

Abstract
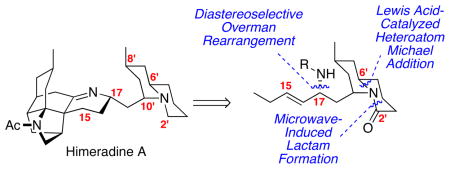
The synthesis of the C15-C17/N1′-C11′ quinolizidine portion of himeradine A is disclosed. An intramolecular, heteroatom Michael addition was employed to established the C6′ stereogenic center with high diastereoselectivity. The quinolizidine ring was constructed using microwave-induced cyclization at the N1′-C2′ position. The C17 stereogenic center was introduced through a diastereoselective Overman rearrangement.
In 2003, Kobayashi and co-workers reported the isolation and structural determination of the alkaloid himeradine A (1) from the club moss Lycopodium chinense in small amounts (2 mg, 0.001%, Figure 1).1 Polycycle 1 was assigned based on extensive 2D NMR techniques (COSY, HOHAHA, HMQC, HMBC and HMQC-HOHAHA); however, they were unable to establish the correlation between the eastern and western halves or the absolute stereochemistry of the molecule. Alkaloid 1 showed modest cytotoxicity against murine lymphoma L1210 cells (IC50 10 μg/mL), but a thorough biological screening of the compound has not been reported. Other members of the lycopodium family have shown intriguing biological activity.2
Figure 1.

Himeradine A and Related Lycopodium Alkaloids.
The heptacyclic himeradine A (1) possesses a challenging array of structural features: ten stereogenic centers including one all-carbon quaternary center, a densely packed pentacyclic western half, the C8′-C10′-trans-disubstituted quinolizidine and the challenging sigma linkage at C11′ (Figure 1). The carbon framework of the western portion is similar to that found in lycopodine (2);3 however, the additional challenge of the C3-C14 linkage increases the complexity of any synthetic endeavor. A related structural scaffold can be found in fastigiatine (3).4 The relative stereochemistries of the eastern quinolizidine ring systems are distinct from typical stereochemical combinations found in related natural products such as cermizine D (4).5 In particular, the axially disposed C10′ moiety creates added synthetic challenges. To date, no synthetic endeavors have been published towards himeradine A.6 Herein, we disclose the synthesis of the eastern portion of himeradine A and the development of a viable model coupling strategy for incorporation of the C15-C17 carbons including the C17 stereogenic center.
Our retrosynthetic strategy is shown in Scheme 1. For the polycyclic western portion of himeradine, we intend to adapt our previously developed route to lycopodine for the construction of the carbon framework.3b, c Compound 5 will be formed via a diastereoselective Overman rearrangement7 of the trichloroimidate derived from alcohol 6. The required C15 stereogenic center will be introduced through an organozinc addition to an α,β-unsaturated aldehyde. This aldehyde will be accessible in turn from a Julia-Kocienski-Blakemore olefination8 with the known 1-phenyl-1H-tetrazol-5-yl (PT) sulfone 89 and aldehyde 9 followed by acid-catalyzed deacetalization with in situ alkene migration. The quinolizidine ring system 9 will be formed via N1′-C2′ lactamization strategy. The piperidine ring system 10 will be constructed from a substrate-controlled, intramolecular heteroatom Michael addition of enal 11.
Scheme 1.

Retrosynthesis.
Synthesis of cyclization precursor 11 is shown in Scheme 2. The key C10′ stereocenter was constructed from a Grignard addition of organomagnesium species 1210 with the known Ellman imine 1311 in good levels of diastereoselectivity and chemical yield. Removal of the sulfoxamine under acid-catalyzed conditions followed by Cbz protection revealed the carbamate 15. Cross metathesis of alkene 15 with crotonaldehyde (16) cleanly provided the enal 11. We have previously shown that cross metathesis of monosubstituted alkenes with α,β-unsaturated carbonyl compounds proceeds in higher chemical yield when the unsaturated carbonyl compound contains a β-methyl substitution (e.g. 16).3b–c,12
Scheme 2.

Synthesis of Cyclization Precursor.
The key Lewis acid-catalyzed intramolecular, heteroatom Michael addition is shown in Scheme 3. We had hypothesized that the C8′ and C10′ stereogenic centers would work in concert to direct to facial attack on an α,β-unsaturated oxonium ion, as shown in possible transition state model 17. The planar carbamate at C10′ should force the CH2OBn moiety into a pseudoaxial position in the transition state in order to minimize steric repulsions with the Cbz moiety.13 We were pleased to see that our hypothesis proved accurate as aldehyde 10 was isolated as the sole diastereomer (>20:1 dr) in excellent chemical yield (98%).
Scheme 3.

