

Summary
The cyclic adenosine monophosphate (cAMP)-dependent signaling pathway directs the expression of several genes involved in diverse neuroendocrine, immune, metabolic, and developmental pathways. The primary effectors of this pathway are members of the cAMP response element binding (CREB) family of transcription factors, in particular the CREB-1 and cAMP response element modulator (CREM). Both these genes encode alternative splice variants that serve as activators or repressors in a context- and position-specific manner. Although the β- chemokine receptor CC chemokine receptor 5 (CCR5) has been identified on progenitor cells in the bone marrow, the regulatory mechanisms orchestrating its expression are not fully understood. Previous reports have identified putative cAMP response elements in the CCR5 promoter and have described a suppressive role of cAMP in CCR5 expression. In this study, the CD34+CD4+CCR5+ human bone marrow progenitor cell line TF-1 was used to investigate the detailed kinetics of CCR5 transcription in response to the elevation of intracellular cAMP levels and the underlying molecular events. We hypothesize that CCR5 transcription follows an asymmetrical sinusoidal pattern in TF-1 cells that parallels a protein kinase A-dependent alternating change in the ratio of activator pCREB-1-α,Δ to repressor pCREM-α,β isoforms. However, elevated CCR5 mRNA levels do not correlate with enhancement in infectivity with respect to the R5 human immunodeficiency virus type 1 (HIV-1) strain. Our results lend critical insight into the precise mechanism governing the cAMP-CCR5 axis in progenitor cells and pose interesting questions regarding its functional role in HIV-1 infection.
Keywords: cAMP, CCR5, HIV-1
1. Introduction
CC chemokine receptor 5 (CCR5) belongs to the family of seven-transmembrane G-protein coupled receptors (GPCRs) and is expressed on several immune cell populations, including but not limited to memory and effector T lymphocytes, monocyte-macrophages, and immature dendritic cells [1]. CCR5 plays a vital role in viral pathogenesis as is evident from its ability to serve as a co-receptor for entry of human immunodeficiency virus type 1 (HIV-1) in target cells [2]. Analogous to the situation with central nervous system microglia [3] and perivascular macrophages [4], it has been proposed that bone marrow macrophages [5] and CD34+ multipotential hematopoietic progenitor cells [6,7] are important cellular reservoirs for HIV-1. Our studies show that, like peripheral blood-derived precursors [8], CD34+ bone marrow progenitors express CCR5 along with the primary receptor CD4, thereby supporting productive infection by the R5-tropic HIV-1 BaL strain (Alexaki and Wigdahl, unpublished results). Transcriptional regulation of CCR5 by transacting factors like NF-κB [9] seems to be cell-type specific [10,11]. On the basis of the identification of a functional cAMP response element (CRE) within the CCR5 promoter, it was recently suggested that the cyclic adenosine monophosphate (cAMP) pathway is involved in repression of CCR5 expression in T lymphocytes, monocyte-derived dendritic cells, and microglia [11]. We used the CD34+CD4+CCR5+ TF-1 human bone marrow progenitor cell line to study the regulation of CCR5 by cAMP signaling and to study the concomitant effects on HIV-1 infection. Our experiments showed that CCR5 transcription follows an asymmetrical sinusoidal pattern in response to an increase in the concentration of intracellular cAMP. To dissect the interplay of downstream transcription factors, western blot analyses were performed to monitor the kinetics of phosphorylated cAMP response element binding (pCREB)-1-α,Δ and phosphorylated cAMP response element modulator (pCREM)-α/β expression, which are cAMP-response transcriptional activators [12] and repressors [13], respectively. Additionally, we investigated the contribution of various protein kinases in the activation of pCREB-1 and, in turn, the upregulation of CCR5. On the basis of our results, we hypothesized that cAMP-induced CCR5 transcription in TF-1 cells is an outcome of the temporal fluctuation of activator and repressor members of the CREB family in the nucleus, with prolonged persistence of activator pCREB-1 during latter time points reflective of the crest in CCR5 mRNA levels. This hypothesis led us to examine further whether an increase in CCR5 transcription would translate into an enhancement in HIV-1 infectivity in these cells.
