

Abstract
Using a computational model, we simulated mitochondrial deoxynucleotide metabolism and mitochondrial DNA replication. Our results indicate that the output from the mitochondrial salvage enzymes alone is inadequate to support a mitochondrial DNA replication duration of as long as 10 hours. We find that an external source of deoxyribonucleoside diphosphates or triphosphates (dNTPs), in addition to those supplied by mitochondrial salvage, is essential for the replication of mitochondrial DNA to complete in the experimentally observed duration of approximately 1 to 2 hours. For meeting a relatively fast replication target of 2 hours, almost two-thirds of the dNTP requirements had to be externally supplied as either deoxyribonucleoside di- or triphosphates, at about equal rates for all four dNTPs. Added monophosphates did not suffice. However, for a replication target of 10 hours, mitochondrial salvage was able to provide for most, but not all, of the total substrate requirements. Still, additional dGTPs and dATPs had to be supplied. Our analysis of the enzyme kinetics also revealed that the majority of enzymes of this pathway prefer substrates that are not precursors (canonical deoxyribonucleosides and deoxyribonucleotides) for mitochondrial DNA replication, such as phosphorylated ribonucleotides, instead of the corresponding deoxyribonucleotides. The kinetic constants for reactions between mitochondrial salvage enzymes and deoxyribonucleotide substrates are physiologically unreasonable for achieving efficient catalysis with the expected in situ concentrations of deoxyribonucleotides.
Author Summary
The powerhouses of human cells, mitochondria, contain DNA that is distinct from the primary genome, the DNA in the nucleus of cells. The mitochondrial genome needs to be replicated often to ensure continued generation of ATP (adenosine triphosphate) which is the energy currency of the cell. Problems with maintenance of mitochondrial DNA, arising from genetic mutations as well as from antiviral drugs, can lead to debilitating diseases that are often fatal in early life and childhood, or reduced compliance to therapy from patients suffering drug toxicity. It is therefore important to understand the processes that contribute to the upkeep of mitochondrial DNA. The activities of a set of enzymes, which together generate the chemical building blocks of mitochondrial DNA, are important in this regard. We used computational methods to analyze the properties of these enzymes. Results from our approach of treating these enzymes as a system rather than studying them one at a time suggest that in most conditions, the activities of the enzymes are not sufficient for completing replication of mitochondrial DNA in the observed duration of around 2 hours. We propose that a source of building blocks in addition to this set of enzymes appears to be essential.
Introduction
Mitochondrial DNA (mtDNA) replication [1], and the mitochondrial nucleoside salvage pathway that generates the precursor deoxyribonucleoside triphosphates (dNTPs) for mitochondrial DNA replication, have generally been believed to function independently of nuclear DNA (nDNA) replication and cytoplasmic nucleotide metabolism. However, the observation associating mutated RRM2B (a p53 inducible ribonucleotide reductase subunit) with mtDNA depletion and at least one observation of mtDNA replication restricted to S phase in DGUOK (deoxyguanosine kinase) deficient cells now make it clear that mtDNA replication and maintenance are not always completely independent of the cytoplasmic state [2], [3]. Older evidence supported the view that mitochondrial nucleotides may be isolated from the corresponding cytoplasmic pools [4], but more recent studies support a metabolic cross-talk between the mitochondria and the cytoplasm and show that nucleotide import from the cytosol very likely contributes to mitochondrial dNTP pools in both cycling and quiescent cells [5], [6]. The mechanism of this import is unknown since the discovery that the carrier SLC25A19 (solute carrier family 25, member 19) actually is a thiamine pyrophosphate transporter and not a deoxyribonucleotide transporter [7]. Similarly, the mitochondrial monophosphate kinases of dG and T deoxyribonucleotides (key elements of the purported salvage pathway) still have not been identified. In the current picture of the mitochondrial nucleoside salvage pathway, DGUOK and TK2 (thymidine kinase 2) are the nucleoside kinases; NT5M (mitochondrial 5′,3′-nucleotidase) is a nucleotidase; CMPK2 (cytidine monophosphate kinase 2), and isoforms of adenylate kinase (AK) are the monophosphate kinases; and NME4 is the major nucleoside diphosphate kinase (Figure 1A). Deoxyribonucleosides (dNs) are converted to dNTPs through three sequential enzyme-catalyzed phosphorylations. This is a complex process with some reactions occurring in parallel for the four deoxyribonucleosides, and some reactions using the same enzyme (for example, the first phosphorylation of dT and dC are both catalyzed by TK2) in addition to the presence (not shown in Figure 1A) of feedback mechanisms (for example, dTTP and dCTP inhibition on TK2 [8]).
Figure 1. Mitochondrial deoxyribonucleoside salvage metabolism.

(A) Biochemical pathway representation of the mitochondrial deoxyribonucleoside salvage metabolism. Deoxyribonucleosides undergo a series of reversible phosphorylations to become deoxyribonucleoside triphosphates (substrates for mtDNA replication). Enzymes that are yet to be identified are represented by question marks. (B) Subcellular localizations of some enzymes important for the production of intra-mitochondrial dNTPs. Arrows denote the flow of substrates between the enzymes. The question marks denote substrate flows that appear to be required by the enzyme localization data, but which seem unreasonable.
The physical structure of the mitochondrion provides another complication that is rarely considered in this context. The mitochondrion has an intermembrane space (between the inner and outer membranes) and a matrix compartment within the inner membrane (Figure 1B). Several contact sites exist between the inner and outer membranes. In addition to the mitochondrial enzymes listed in Figure 1A, the cytoplasmic enzymes Thymidine Phosphorylase (TYMP) and RRM2B are included in the diagram since mutations in these two enzymes are known to cause phenotypes involving defects in mtDNA maintenance [2], [9]. The mtDNA are tethered to the inside of the inner membrane, within the matrix, so it would be expected that the enzymes of the salvage pathway would also be located within the matrix. In the simplest picture of the mitochondrial salvage pathway deoxyribonucleosides are transported through the inner membrane by the ENT (equilibrative nucleoside transporter) and then phosphorylated to dNTPs within the matrix. However, evidence exists to suggest that the AK2 adenylate kinase as well as NME4 nucleoside diphosphate kinase might actually be localized to the mitochondrial intermembrane space [10], [11], not in the matrix. It is possible that other isoforms of these enzymes might localize to the mitochondrial matrix [10], [11]. If not, it is hard to understand how the salvage pathway would function without an unnecessarily complicated transport of deoxyribonucleotides back and forth across the inner membrane (arrows marked with question marks in Figure 1B).
In this paper we analyze the experimentally measured enzyme kinetics of these known enzymes of this pathway. Our analysis of the mitochondrial nucleotide metabolism pathway reveals that the majority of the enzymes of this pathway are not particularly effective in the synthesis of mtDNA precursors (phosphorylated deoxyribonucleosides) either due to the affinities of the enzymes for ribonucleotides and other non-DNA precursors (dI and dUMP for example) or due to a disparity in their affinities for deoxyribonucleotides versus the expected mitochondrial concentrations of those deoxyribonucleotide substrates. Computational simulations of the function of this pathway support our analysis and indicate that a source of deoxyribonucleotides in addition to those provided by mitochondrial salvage is essential to account for the experimentally observed mtDNA replication duration of 1 to 2 hours in cycling cells.
Methods
As far as possible, we restricted our analysis to data from human enzymes. Exceptions are noted below. We assumed Michaelis-Menten kinetics for all enzymes except TK2 which has negatively cooperative kinetics with Hill coefficient less than 1 with thymidine [8].
kcat/Km
Km values were obtained from the literature [8], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22] or the BRENDA database [23]. For most enzymes, we could only find a single report of kinetic parameters. For the nucleoside kinases DGUOK and TK2, we did find multiple reports of kinetic parameters. In these cases, we selected the reference providing the most comprehensive information. To compute kcat values, we first obtained reported Vmax values [8], [12], [14], [15], [16], [18], [19], [20], [21], [22] and molecular weights [13], [22], [23], [24] of the various enzymes from the literature or the BRENDA database. If the enzyme was reported to be a multimer, we added the molecular weights of the subunits to calculate the molecular weight of the holoenzyme. The quantity kcat/Km (M−1 s−1) was calculated from the reported values of Vmax/Km (with units of µmol min−1 mg−1 µM−1) using the following conversion,
where Wenzyme is the enzyme molecular weight. Reported values for Km, Vmax, and the calculated kcat/Km values are provided in Table S1.
