

Abstract
We recently demonstrated that CD1d-restricted NKT cells resident in skin can inhibit CD8 T cell-mediated graft rejection of human papillomavirus (HPV) E7-expressing skin, through an IFN-γ dependent mechanism. Here we examine the role of systemically-derived NKT cells in regulating rejection of skin grafts expressing viral proteins. In lymph nodes draining transplanted skin, antigen-specific CD8 T cell proliferation, cytokine production and cytotoxic activity was impaired by NKT cells. NKT cell suppression was mediated via CD11c+ dendritic cells. Inhibition of CD8 T cell function did not require Foxp3+ regulatory T cells, or NKT cell-secreted IFN-γ, IL-10 or IL-17. Thus, following skin grafting or immunization with HPV-E7 oncoprotein, NKT cells reduce the capacity of draining lymph node resident APC to cross-present antigen to CD8 T cell precursors, as evidenced by impaired expansion and differentiation to antigen-specific CD8 T effector cells. Therefore, in the context of viral antigen challenge in the skin, systemic NKT cells limit the capacity for effective priming of adaptive immunity.
Keywords: NKT cells, CD8 T cells, Human papillomavirus, immunoregulation, skin immunology
INTRODUCTION
Specialized immune regulatory cells are found in chronic skin infections and epithelial tumours and contribute to local mechanisms inhibiting generation and function of immune effector cells targeted at viral- or tumour-specific antigens (1, 2). Recently, using a murine model in which skin expressing human papillomavirus (HPV) 16 E7 oncoprotein as a transgene in basal keratinocytes is grafted to an immune competent host, we have shown that NKT cells present in skin locally suppress the function of systemically generated E7-specific effector CD8 T cells. This functional inhibition of CD8 T cells prevented rejection of E7-expressing skin (3). Local NKT cell-mediated regulation required production of IFN-γ by the NKT cells, and this was induced following CD1d-dependent NKT cell activation by a population of myeloid cells recruited to the skin graft.
In addition to regulation of local immune effector functions, NKT cells possess the capacity to promote or inhibit priming of the adaptive immune response via the release of cytokines that can license APC towards immunogenic or tolerogenic activity (4, 5). We have recently shown that systemically derived NKT cells promote the generation of ovalbumin (OVA)-specific CD8 T cells in skin-draining lymph nodes (DLN) following grafting of skin expressing OVA as a transgene in keratinocytes. NKT cell assistance in priming of CD8 T cells resulted in more effective CD8 T cell mediated rejection of grafted K5mOVA transgenic skin (6). As NKT cells locally inhibit skin graft rejection when the grafted skin expresses E7 as a transgene, we wished to investigate the effects of systemically derived NKT cells on initiation of an adaptive immune response to E7 cross-presented from skin. We demonstrate here that in contrast to the effects of systemically derived NKT cells on priming OVA-specific CD8 T cells following grafting of OVA transgenic skin, NKT cells stimulated by grafting with skin expressing E7, or expressing another viral antigen, the herpes simplex glycoprotein B, inhibit CD8 T cell-mediated rejection of skin grafts. For E7 transgenic skin, we further show that systemically derived NKT cells achieve suppression of graft rejection by inhibiting the ability of APC in the skin DLN to promote proliferation and acquisition of effector functions by antigen-specific CD8 T cells. Unlike the local inhibitory effects of skin resident NKT cells on CD8 T cell effector function, the regulatory function of systemically derived NKT cells is independent of IFN-γ.
MATERIALS AND METHODS
Mice
C57BL/6 mice and HPV16-E7 transgenic C57BL/6 mice (designated K14E7) (3) were obtained from the Animal Resources Centre - ARC (Perth, Australia). K14gB transgenic mice, in which HSV glycoprotein B is driven off the K14 promoter, were kindly provided by F. Carbone (Melbourne, Australia). IFN-γKO mice were purchased from the Jackson Laboratories (Maine, USA), IL-10KO mice were sourced from the Australian National University (Canberra, Australia) and IL-17KO mice were provided by Y. Iwakura (Tokyo, Japan). NKT cell deficient CD1dKO and Jα18KO mice were obtained from M. Smyth (Melbourne, Australia) and maintained locally at the Princess Alexandra Hospital Biologically Research Facility - BRF (Brisbane, Australia). HPV16 E7-specific TCR β chain transgenic C57BL/6 mice (E7TCRβ) were generated in our laboratory by G. Leggatt (7) and crossed with CD45.1 congenic C57BL/6.SJL-Ptprc mice (ARC) to generate mice bearing CD45.1+ E7TCR cells. Depletion of regulatory T cells – DEREG mice were provided by T. Sparwasser (Hannover, Germany) (8). To generate K14E7 transgenic crosses with DEREG mice (K14E7×DEREG), heterozygous K14E7 mice were crossed with DEREG mice to an F1 generation. All mice were housed under specific pathogen-free conditions at the BRF, were sex-matched for all experiments and were used at 6–10 weeks of age. All animal procedures were approved by the University of Queensland Animal Ethics Committee.
Reagents and Flow Cytometry
The HPV16-E7 peptide containing the H-2Db-restricted CTL epitope, with the amino acid sequence RAHYNIVTF (GF001), was purchased from Auspep Pty (Melbourne, Australia) with >80% purity, dissolved in 100% DMSO and stored at −20°C. Anti-mouse monoclonal antibodies (mAb) to CD3 (145-2C11), CD4 (RM4-4), CD8 (53-6.7), CD25 (PC61), CD69 (H1.2F3), CD11c (HL3), CD45.1 (A20), CD45.2 (104), CD40 (HM40-3), CD80 (16-10A1), CD86 (GL-1), TCRβ (H57-597), MHCII (M5/114.15.2), IFN-γ (XMG1.2) and associated isotype control immunoglobulins were purchased from BD Biosciences (CA, USA), eBioscience (CA, USA) and BioLegend (CA, USA). Preparation of α-Galactosylceramide (αGalCer)-loaded CD1d tetramer is described elsewhere (9) and was kindly provided by D. Godfrey (Melbourne, Australia). Cells were stained at pre-determined optimal concentrations of antibody for 30min at 4°C. For intracellular staining of cytokines, permeabilization and fixation of cells was conducted using the BD Cytofix/Cytoperm kit according to the manufacturers’ instructions (BD Biosciences). Monensin (BioLegend, San Diego CA) was added to the cells to inhibit cytokine release from the Golgi/ER complex. Stained cells were acquired on a FACScalibur or FACScanto flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, OR, USA).
