

Abstract
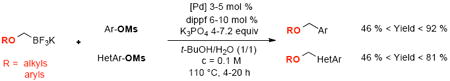
Cross-coupling of mesylated phenol derivatives with various potassium alkoxymethyltrifluoroborates has been achieved. The corresponding aryl- and heteroaryl alkoxymethyl compounds have been obtained with equal facility with both electron-rich and electron-poor substituents on the activated alcohol.
Ethers are encountered in a large variety of natural products including lipids, oxiranes, terpenoïds, and carbohydrate derivatives.1 They are known to be of interest in the pharmaceutical, agrochemical, and other industrial sectors. As an example, the quinolylmethoxyphenyl group is recognized as a pharmacophore in leukotriene inhibitors (Figure 1).2 Ethers are also important in organic synthesis as protecting groups for alcohols.3
Figure 1.
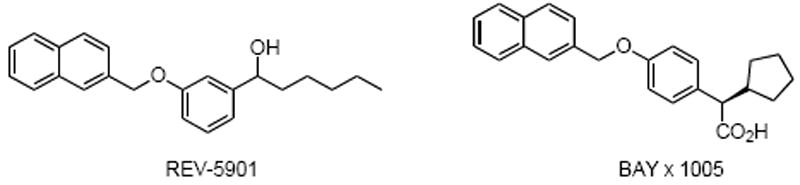
Leukotriene biosynthesis inhibitors.
Cross-coupling reactions of alkoxymethylmetallic species, embodying a dissonant reactivity pattern, would represent one of the more straightforward approaches for the synthesis of these alkyl ether compounds. Unfortunately, this approach is somewhat restricted owing to the dearth of readily available reagents and because Csp2-Csp3 cross-couplings are known to be quite challenging.4
Alkoxymethylstannanes were first reported as nucleophilic partners in the Stille reaction, but their use was limited because of their inherent toxicity and difficulties associated with their purification.5 Recently, more environmentally sound alkoxymethylboron species have been employed in the Suzuki–Miyaura reaction to afford the same scaffolds. For example, methoxymethylboronic acid has been used in large excess (2.3 equiv) in the presence of chloro-1,8-naphthyridine to obtain the corresponding cross-coupled compound with a moderate yield of 45%.6 Prior to that report, our laboratory had efficiently synthesized and cross-coupled air- and moisture-stable potassium alkoxymethyltrifluoroborates with aryl halides in high yields.7 That study was restricted to the use of aryl and heteroaryl chlorides and bromides, but afforded the desired target structures in moderate to good yields without employing a large excess of the organoboron reagent.7a Tanaka extended the scope of the reaction to an intramolecular Suzuki–Miyaura cross-coupling with sodium alkoxymethylaryltrifluoroborates bearing an attendant chloride.8 However, this method, carried out on a very small scale (0.10 mmol), suffered from a lack of generality, with yields ranging from 4 to 71%.
All of these previous reports employed halides as electrophilic partners, but to develop more environmentally sound processes, more attention is currently given to electrophilic alcohol derivatives.9 To our knowledge, no example employing these activating electrophilic partners has been reported in cross-coupling reactions with any alkoxymethylmetallic species. Among all the sulfonated activating groups available, we focused our work on the methanesulfonyl (mesyl) counterpart, which has the advantages of being stable, inexpensive, atom-economical, and easy to handle, even though it is known to be among the least reactive of these nucleofugal species.9i,k,m,n,p We disclose herein the first system for the Csp2-Csp3 Suzuki–Miyaura cross-coupling of aryl- and heteroaryl mesylates with various potassium alkoxymethyltrifluoroborates.
Previous reports from our laboratory underscore the fact that the use of potassium trifluoroborates in cross-coupling of C–O bonded species is facilitated by employing potassium phosphate as a base in a mixture of t-BuOH/H2O (1/1).9i,p Keeping these observations in mind, we screened different palladium and ligand sources with mesylated naphthalene 1 and potassium methoxymethyltrifluoroborate 2a, a partner known to be difficult,10 as model substrates (Table 1). Various alkyl or biaryl (Figure 2) monodentate phosphines (including Buchwald ligands) have been tested without success. RuPhos (II), which was the best ligand when halides were used as electrophilic partners,7a does not afford the desired compound 3a (Table 1, entry 4). After extensive screening, only one (bidentate) ferrocenic ligand, dippf (VII), led to the formation of 3a with a 61% GC yield (Table 1, entry 11). Different palladium sources were also tested, and PdCl2(COD) appeared to be the best catalyst precursor, with 3a obtained with complete conversion and 71% isolated yield after 4 h (Table 1, entry 13).
Table 1.