Substrate Concentrations
Values for the concentrations of the deoxyribonucleoside, deoxyribonucleotide, ribonucleoside, and ribonucleotide substrates were used to calculate ‘(substrate) Concentration/(substrate) Km’ ratios. These values were used for a comparison of activities of the enzyme with different substrates and are not meant to be precise. Instead, rough order-of-magnitude concentration values were used to compare values for this ratio, which often varies by several orders of magnitude within a single enzyme for different substrates. Literature reports suggested that mitochondrial dNTP pools are higher in actively cycling cells compared to quiescent cells [25], [26], [27], [28]. We assumed a 10-fold lower concentration of deoxyribonucleotides in quiescent cells, and chose 10 µM and 1 µM as reasonable representative estimates of mitochondrial dNTP concentrations in cycling and quiescent cells respectively. The basis of these estimates are the concentrations calculated from published values in HeLa cells [29] and quiescent fibroblasts [30] respectively. We used a value of 0.82 ml/g mitochondrial protein [31] to calculate concentrations from the measured pool sizes in HeLa cells, and we used the value of 92.3 µm3 for mitochondrial volume per cell [32] to obtain the concentrations from the measured pool size in quiescent fibroblasts. For simplicity, we assumed ribonucleotides and deoxyribonucleotides to be equally concentrated in the three phosphorylation states (mono, di, or tri-phosphorylated). Again for simplicity we assumed all four nucleotides (dAXP, dCXP, dGXP, dTXP where X = phosphorylation state) to have equal concentrations. Nucleoside concentrations were assumed to be equilibrated between plasma, cytoplasm, and mitochondria and set at a constant 0.5 µM using a reported value for plasma concentration [33]. Lower nucleoside concentrations have also recently been reported [34], [35]. We have kept the higher value in our analysis since this is the most conservative choice. Lower nucleoside concentration values would make the problems that we point out in this analysis even more severe. Ribonucleotide concentrations were assumed to be constant and set at 100 µM, that is, one order of magnitude higher in cycling cells and two orders of magnitude higher in quiescent cells compared to deoxyribonucleotide concentrations. This is a fairly conservative (i.e. low) choice for the ribonucleotide concentrations. For other special cases of substrates (such as dUMP, dI, or IMP) concentrations data are not readily available so we again assumed low concentration values for these substrates. The complete list of assumed concentrations is provided in Table S1.
Inhibitions
In the case that we could not find Ki values of for enzyme inhibitors, we assumed competitive inhibition so that the Ki for the inhibitor was set to be equal to the Km for that chemical as a substrate. Inhibition kinetics data [8], [12], [16], [19], [21], [22], [36] are provided in Table S1.
Computational Simulations
Our group has previously published a computational model of mitochondrial deoxyribonucleotide metabolism [25]. Parameter values for the model were based, whenever available, on published experimental values [8], [12], [14], [16], [17], [19], [20], [21], [22], [23], [24], [25], [36], [37], [38], [39], [40], [41], [42], [43]. As part of the present work, we updated the model to reflect the findings since the original model was defined. We refer the readers to the previous publication for a complete explanation of the basic framework of the model [25]. Briefly, enzymatic reactions were modeled with Michaelis-Menten equations (except TK2, which is modeled by the Hill equation) and rates of change of metabolites were modeled using ordinary differential equations. The updates to the model include adding (e.g. CMPK2) and removing (e.g. SLC25A19 or DNC) pathway components and updated kinetics (e.g. inhibition terms and kinetic constants). The model was written in Mathematica 7. The model files are available as supporting information (Text S1). The model constants are also available as supporting information in plain text (Text S2) and PDF (Text S3).
Deoxynucleoside transport was modeled through the ENT protein as equilibrative between the cytoplasm and mitochondria. Thus, the net rate of deoxynucleoside transport was defined using the Michaelis-Menten equation as follows:
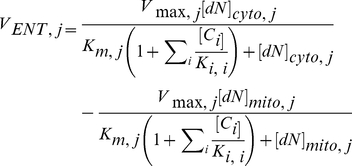 |
where j represents the four deoxynucleoside species (dA, dC, dG, dT) and i represents inhibitors. Vmax and Km were taken to be the same for both directions of transport.
The various enzymatic reactions (i.e., phosphorylations and dephosphorylations) were modeled using the Michaelis-Menten equation. Thus, the reaction velocity was
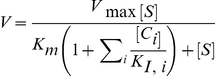 |
where S stands for substrate and [C] stand for the concentration of any competitive inhibitors. For the reaction of dT with TK2, the above equation was modified by raising the Km and [S] terms to the power 0.5 to represent the Hill coefficient.
The model of the mtDNA polymerization process was explained in the previous publication [25]. It models polymerization using fractions of the four deoxynucleotides in the mtDNA sequence, setting the prevalence of each base in the mtDNA light and heavy strands separately to match the prevalence in the rCRS reference sequence [44]. We have modeled mtDNA replication as asynchronous [45] using the locations of the origins of replication of the light strand and the heavy strand.
Differential equations for the concentrations of the various metabolites were defined by adding and subtracting the relevant reaction velocity equations. For example, for dNMPs (deoxyribonucleoside monophosphates), the following differential equation models the rate of change of a particular dNMP:
where NK represents the nucleoside kinase reaction, NT represents the nucleotidase reaction, NMPK represents the forward and reverse monophosphate kinase reactions. The kinetic constants and inhibition parameter values are available in Table S1.
We used this updated model to test the hypothesis that a source of deoxyribonucleotides in addition to intra-mitochondrial salvage is essential for completing mtDNA replication in cycling cells in the experimentally observed time of 1–2 hours [45]. To be conservative, we set the ‘target’ replication time to be 2 hours (requiring an average replication rate = 33136 (nucleotides)/120 (minutes) = ∼276 nucleotides/minute). We ran simulations with a simulation time of 120 minutes (2 hours), with all dynamics including mtDNA replication starting immediately at the beginning of the simulation. We also tested a target replication time of 10 hours (requiring an average replication rate = 33136 (nucleotides)/600 (minutes) = ∼55 nucleotides/minute) – reasoning that in quiescent cells the time constraints for completing mtDNA replication may be more relaxed.
Transport of deoxynucleotides from the cytoplasm to the mitochondrial matrix was modeled in a simple manner, by setting a constant production term of either deoxynucleosides, dNMPs, dNDPs (deoxyribonucleoside diphosphates), or dNTPs. Transport was modeled as occurring at only one phosphorylation level at a time, in order to assess the effectiveness of transport at each level. The essence of our simulation experiments was to test whether mtDNA replication was completed in the target time under varying levels of added molecules, including no addition, of various (A, C, G, T) deoxynucleosides and deoxynucleotides. We note that in principle the additional source of deoxynucleotides in this model does not necessarily have to be import from the cytoplasm, but could also be from other unknown intra-mitochondrial sources. However, considering the evidence that nucleotide transport does occur between the cytoplasm and mitochondria [5], [6], we assume that the additional source we have modeled corresponds to import from the cytoplasm. We tested multiple ‘transport profiles’. A transport profile is composed of simply the rate of the transported deoxynucleosides and deoxynucleotides. For each transport profile, we ran 100 simulations each beginning with a different, randomly selected (with uniform probability) set of initial mitochondrial concentrations of each deoxynucleoside and deoxynucleotide. As an initial test of the level of exogenous precursor transport needed, we set equal rates of import for all four (A, C, G, T) nucleosides (or nucleotides) at a particular phosphorylation level and then let the rate of import vary from 0 to 1200 molecules per minute, in increments of 100. Thus, for example, for testing whether transport of deoxynucleosides alone suffices, we ran 13 sets of 100 simulations. In each of those 13 sets, deoxynucleosides alone were imported at equal rates for each of the four nucleosides, in increments of 100 starting from 0 and up to 1200. Such simulation sets of 13 different import levels were conducted similarly for each phosphorylation level of the four deoxynucleoside species.
The initial conditions of the simulations were set randomly with a uniform distribution over a set range. The allowed range (minimum and maximum) of initial deoxynucleoside concentrations was 0.05 µM to 5 µM and the range of initial deoxynucleotide concentrations was 0.1 µM to 10 µM. We set the concentrations of ribonucleosides, ribonucleotides, and non-canonical deoxynucleosides and deoxynucleotides to be proportional to the randomly selected dN and dNXP concentrations (see Table S1 for details), and held these concentrations (which only acted as inhibitors) constant throughout the time course of the simulation. The simulations were repeated 100 times with varying initial conditions.
We extended the transport analysis further by obtaining the minimum number of molecules of each transported dNTP required for mtDNA replication to be completed in 2 hours (representing cycling cells) or 10 hours (representing quiescent cells). For the simulations to determine the minimum transport profiles, we tested whether the replication rate exceeded 55 (‘quiescent cells’, fixed initial concentrations: dNs = 0.5 micromolar and dNXPs = 1 micromolar) or 276 (‘cycling cells’, fixed initial concentrations: dNs = 0.5 micromolar and dNXPs = 10 micromolar) nucleotides per minute. We started at equal import of all four dNTPs at a rate such that replication would be completed in slightly less than the target time (2 hours or 10 hours). Next, we decreased the import of one dNTP at a time to check whether the target replication rate was observed. We continued this relaxation process until we obtained the minimum transport for each individual deoxyribonucleotide species necessary to support the target replication rate.