2. Materials and methods
2.1. Materials
TF-1 cells were treated with forskolin (Sigma-Aldrich, St. Louis, MO), a diterpene obtained from Coleus forskohlii, to elevate intracellular cAMP concentration. The protein kinase A (PKA)-specific inhibitor H-89 was also purchased from Sigma-Aldrich. Additionally, LY294002 [protein kinase B -specific inhibitor], SB203580 (p38-specific inhibitor), KN-62 (CaMKII-specific inhibitor), and myristoylated protein kinase C (PKC)-ζ (peptide inhibitor) were all obtained from Enzo Life Sciences International, Inc. (formerly Biomol International, LP; Plymouth Meeting, PA). Whole cell lysates and nuclear extracts were prepared in radioimmunoprecipitation assay buffer and NE-PER nuclear extraction reagent (Pierce Biotechnology, Thermo Fisher Scientific, Rockford, IL) treated with Protease Inhibitor Cocktail Set III and Phosphatase Inhibitor Cocktail Set II from Calbiochem (San Diego, CA). Protein concentrations were calculated using the bicinchoninic acid protein assay procedure described by Pierce Biotechnology (Thermo Fisher Scientific, Rockford, IL). Total cellular RNA for the Taqman quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) was prepared from TF-1 cells using TRI Reagent (Sigma-Aldrich).
2.2. Cell culture and treatment procedures
The TF-1 CD34+38+ cell line [American Type Culture Collection (ATCC), Manassas, VA] was grown in Roswell Park Memorial Institute 1640 medium (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Thermo Fisher Scientific, Logan, UT), penicillin (100 U/mL), streptomycin (100 μg/mL), and recombinant human granulocyte/macrophage-colony stimulating factor (2 ng/ml; eBioscience, San Diego, CA). The cells were maintained at 37°C in 5% CO2 at 90% relative humidity.
2.3. Western immunoblot hybridization
TF-1 cells were serum starved for 1 h and then treated with adenyl cyclase activator forskolin for different times (0.5, 1, 3, 6, and 9 h). Whole cell lysates and nuclear extracts were prepared from untreated and forskolin-treated cells, and western immunmoblots were performed to assay levels of CREB-1 and β-actin in the cytoplasmic fraction and of pCREB-1 in the nuclear fraction. An equal amount of protein for all samples was run on a 10% sodium dodecyl sulfate–polyacrylamide gel and transferred to a 0.45-μm Immobilon-P polyvinylidene difluoride membrane followed by probing with a rabbit polyclonal pCREB-1 antibody (Upstate Chemicon, Temecula, CA). CREB-1 was detected using a rabbit polyclonal CREB-1 antibody (Active Motif, Carlsbad, CA). Equal sample loading was confirmed by stripping the membranes using the Restore Western Blot Stripping Buffer (Thermo Fisher Scientific, Rockford, IL) and reblotting for β-actin with a mouse-monoclonal antibody from Sigma-Aldrich. The specific protein bands were then detected using a horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). To identify cellular protein kinases mediating phosphorylation of CREB-1 in response to forskolin, TF-1 cells were pretreated separately with inhibitors of various protein kinases (H-89, LY294002, SB203580, KN-62, and PKC-ζ inhibitor) for 1 h, followed by forskolin exposure for another hour and by preparation of whole cell lysates and nuclear extracts. Western immunoblots for pCREB-1, CREB-1, and β-actin were performed as indicated previously. To investigate the temporal pattern of CREM phosphorylation induced by cAMP, TF-1 cells were treated with forskolin and processed for extract preparation as indicated above. Western immunoblot hybridization used a rabbit polyclonal CREM antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The optimized concentrations of reagents used for treatments were as follows: forskolin (100 μM), H-89 (10 μM), LY294002 (50 μM), KN-62 (10 μM), SB203580 (10 μM), and PKC-ζ inhibitor (10 μM). Western immunoblots were visualized using a chemiluminescent detection procedure (Pierce Biotechnology, Thermo Fisher Scientific, Rockford, IL) as described on a ChemiDoc acquisition/analysis station (Bio-Rad Laboratories, Hercules, CA). Densitometry was performed with an AlphaEase FC software package (Alpha Innotech Corporation, San Leandro, CA) with specific bands being normalized with regard to corresponding β-actin levels and represented graphically as fold over untreated sample.