Optimization of the Conditions
|
| ||||
|---|---|---|---|---|
| entry | [Pd] | ligand | conversionb | yield (%)b |
| 1a | Pd(OAc)2 | Cy3PHBF4 | 23% | / |
| 2 | Pd(OAc)2 | t-Bu3PHBF4 | 76% | / |
| 3 | Pd(OAc)2 | XPhos I | 43% | / |
| 4a | Pd(OAc)2 | RuPhos II | 47% | / |
| 5a | Pd(OAc)2 | SPhos III | 37% | / |
| 6a | Pd(OAc)2 | DPEPhos IV | 39% | / |
| 7 | Pd(OAc)2 | Q-Phos V | 17% | / |
| 8 | Pd(OAc)2 | (S)-TolBinap VI | 23% | / |
| 9 | Pd(OAc)2 | dppm | 21% | <5 |
| 10 | Pd(OAc)2 | dppb | 18% | <5 |
| 11 | Pd(OAc)2 | dippf VII | 100% | 61 |
| 12 | PdCl2(dppe) | / | 5% | / |
| 13 | PdCl2(COD) | dippf | 100% | 75 (71)c |
Time = 1 h.
Relative GC yield using dodecane as the internal standard.
Isolated yield
Figure 2.
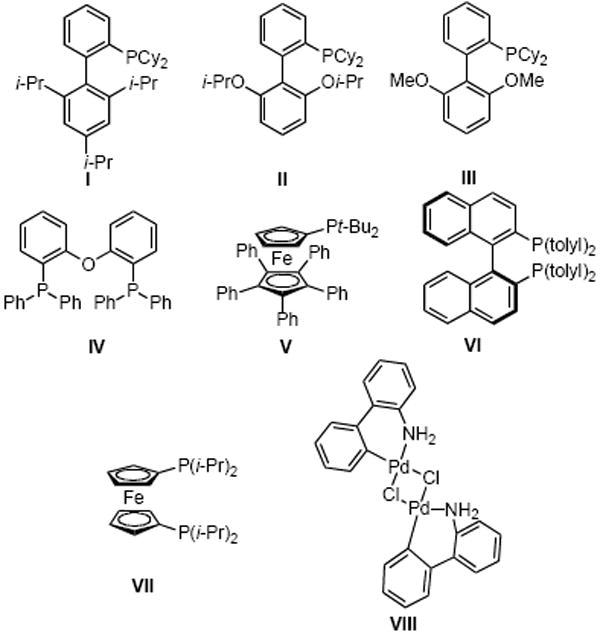
Ligands (I-VII) and palladium dimer precatalyst VIII.
We then investigated the scope of this reaction by varying the nature of the potassium alkoxymethyltrifluoroborates 2 in the presence of the mesylated naphthalene 1 (Table 2). All these nucleophiles 2 have first been prepared starting from the potassium chloromethyltrifluoroborate, instead of its corresponding bromo counterpart as earlier reported,7a to avoid any KBr salt contamination.9p,11 It is important to note that among them, some of the potassium alkoxymethyltrifluoroborates (2b, 2c, 2f, and 2i) bear alcohol protecting groups (Table 2, entries 2, 3, 6 and 9) known to be stable under Suzuki–Miyaura reaction conditions but which can be easily removed to generate the corresponding hydroxymethyl group.
Table 2.
Scope of Potassium Alkoxymethyltrifluoroborates
|
| ||||
|---|---|---|---|---|
| entry | R | yield (%) | ||
| 1 | Me | 2a | 3a | 71a |
| 2 | Bn | 2b | 3b | 82 (79)b |
| 3 | t-Bu | 2c | 3c | 87 |
| 4 | Cyclopentyl | 2d | 3d | 76 |
| 5 | Cyclopropyl-CH2 | 2e | 3e | 90 |
| 6 | TMS-CH2-CH2 | 2f | 3f | 91 |
| 7 | CH3(CH2)3CH(CH3) | 2g | 3g | 79 |
| 8 | Menthyl | 2h | 3h | 72c |
| 9 | PMB | 2i | 3i | 82 |
5 mol % PdCl2(COD), 10 mol % dippf, 110 °C, 4 h.
reaction run on a 4 mmol scale using 2 mol % of PdCl2(COD) and 4 mol % of dippf.
5 mol % palladium precatalyst VIII, 10 mol % dippf, 4 equiv of K3PO4, 110 °C, 4 h.
We applied the previously optimized conditions (Table 1, entry 13) to a broad spectrum of potassium alkoxymethyltrifluoroborates 2 (Table 2).
The cross-coupling reaction is very well tolerant of both acyclic and cyclic substituents on the alkoxymethyl counterpart: the desired compounds 3a-i were obtained with yields ranging from 71% to 91%. The catalyst loading could be reduced significantly by scaling the reaction (Table 2, entry 2). Moreover, even on small scale the catalyst and ligand loadings can be reduced to 3 mol % and 6 mol %, respectively, for almost all the substrates by increasing the reaction time to 20 h. The potassium methoxymethyltrifluoroborate 2a does not follow this trend, affording a lower yield of desired compound 3a, perhaps because of decomposition under the reaction conditions. Of particular note, potassium phenyloxymethyltrifluoroborate does not react well in the cross-coupling, as a complex mixture of compounds were formed in the reaction with naphthyl mesylate (1).