Results
The kcat/Km Ratio
In Michaelis-Menten kinetics, kcat and Km are the basic parameters of an enzyme-substrate reaction pair. The parameter kcat is the number of substrate molecules catalyzed per enzyme molecule per unit time and Km is the substrate concentration at which the reaction proceeds at half-maximal velocity. High kcat and low Km values imply a fast and efficient reaction, and thus, a high kcat/Km ratio indicate that this substrate is catalytically preferred by the enzyme. We searched the literature [8], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22] and databases [23] and gathered the available data on the reaction kinetics of enzymes of mitochondrial nucleotide salvage. Figure 2A shows a plot of kcat/Km values. Each group of bars is for one enzyme, and within each group the bars are arranged from lowest to highest so that the best substrates lie to the right on each plot. For clarity, the substrates that are DNA precursors (presumed to the ‘proper’ substrates of these enzymes) are in green, and non-DNA precursor substrates are in red. The kcat/Km values cover a very wide range and so are plotted on a logarithmic scale.
Figure 2. kcat/Km values of mitochondrial enzymes.

(A) kcat/Km values of mitochondrial salvage enzymes. Each group of bars is for one enzyme, and within each such group the bars are arranged from lowest to highest so that the best substrates lie to the right on each plot. Substrates that are DNA precursors are in green, and non-DNA precursor substrates are in red. 2′-UMP and 3′-UMP refer to uridine 2′ monophosphate and uridine 3′ monophosphate. (B) kcat/Km values of the mitochondrial DNA polymerase POLG, justifying the use of kcat/Km as a measure of substrate preference.
Figure 2A shows that each of these enzymes has significant reactions with non-DNA precursors. More importantly, except for TK2, none of the mitochondrial enzymes have DNA precursors as their preferred substrates, as seen from the fact that the substrates which lie to the right in each group of bars are non-DNA precursors. Prior work [46] has estimated the theoretical maximum of kcat/Km for an enzyme-substrate pair. This maximum is constrained by the diffusion limit, and was estimated to be ∼108 per M per second [46]. Compared to the diffusion limit, the kcat/Km values for reactions of the mitochondrial salvage pathway with DNA precursors are orders of magnitude lower (range = 888 to 5.63×105 per M per second). In summary, in both absolute and relative terms these enzymes of the mitochondrial salvage pathway (with the possible exception of TK2) do not appear to be optimized for discriminating mtDNA precursor substrates from chemically related non-precursor substrates.
To put the kcat/Km results in Figure 2A in perspective, Figure 2B is a plot of kcat/Km for the various substrates of the mitochondrial DNA polymerase gamma (POLG). In contrast to the enzymes of mitochondrial nucleotide metabolism, Figure 2B shows that, as expected, DNA precursors are preferably discriminated by POLG. This is true both absolutely and relatively. The kcat/Km values for the dNTP substrates approach the diffusion limit of ∼108 per M per s, and the values for dNTP substrates are many orders of magnitude larger than the kcat/Km values of the ribonucleotide substrates. GTP and UTP kinetics data are not shown because the POLG kinetics with these potential substrates have not been measured.
The Effects of Substrate Concentration
While the ratio kcat/Km captures the efficiency of a reaction between an enzyme and a substrate, it does not take into account the expected physiological concentration of the substrate, which may vary by several orders of magnitude between ribonucleotide and deoxyribonucleotide substrates. The ratio of ‘(substrate) Concentration/(substrate) Km’ provides information that is complementary to that revealed in the previous section by the ratio kcat/Km. When the substrate concentration is much smaller compared to the Km, the enzyme is sensitive to substrate concentration and can thus operate at a range of velocities. However, the velocities in this range would be smaller than the maximum possible velocity. Depending on the relation between maximum possible velocity and the required rate of enzymatic output, substrate concentrations smaller than Km can be a detriment. This is the case for the mitochondrial salvage enzymes because mtDNA replication has to satisfy certain time constraints. We searched the literature [8], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22] and databases [23] for Km values of the mitochondrial salvage enzymes for various substrates and their expected in situ concentrations. Figures 3A and 4A show a plot of Concentration/Km values for all of the enzyme-substrate pairs for which we could find data. As before, each group of bars is for one enzyme, and within each such group the bars are arranged from lowest to highest value of the ratio. Preferred substrates would be expected to have higher concentrations relative to the reaction Km and thus would fall to the right in each enzyme. Substrates that are DNA precursors are plotted in green, and non-DNA precursor substrates are in red.
Figure 3. Concentration/Km values in ‘cycling cells’ for mitochondrial enzymes.

Concentration/Km values at higher mitochondrial concentrations (‘cycling cells’) of the deoxyribonucleotide substrates (10 µM). (A) Values for mitochondrial salvage enzymes. All reactions involving DNA precursor substrates have Concentration/Km values less than 1, suggesting that none of these reactions would be expected to be running at even half-maximal velocity. (B) Values for POLG and SLC25A19, justifying the principle of using the ratio Concentration/Km as a measure of substrate preference. The Concentration/Km ratio for dNTP substrates for POLG is about an order of magnitude larger than the ratios for reactions with rNTPs. The Concentration/Km ratios of DNA precursor substrates of SLC25A19 are low.
Figure 4. Concentration/Km values in ‘quiescent cells’ for mitochondrial enzymes.

Concentration/Km values at lower mitochondrial concentrations (‘quiescent cells’) of the deoxyribonucleotide substrates (1 µM) for (A) mitochondrial salvage enzymes and (B) POLG and SLC25A19.
Figure 3A shows Concentration/Km values at higher mitochondrial concentrations (‘cycling cells’) of the deoxyribonucleotide substrates (10 µM). The Concentration/Km values for DNA precursor substrates range from 0.001 to 0.19. Thus, none of the reactions involving DNA precursor metabolism in the mitochondria would be running at maximal reaction velocity. In fact, since all reactions involving DNA precursor substrates have Concentration/Km values less than 1, none of these reactions would be expected to be running at even half-maximal velocity. It is apparent that these enzymes of mitochondrial nucleotide metabolism have significant affinities for non-DNA precursors. In many cases, the enzymes have higher affinities for non-DNA precursors than for the DNA precursors. In the case of the nucleoside kinases TK2 and DGUOK, although they have higher affinities for DNA-precursors, there is less than 10-fold difference from their preference of non-DNA precursors. For some reactions, the expected substrate concentrations are orders of magnitude lower than the reaction Km values (range: 0.001 to 0.19). The same trends exist in the values of Concentration/Km assuming lower mitochondrial concentrations of substrates (Figure 4A). Moreover, comparing Figure 4A (low deoxyribonucleotide concentrations) to Figure 3A (high deoxyribonucleotide concentrations), the disparity between DNA precursors and other substrates is more striking with an order of magnitude decrease in the Concentration/Km ratio of the DNA precursor substrates (range: 0.0007 to 0.19). This was expected as we assumed mitochondrial ribonucleotide concentrations to be constant and independent of high or low mitochondrial deoxyribonucleotide concentrations.
To place these enzyme kinetics values in context Figures 3B and 4B show positive and negative examples justifying the principle of using the ratio Concentration/Km as a measure of substrate preference. The Concentration/Km ratio for dNTP substrates for POLG is about an order of magnitude larger than the ratios for reactions with ribonucleoside triphosphates (rNTPs) (Figure 3B). It is noteworthy that POLG is the only enzyme of the mitochondrial ‘salvage’ (DNA replication) pathway whose DNA precursor substrates have expected concentrations that are larger than the enzyme Km values. For our negative example, we considered SLC25A19 (formerly named the deoxynucleotide carrier (DNC), now identified as the thiamine pyrophosphate carrier) [7] to be a suitable choice. In contrast to POLG, the Concentration/Km ratios of DNA precursor substrates of SLC25A19 are low both in the absolute and the relative sense. Dolce et al [13] published data on the Km and Ki values of substrates (we used Ki as a proxy for Km if Km was not reported) that were tested for transport by the SLC25A19 protein, and it is seen in Figures 3B and 4B that DNA precursor substrates (green bars) are not the preferred substrates of this enzyme. Eventually, it was discovered that the function of SLC25A19 had been misinterpreted [7], [47]. When we compare the concentration/Km plots of the mitochondrial nucleotide metabolism (Figures 3A and 4A) to those of POLG and SLC25A19 (Figures 3B and 4B) we observe that the Concentration/Km values of DNA precursors with the enzymes of mitochondrial nucleotide metabolism are at the same level as the Concentration/Km values of the DNA precursors with SLC25A19, even though these DNA precursors are not the physiological substrates of SLC25A19. We note that SLC25A19 was not used a negative example in Figure 2B because the enzyme kinetics values (kcat) were not available for the relevant deoxyribonucleotide or ribonucleotide substrates.
As a side observation, we are intrigued by the fact that at lower dNTP concentrations the Concentration/Km values for rNTPs are essentially equal to those for dNTPs for polymerization by POLG (Figure 4B). This observation reveals that discrimination by POLG in this case is perhaps almost completely dependent on the corresponding reaction Vmax. As the Vmax (or kcat) of rNTPs with POLG are much lower than those for dNTPs, it is possible that in quiescent cells POLG faces more interference by ribonucleotides, thus obstructing the polymerization of deoxyribonucleotides into the DNA molecule being synthesized and at the same time promoting the incorporation of ribonucleoside triphosphates in the DNA strand. This is consistent with the reported incorporation of ribonucleotides in replicating mtDNA [48].