2.4. Isolation of RNA and qRT-PCR
TF-1 cells were serum starved for 1 h; total RNA was isolated from forskolin- (100 μM) treated cells every 3 h for 24 h using TRI reagent. To confirm the role of PKA in cAMP-mediated stimulation of CCR5, cells were pretreated with H-89 for 1 h followed by treatment with forskolin for 15 h and total RNA extraction. Reverse transcription was performed using the SuperScript III First-Strand Synthesis System (Invitrogen Corporation, Carlsbad, CA) on 2 μg of total RNA per sample. qRT-PCR was performed on cDNA samples using Taqman Universal PCR Master Mix (Applied Biosystems Inc., Foster City, CA); gene expression primer/probe assays (Applied Biosystems Inc) were used for the detection of CCR5; and human large ribosomal protein (RPLPO) was used as the normalization control. Augmented mRNA levels in treated samples were analyzed as fold over untreated (calibrator) on an ABI 7300 Thermocycler (Applied Biosystems Inc., Foster City, CA) and expressed in linear scale. Alternatively, forskolin-induced mRNA levels were arbitrarily set as the baseline, and reduction following treatment with H-89 was plotted on a linear scale. All samples were assayed in triplicate.
2.5. HIV-1 p24 ELISA
Cells were seeded into 12-well plates at a density of 0.4 × 106 cells/ml and pretreated for 72, 48, 24, or 0 h with forskolin (100 μM). Cells were then recounted, seeded at a concentration of 1 × 106 cells/ml/well, and exposed to the R5-tropic BaL HIV-1 strain at a titer of 105 TCID50 (Advanced Biotechnologies, Inc., Columbia, MD). At 2 h post infection, cells were washed with 1X phosphate-buffered saline, resuspended in fresh Roswell Park Memorial Institute 1640 medium, and reseeded in 12-well plates. Following incubation for 24 h, the supernatant was collected and assayed for p24 viral core antigen using an Alliance p24 ELISA procedure described by the manufacturer (Perkin Elmer, Waltham, MA). All samples were assayed in triplicate.
3. Results
3.1. Kinetics of cAMP-induced CCR5 transcription
qRT-PCR assays were performed to study the regulation of CCR5 transcription in response to enhanced cAMP levels (Fig. 1). A kinetic analysis performed at 24 h revealed an asymmetrical sinusoidal pattern wherein an initial reduction was followed by recovery and subsequent augmentation of mRNA levels. The full amplitude of the negative curve was observed by 3 h post forskolin stimulation. At 6 h, mRNA levels approximated untreated baseline levels. The peak of the positive curve was observed at 15 h following, which transcript levels declined steadily. The maximum magnitude of the CCR5 decrease and increase in expression was ~1.9-fold and ~2.2-fold, respectively. The results suggest the functional importance of the putative CRE [11] identified in the CCR5 promoter.
Fig. 1.
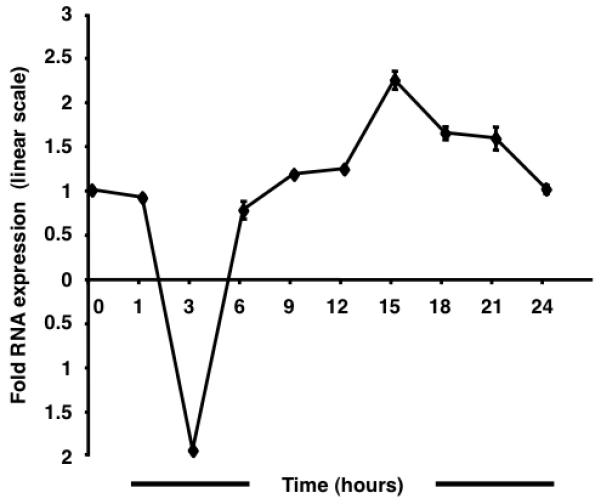
Forskolin stimulation alters CCR5 transcription in TF-1 cells. The sinusoidal wave of CCR5 transcript levels is characterized by an initial trough followed by recovery and an eventual peak at 15 h post stimulation. A 1.9-fold downregulation is followed by a maximal 2.2-fold upregulation. Results are normalized to human large ribosomal protein expression and expressed in linear scale as fold over untreated.
3.2. Implications for susceptibility to R5-tropic HIV-1
To find an indirect association between cAMP-induced elevation in CCR5 transcription and consequent susceptibility of TF-1 cells to infection with HIV-1, assays for measuring replication of the R5 HIV-1 strain BaL were performed. Samples pretreated for different intervals (72, 48, or 24 h) did not exhibit any marked increase in p24 levels (data not shown).