Skin grafting and in vivo cell depletions
The process of grafting donor ear skin onto the flanks of recipient mice, and assessment of graft acceptance versus rejection, is described in detail elsewhere (10, 11). Anti-CD4 (GK1.5), anti-CD8β (53-5.8) and anti-CD25 (PC61) monoclonal antibodies were purified in house from supernatant taken from hybridoma cell line culture, by protein elution using G-protein columns (Thermo Fisher Scientific, Rockford, IL, USA). CD4+, CD8+ or CD25+ cells were depleted from donor and/or host mice, as indicated, prior to skin grafting by i.p. injection of 500µg GK1.5 (×1), 100µg 53-5.8 (×3) or 500µg PC61 (×2) antibodies, respectively. Equal volume of isotype-matched Rat IgG antibody was used for control treatments. Maintenance treatments were given weekly to recipient mice to continue cell depletion for the length of the experiment. Foxp3+ cells were depleted from DEREG and K14E7×DEREG mice by 3 administrations of 1µg diphtheria toxin (DT) in one week, prior to grafting.
Adoptive transfers of NKT cell populations
For bulk reconstitution experiments (providing a source of NKT cells), 5×107 splenocytes isolated from C57BL/6 mice were injected i.v. into Jα18KO recipients 3 days prior to grafting with K14E7 skin. For pure NKT cell transfers, mononuclear cells pooled from liver, thymus and inguinal lymph nodes of WT, IL-10KO, IFNγKO or IL-17KO C57BL/6 mice were sorted by flow cytometry (MoFlo, BD Biosciences) based on dual CD3+ and CD1d-tetramer+ staining. CD3+CD1d-tetramer+ T cell purity following sorting was consistently greater than 90%. For NKT cell reconstitution of Jα18KO recipient mice, 2×105 pure NKT cells were injected i.v. into the tail vein 3 days prior to skin grafting.
In vivo proliferation assay and immunizations
To assess HPV16 E7-specific CD8+ T cell proliferation in vivo, CD45.1+ E7TCRβ splenocytes were labeled with 2.5µM CFSE and injected i.v. (1×107) into the tail vein of WT, CD1dKO or Jα18KO mice. Seven days later, recipient spleens were harvested and CFSE dilution in CD45.1+/CD3+/CD8+ cells was assessed by flow cytometry. Indices of proliferation were generated using ModFit LT software (VSH, Topsham, ME). To determine antigen-specific CD8 T cell responsiveness to soluble antigen challenge, mice were immunized subcutaneously at the tail base with 50µg GF001 peptide, 20µg Keyhole Limpet Hemanocyanin (KLH) (Sigma, NSW, Australia) and 20µg QuilA adjuvant (Soperfos Biosector DK-Vedback, Denmark).
In vitro assays of DC and CD8 T cell function
CD8 T cell cytokine production and cytotoxicity
CD8 T cells were isolated from skin-DLN by MACS separation using CD8 microbeads (Miltenyi Biotec, Germany). For detection of cytokine secretion CD8 T cells were re-stimulated in vitro for 4 hours with 25ng/ml PMA and 1µg/ml ionomycin prior to collecting culture supernatant. Secreted levels of IFN-γ, TNF-α and IL-2 were detected by Th1/Th2 cytometric bead array (CBA), according to the manufacturers’ protocol (BD Biosciences). Samples were analyzed on a FACSarray (BD Biosciences). For IFN-γ ELISPOT, cell suspensions isolated from skin-DLN of immunized or grafted recipients were cultured overnight in complete RPMI medium in the presence of 5ng/ml recombinant mouse IL-2 (BD Biosciences) and with or without addition of 0.01µM GF001 peptide. The IFN-γ ELISPOT procedure has been previously described (12). For assessment of antigen-specific cytotoxicity, cell suspensions isolated from spleens and skin-DLN of immunized mice were cultured in vitro for 5 days with 0.01µM GF001 peptide and 2ng/ml IL-2 to re-stimulate CD8 T cells prior to purification. Isolated CD8 T cells were then co-cultured for 24 hours with GF001-pulsed EL4 cells, used as targets in a standard chromium release assay as previously described (13).
DC functional assay
Dendritic cells were isolated from skin-DLN of grafted or non-grafted mice by FACS, based on dual CD11c+MHCIIhi expression. Purified DC were pulsed for 4 hours with 0.01µM GF001 peptide and co-cultured in vitro with CD8 T cells isolated from E7TCRβ mice (a source of E7-specific CD8 T cells) for 4 days. Antigen-specific IFN-γ production was measured by ELISA of culture supernatant, as previously described (12).
Statistics
Kaplan-Meier plots were used to analyze skin graft survival and a log-rank test was performed to assess the statistical significance of differences between survival curves. For all other data in which statistics were performed, a two-tailed t test or non-parametric Mann-Whitney U test, as indicated, was used for assessment of differences between groups. Differences were considered to be significant when the p value was less than 0.05. Prism (Graphpad Software, La Jolla, CA) software was used to prepare graphs and for statistical analysis.