Substrate Flow through the Salvage Pathway
As an initial analysis of the function of the salvage pathway, we used the Michaelis-Menten equation to calculate reaction rates under assumed substrate concentrations. We ignored the effect of inhibitions. This implies that the reaction rates we calculated (number of substrate molecules catalyzed per enzyme molecule per minute) were the upper-bound of the rates at the estimated concentrations, because inhibitions would act to lower these rates. We call such reaction rates ‘effective velocities’. As we assumed deoxyribonucleoside concentrations to be constant at 0.5 µM and deoxyribonucleotide concentrations to be either 10 µM (high, ‘cycling cells’) or 1 µM (low, ‘quiescent cells’), we obtained two sets of effective velocities for the enzymes of mitochondrial nucleotide metabolism – one approximating the behavior in cycling cells, and one approximating the behavior in quiescent cells.
Figure 5A is a plot of the effective velocities of nucleoside kinases versus NT5M. Remember from Figure 1A that NT5M is the nucleotidase that reverses the action of the nucleoside kinases, so the amount of material fed into the salvage pathway depends in part on the balance between these two groups of enzymes. The substrate dCMP is absent for Figure 5A because no reaction was observed between NT5M and dCMP [19]. Note that the nucleoside concentrations were assumed to be constant, so the high and low concentration rates are only given for NT5M. Because of inhibitions and competing reactions, the deoxyribonucleoside output from NT5M would be much lower than represented here, but it is still instructive to compare objectively the disparity between the forward and reverse reactions at the first phosphorylation level. It is clear that the theoretical maximum velocities (at the assumed concentrations) of NT5M reverse reactions are many-fold higher than the maximum velocities from nucleoside kinases. While the situation is poor for the dG and dT substrates, it is extremely poor for the dA substrate where the reverse reaction has well over an order of magnitude advantage over the forward reaction. Furthermore, NT5M may not be the only nucleotidase in the mitochondria, thus exacerbating this issue [19].
Figure 5. Effective velocities of some of the reactions of mitochondrial nucleotide metabolism.

(A) Effective velocities (number of substrate molecules catalyzed per minute per enzyme molecule) of nucleoside kinase reactions versus NT5M reactions. Theoretical maximum velocities of NT5M reverse reactions are many-fold higher than the maximum velocities from nucleoside kinases. (B) A comparison of the effective velocities for deoxynucleotide substrates for enzymes of the mitochondrial salvage pathway. The horizontal reference line shows the number of nucleotides (approximately 69) of each triphosphate needed per minute on average to complete mtDNA replication in two hours.
In addition to these qualitative comparisons of substrate preferences of mitochondrial nucleotide metabolism enzymes, we analyzed the reaction kinetics further to approximately quantify the flow of substrates through this enzymatic pathway. For simplicity, we ignored the inhibition terms in the Michaelis-Menten equations (inhibitions would further reduce reaction velocities). We could then investigate the effect of kcat, Km, and substrate concentrations on the upper-bound of velocity of the reactions at assumed substrate concentrations and compare the estimated velocities to the expected requirements for completing one round of mtDNA replication in a specified amount of time. It has been reported that one round of mtDNA replication in cell culture takes ∼1–2 hours to complete [45]. To be conservative, we assumed that mtDNA replication takes 2 hours to complete. To replicate 16,568 bases pairs on two mtDNA strands in 2 hours, ∼276 nucleotides are required per minute on average (with the log scale on Figure 5B it is unnecessary to precisely divide this quantity into the specific numbers of dATP, dCTP, dGTP, and dTTP molecules needed for the human mtDNA sequence). Figure 5B shows the effective velocities of some of the enzymes of mitochondrial nucleotide metabolism (DGUOK, TK2, CMPK2 and AK2). These are all the enzymes for which we found data that would enable us to calculate effective velocities. To facilitate comparison across these enzymes, some data for DGUOK and TK2 are repeated in Figure 5B from Figure 5A. As before, nucleoside kinase velocities in Figure 5B are the same for high or low concentration conditions because nucleoside concentrations are assumed to be constant. There exists a many-fold difference in the output of the four dNMPs, with dA nucleosides being fed into the salvage pathway by DGUOK at a rate many orders of magnitude lower than that required to support mtDNA synthesis. Assuming a 2 hour replication duration and an approximately 276/4 nucleotides per minute substrate requirement, the number of molecules of the DGUOK enzyme per mitochondrion required to catalyze the requisite output of dAMP is close to 3000. The poor kinetics of DGUOK with dA is not the only problem with the dA pathway. Although there could be multiple AK isoforms in the mitochondria, some of them are reported to be lacking kinase activity and none of them appear to catalyze dAMP phosphorylation with comparable efficiency to that of AMP phosphorylation [49]. This is verified for AK2 as seen in Figures 2A, 3A, and 4A.
A calculation of dCDP production by CMPK2 at low assumed dCMP concentrations shows that more than 1000 CMPK2 enzymes per mitochondrion would be required to produce the necessary dCMP output per minute (assuming an approximate requirement of 276/4 nucleotides per minute). This result is important considering that CMPK2 expression was undetectable in many tissues [22], thus implying that CMPK2 function may not be essential for the production of mtDNA precursors as has been noted previously [22].
The data on the kinetic parameters of the human mitochondrial nucleoside diphosphate kinase (NME4 in Figure 1) is scarce (Km for dTDP of approximately 1 mM, which is 100 to 1000 times the physiological concentration of dTDP) [17], which is why it is not included in Figure 5B. An NDPK isolated from the pigeon mitochondrial matrix preferred ribonucleoside diphosphates over deoxyribonucleoside diphosphates by several fold [42]. Surprisingly, it appears that both AK2 [11] and NME4 [10] are localized in the mitochondrial intermembrane space, thus suggesting that if their reaction products participate in the mtDNA precursor synthesis, they would then have to be imported into the mitochondrial matrix. Although dAMP is not the preferred substrate for AK2 (Figures 2A, 3A, and 4A), AK2 still has a very fast reaction with dAMP (Figure 5B). Good efficiency with dAMP and the localization of AK2 in the intermembrane space instead of in the mitochondrial matrix seem to contradict each other regarding the role of AK2 in mtDNA precursor synthesis.
Computational Simulations
To test our conclusions and to build upon them, we used an updated computational model to perform simulations of deoxyribonucleotide dynamics and mtDNA replication within the mitochondrion. Our comprehensive computational model allowed us to investigate the dynamics and origins of mitochondrial dNTPs. Our modeling is based on experimentally measured kinetics and model results enable us to quantitatively track the concentrations as well as the balance of the various deoxynucleosides and deoxynucleotides over time within an individual mitochondrion. Furthermore, the mitochondrial salvage pathway is complex and a systems analysis of this pathway as a whole is an important companion to the study of the individual enzyme kinetics.
Figure 6 shows our simulation results. The X-axis represents the number of molecules of each nucleotide supplied to the mitochondrion in the form of a ‘source’ term in the differential equations in addition to the output from salvage within the mitochondrion. Each value on the X-axis is the sum of molecules supplied of all four species. For example, the X-axis value of 400 means that 100 molecules per minute of each of the four (A, C, G, T) species were supplied. The Y-axis represents the average (over 120 minutes of simulation time) mtDNA replication rate that we observed, calculated as number of nucleotides replicated divided by the time taken to replicate them. Initial values for the substrate concentration in the mitochondrion were randomly varied over a set range as described in the Methods section. Each Y value corresponds to the mean of 100 replication rates from 100 simulations with differing initial substrate conditions. The standard deviations were far smaller than the mean values (and are therefore not shown in Figure 6) indicating that the simulation was not sensitive to the initial substrate conditions. We compared the observed replication rates to those required to complete mtDNA replication in 2 hours (‘cycling cells’) or 10 hours (‘quiescent cells’). For the mtDNA length of 33,136 nucleotides (replicating both strands), these would be 33136 (nucleotides)/120 (minutes) and 33136 (nucleotides)/600 (minutes) respectively or approximately 276 nucleotides per minute and 55 nucleotides per minute respectively. Since the mean observed replication rates with no additional nucleotides supplied (0 on the X-axis) fall below the 2 hour line, it is clear that the output from mitochondrial salvage cannot account for an mtDNA replication duration of 2 hours. In fact, even when a 10 hour replication target was set, mitochondrial salvage alone is an inadequate source of dNTPs, though only a slight amount of additional substrate supplied by transport is needed in this case. Next, we note that both deoxynucleoside as well as deoxynucleoside monophosphate import are insufficient to support a 2 hour replication target. Transport of either dNDPs or dNTPs is sufficient to achieve the target replication rate. The profiles of dNDP and dNTP transport are indistinguishable from one another on Figure 6 because of the extremely fast kinetics of NME4.
Figure 6. The effects of nucleotide import into the mitochondrion on the mtDNA replication rate.

Each point is the mean mtDNA replication rate from 100 simulations with different randomly chosen initial concentrations of deoxynucleosides and deoxynucleotides. The X-axis represents the total amount of additional deoxynucleosides or deoxynucleotides supplied (sum total of equal amounts for each of the four species). Additional supply of dN, dNMP, dNDP or dNTP were simulated separately. The dNTP output from mitochondrial salvage alone is insufficient to support a replication rate of as long as 10 hours. Additional supply of dNs and dNMPs was insufficient to support a replication duration of 2 hours indicating that additional dNDPs or dNTPs are essential. The results were essentially identical for supply of either dNDPs or dNTPs.