3.3. Kinetics of CREB-1 and CREM phosphorylation
To gain a better understanding of the molecular events elicited by cAMP, the kinetics of pCREB-1 and pCREM expression in the nucleus of TF-1 cells were observed at regular 3-h intervals post forskolin treatment. Accumulation of activator pCREB-1-α,Δ isoforms (~43 KDa) in the nucleus was triggered as early as 0.5 h (Fig. 2A). Heightened levels of pCREB-1-α,Δ were maintained up to 6 h with maximum levels observed at 1 h. This observation cannot be accredited to a global increase in CREB-1 transcription because the total CREB-1 expression in the cellular lysate remained unchanged (data not shown). The most plausible explanation is the sustained turnover of intracellular cAMP and hence sustained stimulation of CREB-1 phosphorylation [14] by cellular kinases. Interestingly, expression of pCREM-α/β repressor isoforms in the nucleus (~30 KDa) [15] exhibited a bell-shaped curve with progressive amplification up to 1 h (Fig. 2B). This finding is in agreement with reports demonstrating that CREM is an early response gene [16]. Constant levels of the CREM activator isoform-τ (data not shown) confirmed that there was no upregulation of global transcription, similar to that seen with CREB-1. Comparing the protein levels from one time interval to another indicates that the relative change in pCREM exceeds that of pCREB-1 (e.g. 2.04- vs. 1.19-fold upregulation for the 0.5- to 1-h time interval, 2.77- vs. 2.44-fold downregulation for the 3- to 6-h interval). Although pCREB-1 is more abundant in nuclear extracts, it should be noted that pCREM-mediated suppression is achieved at substoichiometric concentrations [17]. Coupled with the progressive increase in the ratio of pCREB-1 to pCREM at latter time points (1.67-fold at 3 h to 3.9-fold at 6 h), this finding might explain the observed temporal pattern of CCR5 transcription.
Fig. 2.
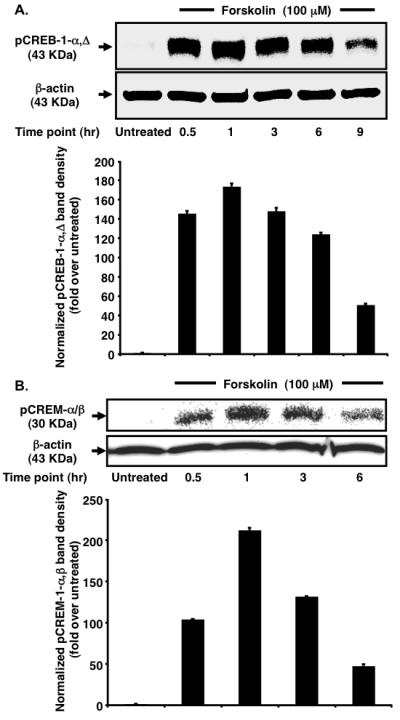
Forskolin stimulation leads to increased nuclear accumulation of pCREB-1α,Δ and pCREM-α/β in TF-1 cells. (A) Analysis of the kinetics of CREB-1 phosphorylation reveals negligible pCREB-1 in the untreated sample. Following forskolin stimulation, maximum pCREB-1 is found at 1 h. Heightened pCREB-1 levels persist up to 6 h followed by dephosphorylation at 9 h. β-actin serves as a normalization control. (B) Analysis of the kinetics of pCREM accumulation reveals a bell-shaped curve. Following stimulation, an exponential increase occurs in nuclear pCREM levels. Maximal accumulation is observed at 1 h. Thereafter, levels drop rapidly up to 6 h. β-actin serves as a loading control.
3.4. Inhibition of CREB-1 phosphorylation by kinase inhibitors
Because cAMP induces multiple protein kinases in a cell type-specific manner [18], we identified the major pathways involved in Ser-133 phosphorylation of CREB-1 [19]. Only PKA-specific inhibitor H-89 abrogated forskolin-induced maximal accumulation of pCREB-1 at 1 h (~2.34-fold reduction), suggesting a role as a central player in the cAMP-PKA-pCREB network (Fig. 3). This finding contradicts studies implicating PKC-ζ in cAMP-induced membrane GPCR elevation [20]. This distinction may be a fallout of the nature of the stimulus (soluble N6,2′-O-dibutyryl-cAMP versus forskolin) used and the particular cell type in question (G2 pre-B ALL progenitors versus TF-1 progenitors). Additionally, because abrogation was not absolute, a potential contribution of other kinases not included in this study cannot be excluded.
Fig. 3.