RESULTS
Host type I NKT cells are critical in the inhibition of K14E7 graft rejection
We have recently reported that a population of NKT cells resident in HPV16-E7 transgenic skin is capable of inhibiting K14E7 graft rejection (3). In addition, a previous report has shown that systemically-derived host NKT cells can regulate rejection of MHC-mismatched skin grafts (14). To address the role of host-derived NKT cells in suppression of K14E7 skin graft rejection, we grafted native K14E7 skin onto NKT cell-deficient CD1dKO recipients. We observed 100% rejection of K14E7 grafts on CD1dKO recipients (Fig. 1A), without rejection of wildtype (WT) C57Bl/6 or littermate E7−CD1d+/− control grafts. K14E7 grafts were also rejected by type I NKT cell-deficient Jα18KO recipients. These findings demonstrate a critical role for host-derived type I, invariant NKT cells in inhibiting HPV16-E7 specific effector T cell functions necessary and sufficient for skin graft rejection (11). To further confirm a role for systemically derived NKT cells in regulating effector functions necessary for graft rejection, we transferred 5×107 splenocytes from WT mice (equating to approximately 4×105 NKT cells) into Jα18KO graft recipients, and showed that this was sufficient to prevent K14E7 graft rejection, as observed in control animals. Only 1 graft out of 8 (12.5%) rejected following NKT cell reconstitution (Fig. 1B), supporting our observation that systemically derived type I NKT cells are, like locally resident NKT cells, fully competent to regulate the effector functions necessary for rejection of K14E7 skin grafts. For skin grafts expressing OVA or human growth hormone (hGH) from a keratin promoter, which unlike E7 grafts are routinely rejected by immunocompetent recipients, graft rejection is mediated by CD8 effector T cells (6, 15). To establish whether CD8 T cells were critical for rejection of K14E7 grafts in NKT cell deficient hosts, we depleted CD1dKO mice of CD8 T cells using an anti-CD8β mAb, prior to grafting with K14E7 skin, and observed a substantial reduction and delay in K14E7 graft rejection during the 6 week period that the CD8 T cells were depleted (Fig. 1C). To confirm that failure to reject K14E7 grafts was due to the absence of CD8 T cells, we permitted CD8 T cell recovery in NKT cell-deficient mice and then placed a second K14E7 graft on the contra-lateral flank of those that failed to reject the primary graft. With recovery of the CD8 T cell population, rejection of second grafts was complete and followed similar kinetics to primary K14E7 graft rejection on non-depleted recipients (Fig. 1C). Interestingly, 4 out of 6 mice that received a second K14E7 graft also rejected their primary graft, suggesting that CD8 T cells induced by a recently placed and hence inflamed skin graft in the absence of NKT cells are capable of rejecting well-healed K14E7 grafts at a distal site. This observation contrasts with our previous findings where passively transferred E7-specific T cells activated by immunization with peptide and adjuvant were unable to achieve rejection of well-healed E7 grafts in WT mice (11).
Figure 1. Host type I NKT cells inhibit K14E7 graft rejection.

(A) Kaplan-Meier survival curves of HPV16 E7-expressing skin grafts (K14E7) transplanted onto naïve WT C57BL/6 and NKT cell-deficient CD1dKO recipients (n=10 mice per group).
(B) Survival of K14E7 grafts on type 1 NKT cell-deficient Jα18KO recipients, some of which were reconstituted 3 days prior to grafting by transfer of 1×107 splenocytes isolated from WT mice, as a source of NKT cells. Transfer of splenocytes from Jα18KO mice were used as controls (n=8 mice per group).
(C) K14E7 skin graft survival on CD1dKO recipients following systemic depletion of CD8 T cells, using anti-mouse CD8β (53-5.8) mAb or similar treatment with an isotype-matched rat IgG mAb. Anti-CD8β mAb treated recipients that failed to reject the primary K14E7 graft, received a 2nd K14E7 graft on the contra-lateral flank on day 42. (n=12 for rat IgG and anti-CD8β mAb treated groups; n=6 for 2nd K14E7 graft group).
(*p<0.0001; #p=0.0001; ns = not statistically different, log-rank test).
Host NKT cell-mediated suppression of adaptive immunity to epithelial antigens is not restricted to HPV16-E7 protein
To determine whether NKT cell mediated regulation of the T effector functions necessary for skin graft rejection was restricted to grafts expressing the E7 protein of HPV16, or was a more general finding, we grafted skin from a K14 transgenic mouse expressing herpes simplex type 2 (HSV2) glycoprotein B in K14+ keratinocytes (K14gB), onto both WT and CD1dKO recipients. K14gB grafts were slowly rejected spontaneously by immunocompetent WT recipients (63% rejection; median survival = 90 days). Rejection of K14gB grafts was significantly accelerated by the absence of NKT cells in CD1dKO recipients (88% rejection; median survival = 46 days) (Fig. 2). Therefore, host NKT cells can regulate immune effector functions directed at multiple viral antigens expressed in skin.
Figure 2. NKT cells suppress rejection of grafts expressing K14-restricted HSV glycoprotein B.

Survival of K14 promoter-driven glycoprotein B (K14gB) grafts on WT C57BL/6 and CD1dKO recipients. C57BL/6 recipients – 63% rejection; median survival of 90 days. CD1dKO recipients – 88% rejection; median survival of 46 days. (*p=0.03; n=8 per group).
Host NKT cell-mediated regulation of K14E7 skin graft rejection is independent of IFN-γ, IL-10 or IL-17 cytokine production and does not require Foxp3+ Tregs
We previously observed that inhibition of K14E7 graft rejection by donor-derived, skin resident NKT cells requires IFN-γ (3). To determine whether immune suppression mediated by systemic host-derived NKT cells is dependent on IFN-γ production by NKT cells, we repopulated NKT cell-deficient Jα18KO mice with IFN-γ+/+ or IFN-γKO NKT cells prior to grafting. NKT cell reconstitution led to complete inhibition of graft rejection in Jα18KO mice, regardless of their capacity for IFN-γ production (Fig. 3A). This observation was extended to other candidate cytokines, including IL-10 and IL-17, as transferred IL-10KO and IL-17KO NKT cells were equally capable of inhibiting graft rejection (Fig. 3A). Therefore, in contrast to suppression of CD8 T effector cell-mediated rejection of skin grafts by skin resident NKT cells, which requires IFN-γ, suppression of the generation of effector function for graft rejection mediated by host resident NKT cells is independent of their ability to produce IFN-γ, IL-10 or IL-17 cytokine production.