Transport of approximately 48 molecules per minute for each of the four nucleotide species was required to complete mtDNA replication in 2 hours. The longer replication time of 10 hours required a transport of 15 dNTP molecules per minute for each of the four nucleotide species. These rates were determined from the simulation by transporting all four nucleotide species at equal rates and with fixed initial concentrations (as described in Methods).We next addressed the question of the minimum transport of each dNTP species necessary to support the target replication. As described in the Methods section, the assumption of equal transport of the four dNTP species was relaxed to find the minimal amount of transport separately for each dNTP species required to meet the mtDNA replication rate goal. To achieve a replication rate of at least 276 nucleotides per minute (‘cycling cells’), 47, 31, 48, and 48 molecules per minute of dTTP, dCTP, dATP, and dGTP were required. Thus, for this condition of relatively fast replication, transport of all four nucleotide species at similar rates is necessary. The total dNTP transport rate sums to 174 nucleotides per minute, a large fraction of the 276 dNTPs per minute consumed by the mtDNA replication. For the slower mtDNA replication with a target of 10 hours, Figure 6 shows that a relatively small amount of transport of dNTP molecules per minute suffices. To achieve the replication rate target of at least 55 nucleotides per minute (representing slow mtDNA replication in ‘quiescent cells’), individual dNTP transport rates of 0, 3, 8, and 15 molecules per minute of dTTP, dCTP, dATP, and dGTP were required. Due to the complexity of the system (a nonlinear one because of feedbacks and inhibitions), slightly different but often practically similar transport profiles were observed to result in similar replication rates. For example, for cycling cells the transport profile of 41, 34, 48, and 41 molecules per minute also achieved the replication rate target. For quiescent cells, the profiles of 2, 2, 8, and 15 and 0, 2, 8, and 15 molecules per minute (practically identical to the transport profile given above) also achieved the replication rate target.
In summary, rapid replication of mtDNA requires a substantial additional source of all four dNTPs (or dNDPs) to supplement the limited kinetics of the mitochondrial salvage pathway. Under the conditions of quiescent cells, the primary requirement is for the transport of dATP and dGTP molecules, and the vast majority of the dNTPs consumed by the mtDNA replication can be provided by the salvage pathway.
Discussion
Kinetic Characteristics of the Mitochondrial Salvage Enzymes and Their Contribution in Producing dNTP Substrates for mtDNA Replication
Based on this analysis of the enzyme kinetics three properties of the mitochondrial nucleoside salvage pathway are thus apparent:
The majority of the enzymes of this pathway are not restricted or specific for metabolism of mtDNA precursors.
The majority of the characterized enzymes prefer non-DNA precursor substrates.
For the majority of substrate-enzyme pairs, the kinetic constants are physiologically unreasonable for achieving efficient catalysis with the expected substrate concentrations in situ.
From the kinetics perspective mitochondrial nucleotide metabolism as defined by this set of enzymes (Figure 1) cannot be expected to be the primary source of dNTP substrates for the rapid replication of mtDNA molecules. Since ribonucleotides exist at higher concentrations than deoxyribonucleotides, enzymes that take both ribonucleotide and deoxyribonucleotide substrates will, in situ, not favor the catalysis of deoxyribonucleotides. This is certainly true for enzymes that possess higher affinities for ribonucleotides, but also for those enzymes that have only slightly better kinetics for deoxyribonucleotides. In these cases ribonucleotide substrates will simply out-compete the deoxyribonucleotides substrates owing to the relative abundance of ribonucleotides.
The Contribution of Cytoplasmic Nucleotide Metabolism in Producing dNTP Substrates for mtDNA Replication
One plausible interpretation of this analysis is that import of cytoplasmic deoxyribonucleotides is the primary source that supplies the direct precursors for the replication of mitochondrial genome while the mitochondrial salvage pathway acts as a back-up metabolism with a minimal role to play in cycling cells. The occurrence of deoxyribonucleotide transport between the mitochondria and cytoplasm and the substantial contribution of cytoplasm deoxynucleotides towards intra-mitochondrial dNTP pools have been demonstrated [5], [6]. Our results make it possible to comment on why this must be so, due to the kinetic properties of the enzymes of mitochondrial salvage. Our results also enable us to conclude that import of deoxyribonucleotides is in fact essential to support an mtDNA replication time of ∼2 hours. Furthermore, simulations based on these enzyme kinetics indicate that this import occurs either at the dNDP or dNTP level. In cells where cytoplasmic deoxynucleotide concentrations are low, mitochondrial salvage would assume a greater role and, in combination with some other supply such as RRM2B mediated reduction of ribonucleotides in the cytoplasm followed by deoxyribonucleotide transport into the mitochondrion, would produce the dNTPs for both the replication of mitochondrial DNA and perhaps repair of nuclear DNA. Possibly, the dNDPs produced by RRM2B activity might first undergo the terminal phosphorylation by NME4 in the intermembrane space (Figure 1B) and may then be imported into the mitochondrion matrix at the dNTP level to combine with the dNTP pool from intra-mitochondrial salvage. Indeed, this is consistent with defects in the mitochondrial salvage pathway having their most severe phenotype in post-mitotic tissues. That mitochondrial salvage has only a back-up role in supporting mtDNA replication is one explanation why DGUOK and TK2 deficiency phenotypes are tissue-restricted and not systemic.
Similarities and Differences between Nucleotide Metabolism in the Cytoplasm and Mitochondria
The kinetic characteristics of the cytoplasmic counterparts of mitochondrial salvage enzymes expose informative parallels and distinctions between the cytoplasmic and mitochondrial pathways of nucleotide metabolism. The good activity of the mitochondrial enzymes (except the nucleoside kinases TK2 and DGUOK) with ribonucleotide substrates implies that these enzymes might play as important a role in ribonucleotide production to support RNA synthesis as they do in supporting DNA synthesis. Based on our analysis, we would argue that future studies of the kinetics of the mitochondrial salvage enzymes would benefit from a broader characterization of the kinetics, particularly the activity of the enzymes with ribonucleotide substrates relative to the activity with deoxyribonucleotide substrates. The majority of the cytoplasmic counterparts also show a preference for non-DNA precursors (such as dUMP) and ribonucleotide substrates [12], [43], [50]. However, the role of nucleoside salvage as a source of dNTPs for nuclear DNA replication is generally assumed to be minimal. In the S-phase of the cell cycle, ribonucleotide reductase irreversibly converts ribonucleoside diphosphates to deoxyribonucleoside diphosphates, and subsequently, deoxyribonucleotides originating from de novo sources proceed to become the predominant precursors to nDNA replication. Thus, ribonucleotide affinities of these cytoplasmic enzymes not only provide the ribonucleoside diphosphates for ribonucleotide reduction but also ensure an adequate supply of RNA substrates. The terminal kinase (NDPK) of the cytoplasmic salvage pathway accepts both ribonucleoside and deoxyribonucleoside diphosphates, and the products can then be appropriately diverted for either RNA or DNA synthesis. Salvage enzymes of thymidine metabolism fit nicely into such a model - examples being the excellent kinetics of TK1 and TK2 with dT, and those of cytoplasmic deoxythymidylate kinase (essential for both salvage and de novo pathways of dTTP synthesis) with dTMP – since thymidine is not an RNA substrate and because of the crucial allosteric control exerted by thymidine nucleotides on ribonucleotide reductase [51] as well as feedback control on mitochondrial TK2 [21]. Such similarities in the enzyme kinetics of the parallel mitochondrial and cytoplasmic metabolisms lead to the question of a ribonucleotide reductase connection to mitochondrial nucleotide metabolism. Such a connection is hinted at by the data supporting a connection between the mitochondrial dNTP pool and the ribonucleotide reductase RRM2B [2], [6]. There has been at least one report of ribonucleotide reductase activity within the mitochondrion [52], though this has never been confirmed as far as we are aware of.
The Identity of the Imported Substrate and the Contribution of Import in Quiescent Cells
Our simulations show that mitochondrial salvage is inadequate to account for the observed replication time of ∼1–2 hours in cycling cells. It is likely that the deficit is supplied by import from the cytoplasm. We propose that deoxyribonucleotide import into the mitochondria not only does occur, but is in fact essential to replicate and maintain mtDNA in cycling cells. Furthermore, in our simulations, import at the monophosphate level was not able to support mtDNA replication under the constraint of a replication duration of 2 hours or less. Our observation that either dNDP or dNTP transport are able to nearly identically support mtDNA replication is due to the extremely fast kinetics of NME4, the nucleoside diphosphate kinase. The fact that the NME4 kinetics for the conversion of dNDP to dNTP are fast lends weight to the hypothesis that transport occurs mainly at the dNDP level, and not at the dNTP level which would bypass the NME4 activity.