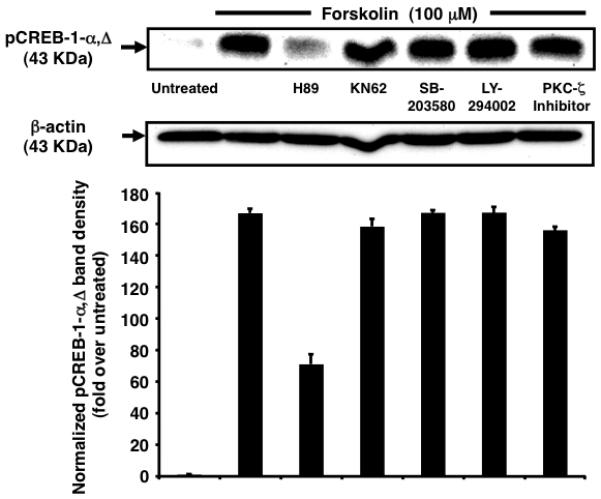
Inhibition of PKA abrogates nuclear accumulation of pCREB-1α,Δ. Nuclear pCREB-1 levels increase in response to forskolin stimulation for 1 h. This response is partly abrogated by pretreatment with H-89 (2.34-fold). pCREB-1 levels are not affected by pretreatment with other kinase inhibitors. Samples treated with inhibitors alone exhibit no significant change in comparison to untreated cells (data not shown).
3.5. Inhibition of CCR5 transcription by selective inhibition of PKA
To determine whether inhibition of the PKA pathway led to a diminution in the peak of CCR5 induction at 15 hour, qRT-PCR assays were performed. In comparison to untreated samples, samples treated with H-89 alone showed a drop of ~1.98 fold, implying that basal level of transcription of CCR5 proceeds through the PKA pathway and is specifically inhibited by H-89. As observed previously, forskolin treated samples exhibited an upregulation of ~2.3 fold in the levels of CCR5 transcript. Additionally, in comparison to forskolin treated samples, H-89 and forskolin dual treated samples exhibited an ~2.2-fold drop in CCR5 mRNA levels, validating that PKA acts in concert with CREB to modulate cAMP-mediated CCR5 transcription (Fig.4).
Fig. 4.
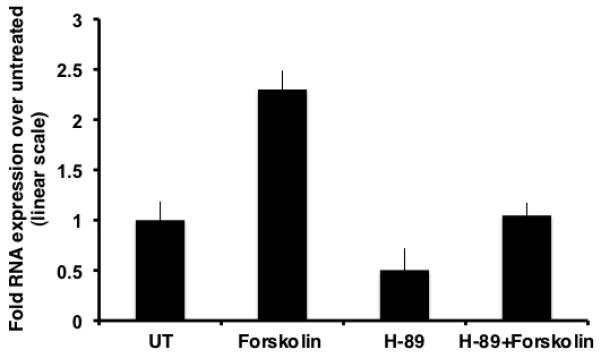
Inhibition of PKA downregulates forskolin-induced CCR5 transcription. Chronic forskolin stimulation for 15 h leads to an induction in CCR5 mRNA levels. This increase is partly inhibited (~50%) by pretreatment with H-89.
4. Discussion and Conclusions
CCR5 has been implicated in the manifestation of various inflammatory and neurodegenerative disorders such as multiple sclerosis, Alzheimer’s disease, and atherosclerosis [21-25]. This association has been attributed in part to an increased recruitment of reactive macrophages (in atherosclerosis) and microglia (in Alzheimer’s disease) to cardiovascular and brain tissues [26] in response to chemical gradients involving the cognate ligands CCL3/MIP-1α, CCL4/MIP-1β, and CCL5/RANTES [27,28], along with the subsequent secretion of proinflammatory mediators by these CCR5+ immune cell populations. CCR5 signaling also may play a vital role in thymic homing of primitive lineage-negative bone marrow cells [29]. To address the effects of elevated cAMP levels on CCR5 transcription, we used the human CD4+/CCR5+ TF-1 bone marrow progenitor cell line. Considerable controversy exists in the literature regarding the ability of transcription factors like GATA-1 and YY-1 to interface with putative cis-acting elements and transactivate the CCR5 promoter. Whereas GATA-1 is thought to stimulate [30] or repress [31] the promoter differentially, YY-1 has been identified as a negative regulator [32]. Because this dichotomy may partly be attributed to cellular specificity, our studies were initially aimed at confirming whether the previously reported inhibitory effects of cAMP on CCR5 transcription [11] were consistent in TF-1 cells. Our experiments revealed that, consistent with the findings of the aforementioned study, the initial phase of CCR5 transcription exhibited a decline. However, the ensuing phase encompassed a recovery and ultimately an increase in the level of transcripts. The modest induction is probably due to the nonconsensus composition (TGAGCAGA) and physical distance [−2187 with respect to ORF(+1)] [11] of the CRE in the promoter, which are key characteristics of other less active CREs [33,34]. In addition, the delayed kinetics of upregulation may be an outcome of an oscillation in the levels of the antagonist pCREM and activator pCREB-1 effector isoforms, which is common to other morphogenetic processes [35]. Because we have established TF-1 cells as a model for HIV-1 infection of progenitor cells in vitro [36], we also examined the link between amplified CCR5 transcription and susceptibility to