Figure 3. NKT cell-mediated suppression is independent of IFN-γ, IL-10 and IL-17 production and Foxp3+ Tregs.
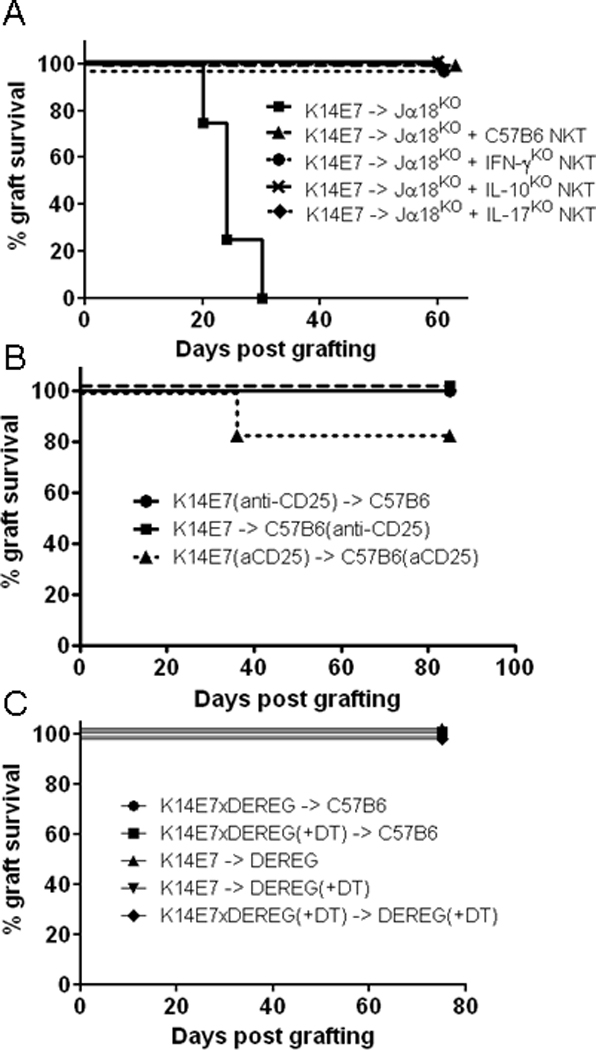
(A) Survival curves of K14E7 grafts on Jα18KO recipients that were reconstituted 3 days prior to grafting by transfer of 2×105 purified NKT cells isolated from either WT C57BL/6, IFN-γKO, IL-10KO or IL-17KO mice (n=4 mice per group).
(B) Survival of K14E7 grafts following depletion of CD25+ cells in donor and/or recipient WT mice, by systemic i.p. administration of anti-mouse CD25 mAb (clone-PC61) (n=6 mice per group).
(C)Survival of skin grafts from K14E7 and K14E7×DEREG mice transplanted onto WT or DEREG recipients, with or without prior treatment of either donor and/or recipient mice with diphtheria toxic (DT) treatments for depletion of Foxp3+ Treg cells (n=6 mice per group).
Host NKT cells could be indirectly mediating suppression of HPV16-E7 graft rejection by CD8 T cells, by enhancing the function of other suppressive or regulatory cell populations in the skin. We therefore investigated the requirement for CD4+CD25+Foxp3+ Tregs in NKT cell-mediated regulation of skin graft rejection. Depletion of Tregs from either donor K14E7 skin or WT recipients, or both, using an anti-CD25 monoclonal antibody did not enable rejection of K14E7 grafts (Fig. 3B). To confirm this finding, we grafted skin from diphtheria toxin (DT) treated DEREG mice, allowing specific depletion of Foxp3+ Tregs without compromising effector cell function. Depletion of Foxp3+ cells from the graft donor or from both donor and host did not enable K14E7 graft rejection (Fig. 3C), confirming that interactions with traditional Foxp3+ Tregs are not critical for host NKT cell-mediated regulation of effector T cell functions necessary for graft rejection.
Host NKT cells suppress the generation of antigen-specific CD8 effector T cells
Rejection of K14E7 skin grafts in NKT cell-deficient recipients is mediated by effector CD8 T cells (Fig. 1). Therefore, we addressed whether NKT cells suppressed host immunity to HPV16-E7 by inhibiting the priming and activation of E7-specific CD8 T cells. Subcutaneous immunization with E7 protein generated less IFN-γ-producing, E7-specific CD8 T cells in skin-DLN in WT mice than in CD1dKO or Jα18KO mice (Fig. 4A). We were unable to detect Ag-specific IFN-γ production by DLN CD8 T cells following grafting with K14E7 skin in mice with or without NKT cells, likely a result of insufficient sensitivity in the assay system. To assess whether NKT cells mediated suppression of Ag-specific CD8 T cell proliferation following grafting, we transferred CFSE-labeled E7-specific CD8 T cells isolated from E7TCRβ transgenic mice to animals receiving K14E7 grafts. In vivo proliferation of transferred CD8 T cells, measured 7 days after grafting, was significantly greater in CD1dKO and Jα18KO recipients than in immune competent WT recipients (Fig. 4B). Taken together, these data show that host type I NKT cells hinder the generation and expansion of HPV16-E7 specific CD8 T cells in skin-DLN when presented with cognate antigen from grafted skin.
Figure 4. Type I NKT cells inhibit the generation of E7-specific CD8 T cells in skin-DLN.

(A) Ag-specific IFN-γ production by CD8 T cells isolated from skin-DLN of WT, CD1dKO and Jα18KO mice 7 days after primary stimulation with either subcutaneous GF001 peptide immunization (E7 Imm) or grafting with K14E7 skin (E7 graft). IFN-γ-producing cells were measured by ELISPOT following overnight re-stimulation with GF001 peptide and IL-2. Δ IFN-γ is the corrected spot forming units (SFU) value after subtracting counts obtained from control wells containing no peptide stimulation. Each dot represents the mean value from triplicate wells from one mouse. (ND = no spots detected) (***p<0.0001, unpaired t test).