Our results are not necessarily in disagreement with previous reports that observed that supplementation with external dA and dG or dAMP and dGMP rescued mtDNA depletion [3], [28]. In those cases, it was undetermined whether these externally supplied substrates changed their phosphorylation level prior to or after entering the mitochondria, or even the cell. In the study conducted by Saada [28], in patient fibroblasts harboring DGUOK defects while dGTP pools were reduced compared to controls, dATP pools were only moderately affected. In this study when these patient fibroblasts were given external supplementation of both deoxyguanosine and deoxyadenosine, mitochondrial dGTP notably increased, while the increase in mitochondrial dATP was less pronounced. Our observation on the inefficient kinetics of DGUOK with dA is consistent with these findings. We are not aware of any studies on the effects of pyrimidine supplementation in TK2 deficiency.
We have chosen a somewhat arbitrary target replication time of 10 hours for the mtDNA in quiescent cells. It has been reported that even in quiescent cells (rat hepatocytes), mitochondrial DNA is subject to rapid turnover [53]. Moreover, it is plausible to suspect that long replication durations might compromise the integrity of either or both the template and the synthesis strand by increasing the probability of damage to the exposed DNA or unfaithful replication (deletions, frameshifting, etc). Therefore, it is possible that the mtDNA replication time may be practically constrained to a shorter duration than 10 hours. In that case deoxynucleotide import could be essential even in quiescent cells. There is a lack of data on mtDNA replication times in quiescent cells, a critically important gap in our knowledge since quiescent cells are the most severely affected cells in most forms of Mitochondrial DNA Depletion Syndromes (MDS).
Could there Be More than One Deoxynucleotide Transporter?
The fact that clinical conditions arising from altered intra-mitochondrial dNTP pools mostly manifest in postmitotic tissues is consistent with our results. The possibility of there being more than one deoxynucleotide transporter, say one for purine deoxynucleotides and one for pyrimidine deoxynucleotides, might explain why mutations in TK2 and DGUOK which are both nucleoside kinases produce different phenotypes. It is plausible that there exists more than one mitochondrial deoxynucleotide transporter whose expression levels, possibly in conjunction with other factors, contribute to tissue specificity of mtDNA depletion syndromes. There have been reports [54], [55] asserting a role of PNC1 (pyrimidine nucleotide carrier encoded by solute carrier family 25, member 33 or SLC25A33) in nucleotide import into mitochondria as well as mitochondrial maintenance. PNC1 was able to transport a variety of metabolites, including purine and pyrimidine ribonucleotides and deoxyribonucleotides, with a preference for UTP. Intra-mitochondrial UTP accumulation decreased in response to siRNA-transfection against PNC1. Mitochondrial ADP, ATP, and GTP levels were not significantly altered but the effect on dNTPs was not investigated. Suppression of PNC1 was associated with reduced mtDNA while overexpressed PNC1 was associated with increased mtDNA relative to controls. Since UTP is a cofactor of the mitochondrial helicase (PEO1 or twinkle), mtDNA levels might have been altered through increased or decreased UTP [54]. It is also possible that these consequences resulted from a lack of RNA primers or lack of mtDNA precursors that might be substrates of PNC1. However, PNC1 mRNA was undetectable in skeletal muscle [55], a tissue that is a target of TK2 defects. Interestingly, ribonucleotide reductase overexpression caused mtDNA depletion in skeletal muscle of mice [56]. Also, per mg protein, PNC1 appeared to transport roughly 1.5 times more UTP compared to dTTP, the next most transported substrate. At this time, the role of PNC1 in transporting deoxyribonucleotides for mtDNA synthesis is inconclusive. Import of radioactively labeled dTMP into mitochondria has been observed [57]. However, it was also observed that a fraction of the labeled dTMP was degraded as well as phosphorylated in the growth medium, leading to the possibility that the transport of phosphorylated states other than the monophosphate may have occurred. A transport activity with preference for dCTP has also been observed [58].
Tissue Specificity of mtDNA Depletion Syndromes
It has been proposed that low basal TK2 expression in muscle renders the tissue vulnerable to TK2 defects, while overlapping substrate specificity of cytosolic dCK prevents mtDNA depletion from mutant DGUOK in tissues where dCK expression is high [59]. While mtDNA defects that have a basis in mutated salvage enzymes might conceivably be rescued by other factors such as overlapping substrate specificity of cytoplasmic enzymes, this hypothesis cannot account for phenotypes relating to POLG defects. Importantly, the fact that phenotypes from mutations in POLG are also tissue-specific and not systemic indicate that other factors, such as rates of mtDNA turnover or energetic demand of tissues might also be a factor in the basis of tissue selectivity. In a recent review, Liya Wang discussed deoxynucleoside salvage enzymes and their association with tissue specific phenotypes of mtDNA depletion [60]. It was hypothesized that since mtDNA turnover rates are different in different tissues and also because dNTP pools show organ-specific differences, it would be expected that the regulation of dNTP pools would also be different for different tissues. Because both muscle and liver have high amounts of mtDNA and also of mtDNA turnover, and since the dTTP pool is lowest in muscle and the dGTP pool is smallest in liver, it was proposed that these tissues would be especially vulnerable to mutations in TK2 and DGUOK respectively. Other contributing factors could include limiting RRM2B, thymidylate synthase, or nucleotide transporter activity. In our opinion, it is probable that there is more than one underlying principle that explains tissue specificity – vulnerability of tissues to mutations might be from a combination of various factors such as transcriptional compensation, turnover rates, energetic demand, etc and that different forms of mtDNA depletion syndromes may trace their etiology to different factors.
The Applicability of Our Results to Different Tissues and Species
Based on their experiments with perfused rat heart, Morris et al concluded that in isolated perfused heart, there is no de novo synthesis of dNTPs [61], stressing the importance of TK2 in rat heart. This could indicate that our observations on the inadequacy of mitochondrial salvage enzymes may not hold across all tissue types. It is also possible that the deoxyribonucleotide pools in rat heart arose in part through salvage mediated by residual TK1 activity. In a recent report of a TK2−/− H126N knockin mouse [62], the authors observed TK1 to be the main thymidine kinase component in heart, compared to TK2 in the brain. In this mouse, phenotypic manifestation of TK2 deficiency was related to TK1 down-regulation and transcriptional compensation. Although by postnatal day 13 both brain and heart had suffered substantial mtDNA depletion, in contrast to brain, heart was spared as respiratory chain proteins were still at normal levels in this organ when assayed at postnatal day 13. This could indicate a difference in the importance of TK2 in the heart tissue of rats compared to mice. A recent report claimed that a cytosolic localization of TK2 is present in many rat tissues [63]. For the knockin mouse [62], a compensatory mechanism involving increased mtDNA transcription through suppression of MTERF3 (mitochondrial transcription termination factor 3) expression was implicated in alleviating some of the effects of mtDNA depletion. It was unclear why dTTP but not dCTP levels were affected and whether cytosolic ribonucleotide reduction had any influence in this observation. In humans, even in quiescent patient fibroblasts with only 5–40% of residual TK2 activity, mitochondrial and cytosolic dTTP pools were unaltered [30]. This finding would be consistent with the possibility that the activities of mitochondrial salvage enzymes may not be strictly necessary even for quiescent cells. Alternatively, it is also possible for there to be practical important differences between species with regard to this metabolism. In their study of the rat heart, Morris et al [61] noted that although known as a substrate of TK2, dU was not converted to dUMP possibly due to ENT1 nucleoside transporter not being localized to mitochondria in rodents, unlike humans, suggesting that dU may not be transported into the mitochondria in rodents. It has been noted that genes involved in MDS (mtDNA depletion syndromes) etiology are essential for life in mouse models [60]. However, the severe phenotype of knock-out mice is not identical to the phenotype in humans [64], although multi-organ phenotypes have come to light in humans also [65]. This divergence could perhaps be due to species differences or because of the complete absence of enzyme activity in knock-out models [64].
Limitations of Our Analysis
One limitation of modeling biochemical pathways is that kinetic parameters as reported in the literature and obtained from recombinant enzymes may not reflect the in situ reality, for instance, if the enzyme conformation is unknowingly affected in the in vitro analysis, or if the assay conditions do not represent the cellular environment adequately. Similarly, we have relied on the literature and our judgment for selecting appropriate concentrations and enzyme copies within the mitochondrion. In our analysis we have assumed a nucleoside concentration of 0.5 µM [33]. There have been reports of nucleoside concentrations of approximately 50-fold lower [34]. Such lower concentrations would have two effects on this analysis. First, the problems that we point out concerning the function of the nucleoside kinases TK2 and DGUOK would be even worse with significantly lower nucleoside concentrations. Second, there is a more subtle problem that the enzyme kinetics for TK2 were measured at much higher substrate concentrations (1 µM to more than 100 µM) [21]. If the true substrate concentrations were on the order of 10 nM, then the kinetics would have to be extrapolated to much lower concentrations, which could introduce additional uncertainty in the kinetic constants. Finally, our estimate of time taken to replicate mtDNA (2 hours) comes from a study of mouse cells [45]. It is worth mentioning that POLG kinetics suggest that polymerization itself is capable of proceeding at a rate much faster than 2 hours [14]. A more comprehensive investigation into mtDNA replication durations in a variety of human cells and particularly in the cell types affected by mtDNA depletion syndromes would thus be very beneficial.
