi: 10.4049/jimmunol.170.10.5188. [DOI] [PubMed] [Google Scholar]
- 30.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 31.Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, Busch DH. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci U S A. 2004;101:5610–5615. doi: 10.1073/pnas.0308054101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bohm W, Thoma S, Leithauser F, Moller P, Schirmbeck R, Reimann J. T cell-mediated, IFN-gamma-facilitated rejection of murine B16 melanomas. J Immunol. 1998;161:897–908. [PubMed] [Google Scholar]
- 34.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 35.Pannellini T, Spadaro M, Di Carlo E, Ambrosino E, Iezzi M, Amici A, Lollini PL, Forni G, Cavallo F, Musiani P. Timely DNA vaccine combined with systemic IL-12 prevents parotid carcinomas before a dominant-negative p53 makes their growth independent of HER-2/neu expression. J Immunol. 2006;176:7695–7703. doi: 10.4049/jimmunol.176.12.7695. [DOI] [PubMed] [Google Scholar]
- 36.Pupa SM, Invernizzi AM, Forti S, Di Carlo E, Musiani P, Nanni P, Lollini PL, Meazza R, Ferrini S, Menard S. Prevention of spontaneous neu-expressing mammary tumor development in mice transgenic for rat proto-neu by DNA vaccination. Gene Ther. 2001;8:75–79. doi: 10.1038/sj.gt.3301360. [DOI] [PubMed] [Google Scholar]
- 37.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. 2005;11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 38.Pham NL, V, Badovinac P, Harty JT. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol. 2009;183:2337–2348. doi: 10.4049/jimmunol.0901203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, Shang L, Goepfert PA, Zajac AJ. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol. 2005;174:5926–5930. doi: 10.4049/jimmunol.174.10.5926. [DOI] [PubMed] [Google Scholar]
- 40.Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol. 2004;16:217–225. doi: 10.1016/j.coi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 42.Bourgeois C, Tanchot C. Mini-review CD4 T cells are required for CD8 T cell memory generation. Eur J Immunol. 2003;33:3225–3231. doi: 10.1002/eji.200324576. [DOI] [PubMed] [Google Scholar]
- 43.Behrens G, Li M, Smith CM, Belz GT, Mintern J, Carbone FR, Heath WR. Helper T cells, dendritic cells and CTL Immunity. Immunol Cell Biol. 2004;82:84–90. doi: 10.1111/j.1440-1711.2004.01211.x. [DOI] [PubMed] [Google Scholar]
- 44.Bourgeois C, Veiga-Fernandes H, Joret AM, Rocha B, Tanchot C. CD8 lethargy in the absence of CD4 help. Eur J Immunol. 2002;32:2199–2207. doi: 10.1002/1521-4141(200208)32:8<2199::AID-IMMU2199>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 45.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 46.Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, Doglioni C, von Kalle C, Naldini L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 47.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, Binder GK, Slepushkin V, Lemiale F, Mascola JR, Bushman FD, Dropulic B, June CH. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 49.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R, Maouche-Chretien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie G, Chretien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]