(B) In vivo proliferation of CFSE-labeled, CD45.1 congenic E7TCR cells 7 days after transfer into WT, CD1dKO or Jα18KO recipients, which were grafted with K14E7 one day after cell transfer. CFSE dilution within the gated CD45.1+/CD3+/CD8+ cell population was converted into indices of proliferation by ModFit software (mean + SEM; n=4) (*p=0.03, Mann-Whitney test).
Results from A and B are representative of two independent experiments.
Dendritic cells isolated from skin-DLN of NKT cell-deficient mice induce greater Ag-specific CD8 T cell activity
To explain the reduction in proliferation of E7-specific T cells in the draining nodes of mice harboring E7 grafts, we hypothesized that NKT cells might inhibit DC maturation and T cell priming activity. To determine the effect of NKT cells on DC function, and to establish whether any effect was dependent on DC activation by grafting, we isolated CD11c+ cells from skin-DLN of grafted or non-grafted WT and CD1dKO mice and assessed their ability to present peptide to stimulate E7TCRβ transgenic CD8 T cells in vitro. Following 4 days co-culture, E7 peptide-pulsed DC from the DLN of mice that had received a graft possessed a substantially greater capacity to stimulate E7-specific IFN-γ production from CD8 T cells, compared to DC isolated from the skin DLN of mice that had not been grafted. In each case, the stimulation was substantially enhanced if the donor was NKT cell deficient (Fig. 5A). This confirms that DC require activation to effectively present antigen and that NKT cells possess the ability to suppress T cell proliferation whether the DC is activated or not. We next investigated whether the decrease in T cell stimulatory capacity of DC that were conditioned in the presence of NKT cells during grafting was due to inadequate DC expression of co-stimulatory molecules. CD86 expression was increased on DC in the DLN of CD1dKO mice grafted with K14E7 skin, and higher when compared to skin grafted WT mice (Fig. 5B). In contrast, CD40 and CD80 expression on DC remained constant when comparing ungrafted and grafted WT and CD1dKO mice. Therefore, abrogated stimulation of CD8 T cells by DC in WT mice does not seem likely to be attributable to NKT cell-mediated suppression of expression of the key DC co-stimulatory molecules. Notably, at the time where DC were isolated from the skin-DLN of grafted mice, no evidence of NKT cell activation was observed in this organ. Expression of CD69 (Fig. 6A) and CD25 (Fig. 6B) on NKT cells in WT mice, as a measure of activation in response to grafting, was not increased following grafting with either control C57BL/6 skin or K14E7 skin. Thus, intrinsic or default function of skin resident NKT cells appears to be regulatory.
Figure 5. Dendritic cells licensed in the presence of NKT cells have suppressed capacity for CD8 T cell priming.

(A) IFN-γ production by CD8+ T cells from E7TCR transgenic mice, measured by ELISA, following 4 days in vitro co-culture with GF001 peptide-pulsed CD11c+ DC that were isolated from skin-DLN of grafted or non-grafted WT C57BL/6 and CD1dKO recipients. M1 and M2 designates mouse 1 and 2 in each group, with each bar representing mean + SEM from triplicate wells.
(B) Mean expression of co-stimulation molecules – CD40 (left), CD80 (middle), CD86 (right) on the surface of CD11c+MHCIIhi DC isolated from skin-DLN of WT and CD1dKO mice, non-grafted or 5 days after grafting with K14E7 skin. (mean + SEM; n=4) (*p<0.05; ns = not statistically different, Mann-Whitney test). Similar results were obtained in 3 independent experiments.
Figure 6. NKT cell activation is not altered following skin grafting.

Expression of CD69 (A) and CD25 (B) on TCRβ+CD1d-tetramer+ NKT cells isolated from skin-DLN of non-grafted WT C57BL/6 mice, or C57BL/6 and K14E7 grafted WT mice on day 7 post-grafting. Overlay histograms show representative expression of CD69 and CD25 on NKT cells from the indicated mice. Expression of CD69 and CD25 on individual mice is displayed as both percentages (upper graphs) and mean fluorescence intensity - MFI (lower graphs). ns = not statistically different, unpaired t test.
Pro-inflammatory cytokine production and Ag-specific cytotoxic activity of CD8 T cells is suppressed following K14E7 skin grafting in the presence of NKT cells
We hypothesized from the observed decrease in generation of IFN-γ-producing Ag-specific CD8 T cells in K14E7 grafted WT mice, when compared to NKT-deficient mice (Fig. 4A) that NKT cells might be inhibiting graft rejection by negatively regulating the acquisition of effector functions by CD8 T effector precursor cells. CD8 T cells isolated from skin-DLN of ungrafted WT and CD1dKO mice possessed equal capacity to secrete pro-inflammatory cytokines, including IFN-γ, TNF-α and IL-2, upon non-specific mitogen stimulation in vitro (Fig. 7A). However, 4 days after grafting with K14E7 skin, CD8 T cells from WT recipients showed significantly reduced capacity for cytokine production when compared with CD8 T cells isolated from NKT cell-deficient CD1dKO recipients (Fig. 7A). Intracellular cytokine staining was performed to determine whether the decrease in IFN-γ secretion by CD8 T cells from the DLN of grafted WT mice, shown by ELISA, was due to a decrease in production by individual CD8 T cells or a reduction in the total number of IFN-γ-producing cells. Expression of IFN-γ in individual CD8 T cells was comparable before and after grafting in WT mice (Fig. 7B), however the percentage of CD8 T cells with IFN-γ-producing capacity was reduced (Fig. 7C), consistent with the impaired proliferation of these cells in NKT populated animals (Fig. 4). Minimal cytokine production of IL-4, IL-5 and IL-10 by CD8 T cells was detected over the same period (data not shown), and so it is unlikely that the CD8 T cells were diverting from a Tc1-like to Tc2-like cytokine profile. Therefore, host NKT cells present during Ag-specific CD8 T cell differentiation in the DLN following K14E7 skin grafting reduce their proliferation and differentiation towards effector cells.
Figure 7. Capacity for CD8 T cell pro-inflammatory cytokine production and cytotoxic activity in skin-DLN is reduced in the presence of NKT cells.