For simplicity, we assumed that only one mtDNA molecule is replicating at any given time in a particular mitochondrion. If two or more mtDNA molecules were replicating simultaneously, then the deficit in the required dNTPs would be even larger than our analysis indicates. It should also be noted that mitochondria are very dynamic and undergo continuous fusion and fission. However, the effects of fusion and fission on the mitochondrial dNTP content would most likely average out. While fusion of two mitochondria would result in a larger dNTP pool (measured as number of molecules per organelle), fission would result in a smaller dNTP pool.
Summary
Since the known elements of the mitochondrial salvage pathway do not have sufficient enzyme kinetics to support mtDNA replication in the observed duration of ∼1–2 hours, then, an alternative source of mtDNA precursors must be essential. Despite the intensive focus of research on this pathway associated with mitochondrial depletion syndromes, it seems likely that our knowledge of mitochondrial nucleotide metabolism is still incomplete and that this pathway might need to be considerably expanded in the future to include new enzymes, mechanisms, nucleotide transporters and modes of regulation.
Supporting Information
kcat/Km, Concentration/Km, and inhibition data for the enzymes.
(XLS)
File containing the Mathematica simulation code in plain text file format.
(TXT)
Mathematica-readable file containing simulation parameters for the Mathematica simulation.
(TXT)
Simulation parameters and references in PDF format.
(PDF)
Footnotes
The authors have declared that no competing interests exist.
This work was supported by the National Institutes of Health through grant GM073744. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bogenhagen D, Clayton DA. Mouse L-Cell Mitochondrial-DNA Molecules Are Selected Randomly for Replication Throughout Cell-Cycle. Cell. 1977;11:719–727. doi: 10.1016/0092-8674(77)90286-0. [DOI] [PubMed] [Google Scholar]
- 2.Bourdon A, Minai L, Serre V, Jais J-P, Sarzi E, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 3.Taanman JW, Muddle JR, Muntau AC. Mitochondrial DNA depletion can be prevented by dGMP and dAMP supplementation in a resting culture of deoxyguanosine kinase-deficient fibroblasts. Hum Mol Genet. 2003;12:1839–1845. doi: 10.1093/hmg/ddg192. [DOI] [PubMed] [Google Scholar]
- 4.Berk AJ, Clayton DA. A Genetically Distinct Thymidine Kinase in Mammalian Mitochondria. Exclusive Labeling of Mitochondrial Deoxyribonucleic Acid. J Biol Chem. 1973;248:2722–2729. [PubMed] [Google Scholar]
- 5.Leanza L, Ferraro P, Reichard P, Bianchi V. Metabolic Interrelations within Guanine Deoxynucleotide Pools for Mitochondrial and Nuclear DNA Maintenance. J Biol Chem. 2008;283:16437–16445. doi: 10.1074/jbc.M801572200. [DOI] [PubMed] [Google Scholar]
- 6.Rampazzo C, Fabris S, Franzolin E, Crovatto K, Frangini M, et al. Mitochondrial Thymidine Kinase and the Enzymatic Network Regulating Thymidine Triphosphate Pools in Cultured Human Cells. J Biol Chem. 2007;282:34758–34769. doi: 10.1074/jbc.M705923200. [DOI] [PubMed] [Google Scholar]
- 7.Lindhurst MJ, Fiermonte G, Song SW, Struys E, De Leonardis F, et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci U S A. 2006;103:15927–15932. doi: 10.1073/pnas.0607661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munch-Petersen B, Cloos L, Tyrsted G, Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem. 1991;266:9032–9038. [PubMed] [Google Scholar]
- 9.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 10.Tokarska-Schlattner M, Boissan M, Munier A, Borot C, Mailleau C, et al. The nucleoside diphosphate kinase D (NM23-H4) binds the inner mitochondrial membrane with high affinity to cardiolipin and couples nucleotide transfer with respiration. J Biol Chem. 2008;283:26198–26207. doi: 10.1074/jbc.M803132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nobumoto M, Yamada M, Song SC, Inouye S, Nakazawa A. Mechanism of mitochondrial import of adenylate kinase isozymes. J Biochem (Tokyo) 1998;123:128–135. doi: 10.1093/oxfordjournals.jbchem.a021899. [DOI] [PubMed] [Google Scholar]
- 12.Alexandre JAC, Roy B, Topalis D, Pochet S, Perigaud C, et al. Enantioselectivity of human AMP, dTMP and UMP-CMP kinases. Nucleic Acids Res. 2007;35:4895–4904. doi: 10.1093/nar/gkm479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolce V, Fiermonte G, Runswick MJ, Palmieri F, Walker JE. The human mitochondrial deoxynucleotide carrier and its role in the toxicity of nucleoside antivirals. Proc Natl Acad Sci U S A. 2001;98:2284–2288. doi: 10.1073/pnas.031430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson AA, Johnson KA. Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J Biol Chem. 2001;276:38090–38096. doi: 10.1074/jbc.M106045200. [DOI] [PubMed] [Google Scholar]
- 15.Lam W, Chen CS, Ruan SL, Leung CH, Cheng YC. Expression of deoxynucleotide carrier is not associated with the mitochondrial DNA depletion caused by anti-HIV dideoxynucleoside analogs and mitochondrial dNTP uptake. Mol Pharmacol. 2005;67:408–416. doi: 10.1124/mol.104.007120. [DOI] [PubMed] [Google Scholar]
- 16.Mazzon C, Rampazzo C, Scaini MC, Gallinaro L, Karlsson A, et al. Cytosolic and mitochondrial deoxyribonucleotidases: activity with substrate analogs, inhibitors and implications for therapy. Biochem Pharmacol. 2003;66:471–479. doi: 10.1016/s0006-2952(03)00290-9. [DOI] [PubMed] [Google Scholar]
- 17.Milon L, Meyer P, Chiadmi M, Munier A, Johansson M, et al. The Human nm23-H4 Gene Product Is a Mitochondrial Nucleoside Diphosphate Kinase. J Biol Chem. 2000;275:14264–14272. doi: 10.1074/jbc.275.19.14264. [DOI] [PubMed] [Google Scholar]
- 18.Murakami E, Feng JY, Lee H, Hanes J, Johnson KA, et al. Characterization of novel reverse transcriptase and other RNA-associated catalytic activities by human DNA polymerase gamma - Importance in mitochondrial DNA replication. J Biol Chem. 2003;278:36403–36409. doi: 10.1074/jbc.M306236200. [DOI] [PubMed] [Google Scholar]
- 19.Rampazzo C, Gallinaro L, Milanesi E, Frigimelica E, Reichard P, et al. A deoxyribonucleotidase in mitochondria: Involvement in regulation of dNTP pools and possible link to genetic disease. Proc Natl Acad Sci U S A. 2000;97:8239–8244. doi: 10.1073/pnas.97.15.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sjoberg AH, Wang L, Eriksson S. Substrate Specificity of Human Recombinant Mitochondrial Deoxyguanosine Kinase with Cytostatic and Antiviral Purine and Pyrimidine Analogs. Mol Pharmacol. 1998;53:270–273. doi: 10.1124/mol.53.2.270. [DOI] [PubMed] [Google Scholar]
- 21.Wang LY, Saada A, Eriksson S. Kinetic properties of mutant human thymidine kinase 2 suggest a mechanism for mitochondrial DNA depletion myopathy. J Biol Chem. 2003;278:6963–6968. doi: 10.1074/jbc.M206143200. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Johansson M, Karlsson A. Human UMP-CMP Kinase 2, a Novel Nucleoside Monophosphate Kinase Localized in Mitochondria. J Biol Chem. 2008;283:1563–1571. doi: 10.1074/jbc.M707997200. [DOI] [PubMed] [Google Scholar]
- 23.Chang A, Scheer M, Grote A, Schomburg I, Schomburg D. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res. 2009;37:D588–D592. doi: 10.1093/nar/gkn820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunsucker SA, Mitchell BS, Spychala J. The 5 ′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther. 2005;107:1–30. doi: 10.1016/j.pharmthera.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Bradshaw PC, Samuels DC. A computational model of mitochondrial deoxynucleotide metabolism and DNA replication. Am J Physiol-Cell Physiol. 2005;288:C989–C1002. doi: 10.1152/ajpcell.00530.2004. [DOI] [PubMed] [Google Scholar]
- 26.Ferraro P, Pontarin G, Crocco L, Fabris S, Reichard P, et al. Mitochondrial deoxynucleotide pools in quiescent fibroblasts - A possible model for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). J Biol Chem. 2005;280:24472–24480. doi: 10.1074/jbc.M502869200. [DOI] [PubMed] [Google Scholar]
- 27.Pontarin G, Ferraro P, Valentin ML, Hirano M, Reichard P, et al. Mitochondrial DNA depletion and thymidine phosphate pool dynamics in a cellular model of mitochondrial neurogastrointestinal encephalomyopathy. J Biol Chem. 2006;281:22720–22728. doi: 10.1074/jbc.M604498200. [DOI] [PubMed] [Google Scholar]