Intramolecular Heteroatom Michael Addition.
The next hurdle was the construction of the quinolizodine ring system (Scheme 4). Wittig olefination of aldehyde 10 provided the α,β-unsaturated ester 21 in excellent yield. We had anticipated that hydrogenation of 21 would not only reduce the alkene, but also induce Cbz deprotection and debenzylation at C11′. To our surprise, only two of the three predicted events occurred – delivering the benzyl ether 22 as the sole product in high yield. Lactam formation of 22 (or its corresponding carboxylic acid) under a variety of conditions proved unusually challenging. Fortunately, we ultimately identified that thermolysis under microwave irradiation cleanly induced lactam formation in high yield. The synthetic challenge with this bond formation may be due the required placement of the C10′ substituent in the axial orientation to facilitate C2′-N1′ bond formation. Interestingly, heating of amine 22 in refluxing xylenes (approximately 140 °C) using an oil bath provided the product 23 in significantly lower yield (40% conversion after 48 h). Microwave irradiation’s ability to provide purely rotational energy transfer14 may facilitate this transformation more efficiently than standard thermal conditions – thereby inducing rotation of the sidearm into close proximity for the 2° amine to attack the C2′ ester. Next, hydrogenation of the benzyl ether 23 under essentially the same conditions as employed previously with compound 21 provided the free alcohol 24. One possible explanation for the divergent reactivity (compounds 21 vs. 23) may be the presence of the free amine in compound 22, which may poison the catalyst. The stereochemistry of the newly formed quinolizidine ring system was conclusively established by x-ray crystallographic analysis on the thiolactam 25.15 Oxidation of 1° alcohol 24 under buffered Dess-Martin conditions revealed the aldehyde 9. We had feared the aldehyde 9 might be prone to epimerization; however, 9 appeared to be configurationally stable at C10′ – likely due to the presence of the lactam carbonyl again disfavoring placement of the C10′ substituent in an equatorial position due to A1,3 strain.13
Scheme 4.

Synthesis of the Quinolizidine Core.
With the aldehyde 9 in hand, we shifted our attention to the incorporation of the C15-C17 carbon atoms and the C17 stereogenic center (Scheme 5). Olefination of aldehyde 9 with the known PT sulfone 89 gave the desired β,γ-unsaturated acetal in good yield with an inconsequential 14:1 E:Z selectivity. Treatment of acetal 7 under aqueous acidic conditions induced both acetal deprotection and alkene isomerization to cleanly provide the desired α,β-unsaturated aldehyde 26 with excellent E:Z selectivity (>20:1). N-Methyl diphenylprolinol catalyzed addition of Et2Zn to the aldehyde 26 revealed the allylic alcohol 27 in 10:1 dr.16,17 Formation of the trichloroimidate was accomplished using DBU as the base followed by thermolysis at 90°C in the presence of K2CO3 to cleanly generate the rearranged amide 5. The presence of the carbonate base appeared to prevent decomposition under the reaction conditions.18
Scheme 5.