(A) Secretion of IFN-γ (top graph), IL-2 (middle graph) and TNF-α (bottom graph) by CD8 T cells isolated from skin-DLN of K14E7 grafted or non-grafted WT and CD1dKO mice, measured by cytometric bead array following a 4 hour period of PMA/Ionomycin re-stimulation in vitro. Each dot represents the mean value from triplicate wells from one mouse. (#p=0.01; *p=0.001; ^p=0.0005; ns = not statistically different, unpaired t test).
(B) Representative flow cytometry dot plots showing intracellular IFN-γ staining in CD8 T cells from non-grafted and grafted WT mice, following PMA/Ionomycin stimulation. Mean fluorescence intensity (MFI) values shown are of IFN-γ expression from the gated population.
(C) Percentage of CD8 T cells isolated from skin-DLN of K14E7 grafted or non-grafted WT mice producing IFN-γ following PMA/Ionomycin stimulation. Each dot represents intracellular staining from one mouse. Data was pooled from two independent experiments (***p=0.0001, unpaired t test).
(D) Percentage lysis of GF001-pulsed EL4 target cells after 24hrs co-culture with CD8 T cells (effectors) at 3 effector/target (E:T) ratios, measured in an in vitro chromium-release assay. CD8 effector T cells were generated in WT and CD1dKO mice by s.c. immunization with GF001+KLH+QuilA and isolated from spleen and inguinal LN. (mean + SEM; n=6) (*p<0.05, **p<0.005, ns = not statistically different, unpaired t test).
To determine if CD8 T cells generated in the presence of NKT cells demonstrated decreased cytotoxic potential, we measured cytotoxic activity of CD8 T cells induced in WT and CD1dKO mice following immunization. WT and CD1dKO mice immunized with E7 peptide and adjuvant, after a period of antigen re-stimulation in vitro, demonstrated CD8 T cell-mediated and Ag-specific killing of peptide-pulsed targets, and CD8 T cells from CD1dKO animals displayed significantly enhanced killing (Fig. 7D). Enhanced cytotoxicity was not dependent on increased production of cytotoxic granules, as intracellular stores of perforin and granzyme B were comparable between activated CD8 T cells from WT and CD1dKO mice (data not shown). NKT cells activated by grafting or local inflammatory stimuli associated with immunization can therefore prevent the acquisition of full cytotoxic functionality of Ag-specific CD8 T cells exposed to HPV16-E7 antigen cross-presented in the skin DLN, and this is achieved by NKT cell-mediated inhibition of the capacity of APC to promote division and differentiation of CD8 effector T cells from Ag-specific precursors.
DISCUSSION
In a recent study we reported a critical role for skin resident NKT cells and IFN-γ in establishing a local immune suppressive environment in skin grafts. Immune competent host animals simultaneously challenged with an NKT populated and an adjacent NKT deplete graft, each expressing the E7 protein of HPV16 from a keratin 14 promoter, rejected only the NKT deplete graft (3). These data demonstrate that immune effector cells, generated by cross-priming of graft-derived antigen in the DLN, cannot acquire effector functions necessary for graft rejection in the presence of skin donor derived NKT cells. The present study was undertaken to investigate the effects of systemic NKT cells on the generation and differentiation of Ag-specific CD8 effector T cells following cross-priming from antigen expressed in grafted skin. We demonstrate here that systemically derived type I NKT cells also inhibit generation of CD8 effector T cells capable of mediating rejection of grafts. We show further that this is attributable to suppression, by NKT cells in the DLN, of generation and differentiation of Ag-specific CD8 T cells in response to antigen cross-presented by DLN APC.
NKT cells in K14E7 transgenic skin, although sufficient to prevent local CD8 T cell mediated graft rejection in a graft primed animal (3), did not inhibit Ag-specific priming and hence rejection of K14E7 grafts by CD1dKO or Jα18KO recipients, indicating a further distinct role for host NKT cells in regulation of local priming to cross-presented antigen. Reconstitution of Jα18KO recipients with NKT cells was sufficient to restore immune suppression supporting a direct contribution of type I, invariant NKT cells. This observation supports findings in a skin transplant model by Oh et al., 2005 who reported that, in CD1dKO mice transplanted with sex-mismatched grafts, rejection occurred in an accelerated rate compared to WT recipients (14). We previously demonstrated that donor- and then host-derived NKT cells and IFN-γ locally suppress CD8 T cell-mediated graft rejection (3). Matsumoto et al., 2004 observed that immune suppression to HPV16-E7 can be overcome by transferring an enriched population of E7-specific CD8 T cells, activated in situ by simultaneous immunization with E7 protein (11). In addition, we show in this study that CD8 T cells, although not an absolute requirement for rejection in some mice, maximize the rate and probability of rejection of K14E7 grafts in the absence of NKT cell-mediated suppression in CD1dKO mice (Fig. 1C). Based on this evidence for the requirement of CD8 T cells in rejection of K14E7 grafts, we hypothesized that host NKT cells may also be acting to inhibit graft rejection by suppressing the priming of E7-specific CD8 T cells in the skin-DLN.
Generally, NKT cells have been associated with enhanced priming of Ag-specific T cells, particularly when NKT cells and CD8 T cells are simultaneously co-activated by the same dendritic cell (16, 17). In fact, we have shown in a skin grafting model incorporating OVA protein expressed by a transgene in epithelial keratinocytes (K5mOVA mouse), that NKT cells can enhance CD8 T cell-dependent rejection of OVA-expressing grafts (6). In contrast, here we have demonstrated that in response to skin-derived viral antigens that require additional CD4 T cell help for graft rejection, NKT cells inhibit efficient priming of endogenous Ag-specific T cells, leading to inhibition of graft rejection. Therefore, the resulting NKT effect on CD8 T cell priming (activating versus inhibitory) may be dependent on type of antigen and linked to the requirement for CD4 helper epitopes. In addition, skin grafts which express both E7 and OVA proteins were rejected rapidly in wildtype mice and time to rejection was not altered in NKT-deficient mice (Supplementary Figure 1), further suggesting that antigen ‘dominance’ and T cell requirements for graft rejection alter the role for NKT cell regulation.