The introduction of the menthyl group (Table 2, entry 8) required the use of 5 mol % of catalyst for the reaction to go to completion and afforded 3h with 65% yield. By switching the palladium source from PdCl2(COD) to a palladium dimer precatalyst VIII developed by Buchwald (Figure 2) and reducing the amount of base to 4 equiv, we were able to increase the yield of 3h to 72%.12 This air-stable precatalyst is known to form the catalytically active species by cleavage of the dimer in favor of the ligand insertion under conditions where protodeboronation is slowed.13
We next pursued an examination of electron-rich and electron-poor aryl mesylates as electrophilic partners (Table 3). Encouraged by the previous result with the menthyl species (Table 2, entry 8), we tested the two optimized conditions [PdCl2(COD) versus palladium precatalyst VIII with 7.2 or 4 equiv of K3PO4, respectively] and it has been observed that almost all the yields increased by 10-15% by using the latter set of conditions. The reaction seems to proceed well without being influenced by the electronic density on the aryl ring, and the desired compounds 4a-j can be obtained with yields up to 92%. Moreover, the reaction is compatible with a large variety of substituents such as ethers (4b-d), ketones (4f-h), as well as trifluoromethyl (4i) and nitrile (4j) groups (Table 3, entries 2-4, 6-10). It has also been noticed that purifying the compounds under basic alumina instead of silica gel increases almost all the yields by 10% (Table 3, entries 1, 3, 5-8, 10).
Table 3.
Scope of Functionalized Aryl Mesylates
 | |||
|---|---|---|---|
| entry | Ar-OMs | yield (%) | |
| 1 |
|
4a | 92 |
| 2 |
|
4b | 60 |
| 3 |
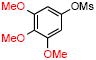
|
4c | 78 |
| 4 |
|
4d | 64 |
| 5 |
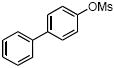
|
4e | 77 |
| 6 |
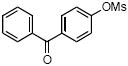
|
4f | 55 |
| 7 |
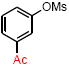
|
4g | 67 |
| 8 |
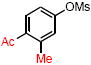
|
4h | 71 |
| 9 |
|
4i | 69 |
| 10 |
|
4j | 46 |
We further extended the scope of the reaction to more difficult heteroaryl mesylate partners (Table 4). It has already been demonstrated that cross-couplings of heteroaryl halides with potassium alkoxymethyltrifluoroborates remains very challenging because of their lack of reactivity.7a Using the least reactive electrophilic species, we were pleased to be able to couple several heteroaryl mesylates with moderate to good yields. Mesylated quinoline, dibenzothiophene and isoquinoline provided the alkoxymethyl compounds 5a, d, e with yields ranging from 57% to 81% (Table 4, entries 1, 4 and 5). To favor the completion of the reaction for the dibenzofuran and the benzothiazole, 5 mol % of precatalyst were used to afford 5b and 5c with 69% and 46% yields, respectively (Table 4, entries 2 and 3).
Table 4.
Scope of Heteroaryl Mesylates
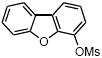
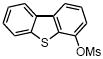
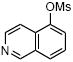
5 mol % palladium precatalyst VIII, 10 mol % dippf.
In summary, we have developed an efficient method that enables the cross-coupling of various potassium alkoxymethyltrifluoroborates with mesylated partners through C–O activation in very high yields. When desired, hydroxymethyl compounds should be easily accessed by following this procedure utilizing alkoxymethyl derviatives that are easily cleaved to the corresponding alcohol. A large array of functionalized aryl electrophilic partners has been successfully engaged in the cross-coupling to obtain the desired compounds with good yields. Moreover, several heteroaryl mesylates have also proven to be suitable electrophilic partners in this reaction even though the scope of the reaction is more limited. This method represents a complementary way to generate an alkoxymethyl linkage, which is known to be encountered in compounds of biological interest.
Supplementary Material
Acknowledgments
This research was supported by a National Priorities Research Program (NPRP) grant from the Qatar National Research Fund (Grant no. 08-035-1-008) and the NIGMS (R01 GM-081376). We acknowledge Dr. Matthew Tudge (Merck Research Laboratories) for a donation of biphenyl palladium dimer VIII and Johnson Matthey for its donation of PdCl2(COD). Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data. Dr. Virginie Colombel and Dr. Valerie Braz (University of Pennsylvania) are acknowledged for making some potassium alkoxymethyltrifluoroborates.