- 28.Saada A. Mitochondrial deoxyribonucleotide pools in deoxyguanosine kinase deficiency. Mol Genet Metab. 2008;95:169–173. doi: 10.1016/j.ymgme.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Song S, Wheeler LJ, Mathews CK. Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J Biol Chem. 2003;278:43893–43896. doi: 10.1074/jbc.C300401200. [DOI] [PubMed] [Google Scholar]
- 30.Frangini M, Rampazzo C, Franzolin E, Lara M-C, Vilà MR, et al. Unchanged thymidine triphosphate pools and thymidine metabolism in two lines of thymidine kinase 2-mutated fibroblasts. FEBS J. 2009;276:1104–1113. doi: 10.1111/j.1742-4658.2008.06853.x. [DOI] [PubMed] [Google Scholar]
- 31.Vinnakota KC, Bassingthwaighte JB. Myocardial density and composition: a basis for calculating intracellular metabolite concentrations. Am J Physiol Heart Circ Physiol. 2004;286:H1742–1749. doi: 10.1152/ajpheart.00478.2003. [DOI] [PubMed] [Google Scholar]
- 32.Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol. 1977;74:468–491. doi: 10.1083/jcb.74.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 34.Li KM, Rivory LP, Hoskins J, Sharma R, Clarke SJ. Altered deoxyuridine and thymidine in plasma following capecitabine treatment in colorectal cancer patients. British Journal of Clinical Pharmacology. 2007;63:67–74. doi: 10.1111/j.1365-2125.2006.02710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honeywell R, van Groeningen CJ, Laan AC, Strocchi E, Ruiter R, et al. Analysis of deoxycytidine accumulation in gemcitabine treated patients. Nucleosides Nucleotides Nucleic Acids. 2006;25:1225–1232. doi: 10.1080/15257770600894642. [DOI] [PubMed] [Google Scholar]
- 36.Sjoberg AH, Wang LY, Eriksson S. Antiviral guanosine analogs as substrates for deoxyguanosine kinase: Implications for chemotherapy. Antimicrob Agents Chemother. 2001;45:739–742. doi: 10.1128/AAC.45.3.739-742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Munch-Petersen B, Sjoberg AH, Hellman U, Bergman T, et al. Human thymidine kinase 2: molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 1999;443:170–174. doi: 10.1016/s0014-5793(98)01711-6. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y-L, Lin D-W, Chang Z-F. Identification of a putative human mitochondrial thymidine monophosphate kinase associated with monocytic/macrophage terminal differentiation. Genes Cells. 2008;13:679–689. doi: 10.1111/j.1365-2443.2008.01197.x. [DOI] [PubMed] [Google Scholar]
- 40.Jimenez A, Pubill D, Pallas M, Camins A, Llado S, et al. Further characterization of an adenosine transport system in the mitochondrial fraction of rat testis. Eur J Pharmacol. 2000;398:31–39. doi: 10.1016/s0014-2999(00)00297-1. [DOI] [PubMed] [Google Scholar]
- 41.Watkins LF, Lewis RA. The metabolism of deoxyguanosine in mitochondria - characterization of the uptake process. Mol Cell Biochem. 1987;77:71–77. doi: 10.1007/BF00230152. [DOI] [PubMed] [Google Scholar]
- 42.Lambeth DO, Mehus JG, Ivey MA, Milavetz BI. Characterization and Cloning of a Nucleoside-diphosphate Kinase Targeted to Matrix of Mitochondria in Pigeon. J Biol Chem. 1997;272:24604–24611. doi: 10.1074/jbc.272.39.24604. [DOI] [PubMed] [Google Scholar]
- 43.Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of deoxycytidine analog monophosphates by UMP-CMP kinase: Molecular characterization of the human enzyme. Mol Pharmacol. 1999;56:562–569. doi: 10.1124/mol.56.3.562. [DOI] [PubMed] [Google Scholar]
- 44.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147–147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 45.Clayton DA. Replication of Animal Mitochondrial-DNA. Cell. 1982;28:693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 46.Samson R, Deutch JM. Diffusion-controlled reaction rate to a buried active site. The Journal of Chemical Physics. 1978;68:285–290. [Google Scholar]
- 47.Kang J, Samuels DC. The evidence that the DNC (SLC25A19) is not the mitochondrial deoxyribonucleotide carrier. Mitochondrion. 2008;8:103–108. doi: 10.1016/j.mito.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Yasukawa T, Reyes A, Cluett TJ, Yang MY, Bowmaker M, et al. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006;25:5358–5371. doi: 10.1038/sj.emboj.7601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther. 2000;87:189–198. doi: 10.1016/s0163-7258(00)00048-6. [DOI] [PubMed] [Google Scholar]
- 50.Gallois-Montbrun S, Veron M, Deville-Bonne D. Antiviral nucleoside analogs phosphorylation by nucleoside diphosphate kinase. Mini-Rev Med Chem. 2004;4:361–369. doi: 10.2174/1389557043403990. [DOI] [PubMed] [Google Scholar]
- 51.Hu CM, Chang ZF. Mitotic control of dTTP pool: a necessity or coincidence? J Biomed Sci. 2007;14:491–497. doi: 10.1007/s11373-007-9175-1. [DOI] [PubMed] [Google Scholar]
- 52.Young P, Leeds JM, Slabaugh MB, Mathews CK. Ribonucleotide Reductase: Evidence for Specific Association with HeLa-Cell Mitochondria. Biochem Biophys Res Commun. 1994;203:46–52. doi: 10.1006/bbrc.1994.2146. [DOI] [PubMed] [Google Scholar]
- 53.Kai Y, Takamatsu C, Tokuda K, Okamoto M, Irita K, et al. Rapid and random turnover of mitochondrial DNA in rat hepatocytes of primary culture. Mitochondrion. 2006;6:299–304. doi: 10.1016/j.mito.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Favre C, Zhdanov A, Leahy M, Papkovsky D, O'Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene. 2010;29:3964–3976. doi: 10.1038/onc.2010.146. [DOI] [PubMed] [Google Scholar]
- 55.Floyd S, Favre C, Lasorsa FM, Leahy M, Trigiante G, et al. The insulin-like growth Factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell. 2007;18:3545–3555. doi: 10.1091/mbc.E06-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, et al. Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Res. 2010;38:8208–8218. doi: 10.1093/nar/gkq735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferraro P, Nicolosi L, Bernardi P, Reichard P, Bianchi V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc Natl Acad Sci U S A. 2006;103:18586–18591. doi: 10.1073/pnas.0609020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bridges EG, Jiang Z, Cheng Y-c. Characterization of a dCTP Transport Activity Reconstituted from Human Mitochondria. J Biol Chem. 1999;274:4620–4625. doi: 10.1074/jbc.274.8.4620. [DOI] [PubMed] [Google Scholar]
- 59.Saada-Reisch A. Deoxyribonucleoside kinases in mitochondrial DNA depletion. Nucleosides Nucleotides & Nucleic Acids. 2004;23:1205–1215. doi: 10.1081/NCN-200027480. [DOI] [PubMed] [Google Scholar]
- 60.Wang L. Deoxynucleoside Salvage Enzymes and Tissue Specific Mitochondrial DNA Depletion. Nucleosides Nucleotides & Nucleic Acids. 2010;29:370–381. doi: 10.1080/15257771003729732. [DOI] [PubMed] [Google Scholar]
- 61.Morris GW, Iams TA, Slepchenko KG, McKee EE. Origin of pyrimidine deoxyribonucleotide pools in perfused rat heart: implications for 3 ′-azido-3 ′-deoxythymidine-dependent cardiotoxicity. Biochem J. 2009;422:513–520. doi: 10.1042/BJ20082427. [DOI] [PubMed] [Google Scholar]
- 62.Dorado B, Area E, Akman HO, Hirano M. Onset and organ specificity of Tk2 deficiency depends on Tk1 down-regulation and transcriptional compensation. Hum Mol Genet. 2011;20:155–164. doi: 10.1093/hmg/ddq453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang L, Eriksson S. Tissue Specific Distribution of Pyrimidine Deoxynucleoside Salvage Enzymes Shed Light on the Mechanism of Mitochondrial DNA Depletion. Nucleosides Nucleotides & Nucleic Acids. 2010;29:400–403. doi: 10.1080/15257771003741042. [DOI] [PubMed] [Google Scholar]
- 64.Bartesaghi S, Betts-Henderson J, Cain K, Dinsdale D, Zhou XS, et al. Loss of thymidine kinase 2 alters neuronal bioenergetics and leads to neurodegeneration. Hum Mol Genet. 2010;19:1669–1677. doi: 10.1093/hmg/ddq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gotz A, Isohanni P, Pihko H, Paetau A, Herva R, et al. Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain. 2008;131:2841–2850. doi: 10.1093/brain/awn236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
kcat/Km, Concentration/Km, and inhibition data for the enzymes.
(XLS)
File containing the Mathematica simulation code in plain text file format.
(TXT)
Mathematica-readable file containing simulation parameters for the Mathematica simulation.
(TXT)
Simulation parameters and references in PDF format.
(PDF)