These findings support observations in a recent report by Guillonneau et al., 2009 who showed diminished effector T cell generation to an influenza virus following NKT cell activation by α-GalCer (18). In our model, NKT cell-mediated suppression of effector CD8 T cell priming and graft rejection occurs in the absence of exogenous NKT-ligand, is not altered by local or systemic administration of α-GalCer (data not shown), and does not appear to be associated with expression of the conventional NKT cell activation markets. It likely reflects NKT cell-mediated modulation of DC in the skin-DLN. Activated CD11c+ DC isolated from skin-DLN of WT mice grafted with K14E7 skin were impaired in their capacity to induce E7-specific CD8 T cell responses in vitro, compared to DC isolated from NKT-deficient CD1dKO recipients. Apart from CD86, which was elevated in grafted CD1dKO recipients, the expression of co-stimulatory molecules was comparable on DC isolated from WT and CD1dKO mice, indicating that NKT cells were not suppressing DC activity by directly inhibiting co-stimulation. In NOD mice, activation of NKT cells by injection of α-GalCer was associated with the appearance of tolerogenic DC that prevented the development of diabetes by suppressing pathogenic T cell responses (4, 19). In addition, a study of human NKT cells revealed that APC instructed by NKT cells acquired a tolerogenic phenotype and silenced downstream T cell responses, including IFN-γ production and proliferation (20). NKT cell licensing of DC towards a suppressive or tolerogenic phenotype in skin-DLN of K14E7 grafted mice, in the absence of exogenous ligand stimulation, might be acting via similar mechanisms and this is currently under investigation.
CD8 T cells from skin-DLN of K14E7 grafted mice, generated in the presence of NKT cells, possessed reduced capacity to produce the pro-inflammatory cytokines IFN-γ, TNF-α and IL-2. Reduced secretion levels of these cytokines was not due to lower cytokine production on a per cell basis, as intracellular expression was comparable in CD8 T cells from NKT-sufficient and NKT-deficient mice. Instead, lower secretion was associated with a reduced proportion of CD8 T cells with the capacity for pro-inflammatory cytokine production. Absence of anti-inflammatory cytokine secretion (IL-4, IL-5, IL-10) indicates that these NKT cell-instructed CD8 T cells were not being diverted to a Tc2-like phenotype. It is possible that differentiation of CD8 T cells into pro-inflammatory effector cells, in response to grafting stimulus, is being suppressed by NKT cells. Inhibition of CD4 T cells differentiation by NKT cells has been reported previously in a type 1 diabetes mouse model (21, 22). Vα14 transgenic NOD mice, containing enhanced numbers of NKT cells, are resistant to development of diabetes. This was attributed to NKT cell-mediated inhibition of the expansion and differentiation of pathogenic anti-β cell CD4 T cells (21). In addition, or alternatively, to a defect in differentiation, CD8 T cells might be rendered anergic or dysfunctional upon inhibitory signals received from NKT cells. This may explain why cytokine production by CD8 T cells from grafted WT mice was refractory even to in vitro mitogen stimulation. In association with a defect in pro-inflammatory cytokine production, Ag-specific CD8 T cells generated to E7 protein in the presence of NKT cells also possessed impaired cytotoxic activity.
The mechanism(s) by which NKT cells inhibit DC-mediated priming of E7-specific CD8 T cells and differentiation into cytotoxic effector cells is currently unknown. We have shown that NKT cell-mediated suppression in skin-DLN is independent of Foxp3+ Treg cells and does not involve production of IFN-γ, IL-10 or IL-17 from NKT cells in contrast to the local suppression of effector T cell function in skin which depends on IFN-γ (3). Alternatively, there may be redundancy in the NKT cell regulatory mechanisms contributing to APC signaling. Other cytokines produced by NKT cells may be implicated in the immunosuppression, for example IL-4, IL-13 and TGF-β (23, 24), however cell contacts are likely to be critical (22). Future work will address the relative contribution of cell contacts versus soluble mediators in NKT cell-mediated immune suppression.
Collectively, results from this study along with our recently published work (3) demonstrate that NKT cells possess diverse immune suppressive functions in response to skin-derived viral antigen. They infiltrate into inflamed skin to inhibit CD8 effector T cell function locally and also suppress the generation of Ag-specific CD8 T cells in the associated skin-DLN. Therefore, NKT cells are capable of suppressing CD8 T cell immunity both in the priming phase in secondary lymphoid organs and the effector response at the local site. NKT cell-mediated suppression of rejection of skin grafts expressing viral HSV glycoprotein B in epithelial cells indicates that NKT cell inhibition of adaptive immunity to skin-derived viral antigens is not restricted to HPV oncoproteins and may be more generally associated with viral proteins expressed in epithelium. Inhibition of virus-specific CD8 T cell priming and effector function by NKT cells may be a potential means of virus evasion of the immune system leading to chronic viral persistence and development of pre-cancers and malignancy. Future studies investigating the molecular signals released by epithelial cells harboring viral proteins that induce immunosuppressive activity by NKT cells will be imperative in understanding the host/pathogen interactions leading to immune escape, associated chronic viral infection and cancer development.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Kon Kyparissoudis for generating the α-GalCer-loaded mouse CD1d tetramer, Shanu Sinha for genotyping of mice and the staff of the Biological Research Facility at PAH for excellent technical assistance and animal care.
Grant Support
This work was supported by program grant 080214 from the National Health and Medical Research Council of Australia, NCI grant RFA-CA-08-018, and funding from the Australian Cancer Research Foundation and the Lions Medical Research Foundation. S.M. was recipient of Balzan Foundation fellowship, and I.H.F. was recipient of Queensland Government Premiers Fellowship.