Footnotes
Supporting Information Available. Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dominguez de Maria P, Van Gemert RW, Straathof AJJ, Hanefeld U. Nat Prod Rep. 2010;27:370–392. doi: 10.1039/b809416k. [DOI] [PubMed] [Google Scholar]
- 2.Kolasa T, Gunn DE, Bhatia P, Woods KW, Gane T, Stewart AO, Bouska JB, Harris RR, Hulkower KI, Malo PE, Bell RL, Carter GW, Brooks CDW. J Med Chem. 2000;43:690–705. doi: 10.1021/jm9904102. [DOI] [PubMed] [Google Scholar]
- 3.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. John Wiley; New York: 1999. [Google Scholar]
- 4.(a) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doucet H. Eur J Org Chem. 2008;12:2013–2030. [Google Scholar]; (c) Mitchell TA, Bode JW. J Am Chem Soc. 2009;131:18057–18059. doi: 10.1021/ja906514s. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kosugi M, Sumiya T, Ohhashi K, Sano H, Migita T. Chem Lett. 1985:997–998. [Google Scholar]; (b) Majeed AJ, Antonsen O, Benneche T, Undheim K. Tetrahedron. 1989;45:993–1006. [Google Scholar]; (c) Ferezou JP, Julia M, Yun L, Liu LW, Pancrazi A. Synlett. 1991:53–56. [Google Scholar]
- 6.Salmon R, Mitchell G, Morris JA. 2009:WO091115788. [Google Scholar]
- 7.(a) Molander GA, Canturk B. Org Lett. 2008;10:2135–2138. doi: 10.1021/ol800532p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molander GA, Sandrock DL. J Am Chem Soc. 2008;130:15792–15793. doi: 10.1021/ja807076d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka K, Murai N, Shirotori S, Nagao S, Watanabe Y. 2008:WO08032702. [Google Scholar]
- 9.For recent examples using activated alcohols as electrophilic partners: Quasdorf KW, Tian X, Garg NK. J Am Chem Soc. 2008;130:14422–14423. doi: 10.1021/ja806244b.. Li Z, Zhang SL, Fu Y, Guo QX, Liu L. J Am Chem Soc. 2009;131:8815–8823. doi: 10.1021/ja810157e.. Guan BT, Wang Y, Li BJ, Yu DG, Shi ZJ. J Am Chem Soc. 2008;130:14468–14470. doi: 10.1021/ja8056503.. Quasdorf KW, Riener M, Petrova KV, Garg NK. J Am Chem Soc. 2009;131:17748–17749. doi: 10.1021/ja906477r.. Antoft-Finch A, Blackburn T, Snieckus V. J Am Chem Soc. 2009;131:17750–17752. doi: 10.1021/ja907700e.. Xu L, Li BJ, Wu ZH, Lu XY, Guan BT, Wang BQ, Zhao KQ, Shi ZJ. Org Lett. 2010;12:884–887. doi: 10.1021/ol9029534.. Tobisu M, Shimasaki T, Chatani N. Angew Chem Int Ed. 2008;47:4866–4869. doi: 10.1002/anie.200801447.. Lipshutz BH, Butler T, Swift E. Org Lett. 2008;10:697–700. doi: 10.1021/ol702453q.. Molander GA, Beaumard F. Org Lett. 2010;12:4022–4025. doi: 10.1021/ol101592r.. Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK, Percec V. Chem Rev. 2011;3:1346–1416. doi: 10.1021/cr100259t.. So CM, Lau CP, Kwong FY. Angew Chem Int Ed. 2008;47:8059–8063. doi: 10.1002/anie.200803193.. So CM, Lau CP, Chan ASC, Kwong FY. J Org Chem. 2008;73:7731–7734. doi: 10.1021/jo8014819.. Bhayana B, Fors BP, Buchwald SL. Org Lett. 2009;11:3954–3957. doi: 10.1021/ol9015892.. Chow WK, So CM, Lau CP, Kwong FY. J Org Chem. 2010;75:5109–5112. doi: 10.1021/jo100846t.. Kuroda JI, Inamoto K, Hiroya K, Doi T. Eur J Org Chem. 2009:2251–2261.. Molander GA, Beaumard F. Org Lett. 2011;13:1242–1245. doi: 10.1021/ol200128y.
- 10.The Suzuki-Miyaura cross-couplings between 4-chorobenzonitrile or 4-chloroanisole and 2a undergo with low yields of 34% and 41%, respectively.
- 11.(a) Raushel J, Sandrock DL, Josyula KV, Pakyz V, Molander GA. J Org Chem. 2011;8:2762–2769. doi: 10.1021/jo2001066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molander GA, Colombel V, Braz VA. Org Lett. 2011;7:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pd(0) species such as Pd2dba3 were inefficent in this reaction.
- 13.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.