Incorporation of the C15-C17 Portion.
In summary, the synthesis of the C15-C17/N1′-C11′ quinolizidine core of himeradine A has been accomplished. Key steps in the synthetic sequence include a diastereoselective Overman rearrangement, a substrate-controlled, intramolecular heteroatom Michael addition and a microwave-induced lactam formation. Further studies towards the total synthesis of himeradine A and other related lycopodium alkaloids will be reported in due course.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (NIH) (GM63723). The National Science Foundation (CHE-0722319) and the Murdock Charitable Trust (2005265) are acknowledged for their support of the NMR facility. We thank Dr. Lev N. Zakharov (OSU and University of Oregon) for X-ray crystallographic analysis of compound 25, Professor Max Deinzer and Dr. Jeff Morré (OSU) for mass spectra data. Finally, the authors are grateful to Professor James D. White (OSU) and Dr. Roger Hanselmann (Rib-X Pharmaceuticals) for their helpful discussions.
Footnotes
Supporting Information Available Complete experimental procedures are provided, including 1H and 13C spectra, of all new compounds. X-ray crystallographic data (CIF) for compound 25 is also provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Morita H, Hirasawa Y, Kobayashi J. J Org Chem. 2003;68:4563–4566. doi: 10.1021/jo034294t. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ma X, Gang DR. Nat Prod Rep. 2004;21:752–772. doi: 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi J, Morita H. Alkaloids. 2005;61:1–57. doi: 10.1016/s1099-4831(05)61001-2. [DOI] [PubMed] [Google Scholar]; (c) Hirasawa Y, Kobayashi J, Morita H. Heterocycles. 2009;77:679–729. [Google Scholar]
- 3.Structure Determination: Ayer WA, Iverach GG. Tetrahedron Lett. 1962;3:87–92. doi: 10.1016/s0040-4039(00)72888-7.Enantioselective total syntheses: Yang H, Carter RG, Zakharov LN. J Am Chem Soc. 2008;130:9238–9239. doi: 10.1021/ja803613w.Yang H, Carter RG. J Org Chem. 2010;75:4929–4938. doi: 10.1021/jo100916x.Racemic Total Syntheses: Stork G, Kretchmer RA, Schlessinger RH. J Am Chem Soc. 1968;90:1647–1648. doi: 10.1021/ja01008a042.Ayer WA, Bowman WR, Joseph TC, Smith P. J Am Chem Soc. 1968;90:1648–1650. doi: 10.1021/ja01008a043.Kim S, Bando Y, Horii Z. Tetrahedron Lett. 1978:2293–2294.Heathcock CH, Kleinman EF, Binkley ES. J Am Chem Soc. 1982;104:1054–1068.Schumann D, Mueller HJ, Naumann A. Liebigs Ann Chem. 1982:1700–1705.Kraus GA, Hon YS. Heterocycles. 1987;25:377–386.Grieco PA, Dai Y. J Am Chem Soc. 1998;120:5128–5129.Formal Syntheses: Padwa A, Brodney MA, Marino JP, Jr, Sheehan SM. J Org Chem. 1997;62:78–87. doi: 10.1021/jo960829p.Mori M, Hori K, Akashi M, Hori M, Sato Y, Nishida M. Angew Chem Int Ed. 1998;37:637–638. doi: 10.1002/(SICI)1521-3773(19980316)37:5<636::AID-ANIE636>3.0.CO;2-X.
- 4.Structure Determination: Gerard RV, MacLean DB, Fagianni R, Lock CJ. Can J Chem. 1986;64:943–949.Gerard RV, MacLean DB. Phytochemistry. 1986;25:1143–1150.Total Synthesis: Liau BB, Shair MD. J Am Chem Soc. 2010;132:9594–9595. doi: 10.1021/ja104575h.
- 5.Structure Determination: Morita H, Hiraswa Y, Shinzato T, Kobayashi J. Tetrahedron. 2004;60:7015–7023.Total Synthesis: Nishikawa Y, Kitajima M, Takayama H. Org Lett. 2008;10:1987–1990. doi: 10.1021/ol800574v.Nishikawa Y, Kitajima M, Kogure N, Takayama H. Tetrahedron. 2009;65:1608–1617.
- 6.(a) Collett ND, Carter RG. 241st National American Chemical Society Meeting; Anaheim, CA. March 27–31, 2011; p. ORGN-400. [Google Scholar]; (b) Saha M, Carter RG. 241st National American Chemical Society Meeting; Anaheim, CA. March 27–31, 2011; p. ORGN-263. [Google Scholar]
- 7.(a) Overman LEJ. Am Chem Soc. 1974;96:597–599. [Google Scholar]; (b) Overman LE, Carpenter NE. Org React. 2005;66:1–107. [Google Scholar]; (c) Kitamoto K, Sampei M, Nakayama Y, Sato T, Chida N. Org Lett. 2010;12:5756–5759. doi: 10.1021/ol1026602. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kocienski PJ, Bell A, Blakemore PR. Synlett. 2000:365–366. [Google Scholar]; (b) Blakemore PR. J Chem Soc, Perkin Trans. 2002;1:2563–2585. [Google Scholar]
- 9.Lear MJ, Hirama M. Tetrahedron Lett. 1999;40:4897–900. [Google Scholar]
- 10.Comins DL, Libby AH, Al-awar RS, Foti CJ. J Org Chem. 1999;64:2184–2185. [Google Scholar]
- 11.Tang TP, Volkman SK, Ellman JA. J Org Chem. 2001;66:8772–8778. doi: 10.1021/jo0156868. [DOI] [PubMed] [Google Scholar]
- 12.Carlson EK, Rathbone LK, Yang H, Collett ND, Carter RG. J Org Chem. 2008;73:5155–5158. doi: 10.1021/jo800749t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Watson PS, Jiang B, Scott B. Org Lett. 2000;2:3679–3681. doi: 10.1021/ol006589o. [DOI] [PubMed] [Google Scholar]; (b) Martin R, Murruzzu C, Pericas MA, Riera A. J Org Chem. 2005;70:2325–2328. doi: 10.1021/jo048172s. [DOI] [PubMed] [Google Scholar]; (c) Coombs TC, Lushington GH, Douglas J, Aubé J. Angew Chem Int Ed. 2011;50:2734–3737. doi: 10.1002/anie.201007133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loupy A, editor. Microwaves in Organic Synthesis. Wiley-VCH Verlag GmbH & Co; Weinheim: 2002. pp. 1–73. [Google Scholar]
- 15.See supporting information of crystallographic information.
- 16.(a) Soai K, Ookawa A, Kaba T, Ogawa K. J Am Chem Soc. 1987;109:7111–7115. [Google Scholar]; (b) Shibata T, Tabira H, Soai K. J Chem Soc Perkin Trans. 1998;2:177–178. [Google Scholar]; (c) Nishiyama T, Kusumoto Y, Okumura K, Hara K, Kusaba S, Hirata K, Kamiya Y, Isobe M, Nakano K, Kotsuki H, Ichikawa Y. Chem Eur J. 2010;16:600–610. doi: 10.1002/chem.200901745. [DOI] [PubMed] [Google Scholar]
- 17.Stereochemical assignment at C15 was established via advanced Mosher ester analysis as detailed in the supporting information. Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096.
- 18.Nishikawa T, Asai M, Ohyabu N, Isobe M. J Org Chem. 1998;63:188–192. doi: 10.1021/jo9713924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.