Abbreviations
- HPV
Human papillomavirus
- DLN
draining lymph nodes
- DT
diphtheria toxin
- WT
wildtype
- DC
dendritic cell(s)
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Bhat P, Mattarollo SR, Gosmann C, Frazer IH, Leggatt GR. Regulation of immune responses to HPV infection and during HPV-directed immunotherapy. Immunol Rev. 2011;239:85–98. doi: 10.1111/j.1600-065X.2010.00966.x. [DOI] [PubMed] [Google Scholar]
- 2.Frazer IH, Leggatt GR, Mattarollo SR. Prevention and treatment of papillomavirus-related cancers through immunization. Annu Rev Immunol. 2011;29:111–138. doi: 10.1146/annurev-immunol-031210-101308. [DOI] [PubMed] [Google Scholar]
- 3.Mattarollo SR, Rahimpour A, Choyce A, Godfrey DI, Leggatt GR, Frazer IH. Invariant NKT cells in hyperplastic skin induce a local immune suppressive environment by IFN-gamma production. J Immunol. 2010;184:1242–1250. doi: 10.4049/jimmunol.0902191. [DOI] [PubMed] [Google Scholar]
- 4.Chen YG, Choisy-Rossi CM, Holl TM, Chapman HD, Besra GS, Porcelli SA, Shaffer DJ, Roopenian D, Wilson SB, Serreze DV. Activated NKT Cells Inhibit Autoimmune Diabetes through Tolerogenic Recruitment of Dendritic Cells to Pancreatic Lymph Nodes. J Immunol. 2005;174:1196–1204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 5.Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. 2009;182:1818–1828. doi: 10.4049/jimmunol.0802430. [DOI] [PubMed] [Google Scholar]
- 6.Mattarollo SR, Yong M, Tan L, Frazer IH, Leggatt GR. Secretion of IFN-gamma but not IL-17 by CD1d-restricted NKT cells enhances rejection of skin grafts expressing epithelial cell-derived antigen. J Immunol. 2010;184:5663–5669. doi: 10.4049/jimmunol.0903730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayan S, Choyce A, Linedale R, Saunders NA, Dahler A, Chan E, Fernando GJ, Frazer IH, Leggatt GR. Epithelial expression of human papillomavirus type 16 E7 protein results in peripheral CD8 T-cell suppression mediated by CD4+CD25+ T cells. Eur J Immunol. 2009;39:481–490. doi: 10.1002/eji.200838527. [DOI] [PubMed] [Google Scholar]
- 8.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, Koezuka Y, Kronenberg M. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. 2000;192:741–754. doi: 10.1084/jem.192.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunn LA, Evander M, Tindle RW, Bulloch AL, De Kluyver RL, Fernando GJ, Lambert PF, Frazer IH. Presentation of the HPV16E7 protein by skin grafts is insufficient to allow graft rejection in an E7-primed animal. Virology. 1997;235:94–103. doi: 10.1006/viro.1997.8650. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto K, Leggatt GR, Zhong J, Liu X, de Kluyver RL, Peters T, Fernando GJ, Liem A, Lambert PF, Frazer IH. Impaired antigen presentation and effectiveness of combined active/passive immunotherapy for epithelial tumors. J Natl Cancer Inst. 2004;96:1611–1619. doi: 10.1093/jnci/djh301. [DOI] [PubMed] [Google Scholar]
- 12.Narayan S, Choyce A, Fernando GJ, Leggatt GR. Secondary immunisation with high-dose heterologous peptide leads to CD8 T cell populations with reduced functional avidity. Eur J Immunol. 2007;37:406–415. doi: 10.1002/eji.200535688. [DOI] [PubMed] [Google Scholar]
- 13.Leggatt GR, Narayan S, Fernando GJ, Frazer IH. Changes to peptide structure, not concentration, contribute to expansion of the lowest avidity cytotoxic T lymphocytes. J Leukoc Biol. 2004;76:787–795. doi: 10.1189/jlb.0104026. [DOI] [PubMed] [Google Scholar]
- 14.Oh K, Kim S, Park SH, Gu H, Roopenian D, Chung DH, Kim YS, Lee DS. Direct regulatory role of NKT cells in allogeneic graft survival is dependent on the quantitative strength of antigenicity. J Immunol. 2005;174:2030–2036. doi: 10.4049/jimmunol.174.4.2030. [DOI] [PubMed] [Google Scholar]
- 15.Broom JK, Lew AM, Azukizawa H, Kenna TJ, Leggatt GR, Frazer IH. Antigen-specific CD4 cells assist CD8 T-effector cells in eliminating keratinocytes. J Invest Dermatol. 2010;130:1581–1589. doi: 10.1038/jid.2010.17. [DOI] [PubMed] [Google Scholar]
- 16.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 18.Guillonneau C, Mintern JD, Hubert FX, Hurt AC, Besra GS, Porcelli S, Barr IG, Doherty PC, Godfrey DI, Turner SJ. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc Natl Acad Sci U S A. 2009;106:3330–3335. doi: 10.1073/pnas.0813309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naumov YN, Bahjat KS, Gausling R, Abraham R, Exley MA, Koezuka Y, Balk SB, Strominger JL, Clare-Salzer M, Wilson SB. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci U S A. 2001;98:13838–13843. doi: 10.1073/pnas.251531798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hegde S, Jankowska-Gan E, Roenneburg DA, Torrealba J, Burlingham WJ, Gumperz JE. Human NKT cells promote monocyte differentiation into suppressive myeloid antigen-presenting cells. J Leukoc Biol. 2009;86:757–768. doi: 10.1189/jlb.0209059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beaudoin L, Laloux V, Novak J, Lucas B, Lehuen A. NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity. 2002;17:725–736. doi: 10.1016/s1074-7613(02)00473-9. [DOI] [PubMed] [Google Scholar]
- 22.Novak J, Beaudoin L, Griseri T, Lehuen A. Inhibition of T cell differentiation into effectors by NKT cells requires cell contacts. J Immunol. 2005;174:1954–1961. doi: 10.4049/jimmunol.174.4.1954. [DOI] [PubMed] [Google Scholar]
- 23.Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, Carbone DP, Paul WE, Berzofsky JA. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol. 2000;1:515–520. doi: 10.1038/82771. [DOI] [PubMed] [Google Scholar]
- 24.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.