HIV-1. Lack of a positive correlation is consistent with the hypotheses that the surface level of CCR5 is critical for infectivity by R5-tropic HIV-1 [37] and that low levels of CCR5 on CD34+ progenitors [38] may not be adequate for supporting robust infection. Our subsequent experiments were aimed at identifying possible cellular pathways involved in facilitating such a response and involved using series of protein-kinase inhibitors. Interestingly, contrary to published studies describing the involvement of different cellular kinases, pCREB-1 accumulation and CCR5 transcription in TF-1 cells were found to be exclusively mediated by PKA. Studies performed herein characterize key molecular events coupled to intracellular cAMP augmentation that govern temporal expression of CCR5 in bone marrow progenitor cells. Further investigation is needed to establish the tissue-specific contribution of this stimulatory pathway in CCR5-mediated normal and aberrant physiological processes.
Acknowledgements
These studies were funded in part by the Public Health Service, National Institutes of Health, through grants (B. Wigdahl, Principal Investigator) from the National Institute of Neurological Disorders and Stroke (NS32092 and NS46263) and the National Institute of Drug Abuse (DA19807). Dr. Michael Nonnemacher was supported by faculty development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- CCR5
CC chemokine receptor 5
- CRE
cAMP response element
- CREB
cAMP response element binding
- CREM
cAMP response element modulator
- GPCR
G-protein coupled receptors
- HIV-1
human immunodeficiency virus type 1
- PKA
protein kinase A
- PKC
protein kinase C
- qRT-PCR
quantitative real-time reverse transcriptase polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Blanpain C, Libert F, Vassart G, Parmentier M. CCR5 and HIV infection. Receptors Channels. 2002;8:19–31. [PubMed] [Google Scholar]
- [2].Zhang YJ, Moore JP. Will multiple coreceptors need to be targeted by inhibitors of human immunodeficiency virus type 1 entry? J Virol. 1999;73:3443–8. doi: 10.1128/jvi.73.4.3443-3448.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–9. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- [4].Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–62. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- [5].Gill V, Shattock RJ, Freeman AR, Robinson G, Griffin GE, Gordon-Smith EC, et al. Macrophages are the major target cell for HIV infection in long-term marrow culture and demonstrate dual susceptibility to lymphocytotropic and monocytotropic strains of HIV-1. Br J Haematol. 1996;93:30–7. doi: 10.1046/j.1365-2141.1996.4801017.x. [DOI] [PubMed] [Google Scholar]
- [6].Folks TM, Kessler SW, Orenstein JM, Justement JS, Jaffe ES, Fauci AS. Infection and replication of HIV-1 in purified progenitor cells of normal human bone marrow. Science. 1988;242:919–22. doi: 10.1126/science.2460922. [DOI] [PubMed] [Google Scholar]
- [7].Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell Jt, Bixby D, Savona MR, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 16:446–51. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ruiz ME, Cicala C, Arthos J, Kinter A, Catanzaro AT, Adelsberger J, et al. Peripheral blood-derived CD34+ progenitor cells: CXC chemokine receptor 4 and CC chemokine receptor 5 expression and infection by HIV. J Immunol. 1998;161:4169–76. [PubMed] [Google Scholar]
- [9].Guignard F, Combadiere C, Tiffany HL, Murphy PM. Gene organization and promoter function for CC chemokine receptor 5 (CCR5) J Immunol. 1998;160:985–92. [PubMed] [Google Scholar]
- [10].Liu R, Zhao X, Gurney TA, Landau NR. Functional analysis of the proximal CCR5 promoter. AIDS Res Hum Retroviruses. 1998;14:1509–19. doi: 10.1089/aid.1998.14.1509. [DOI] [PubMed] [Google Scholar]
- [11].Kuipers HF, Biesta PJ, Montagne LJ, van Haastert ES, van der Valk P, van den Elsen PJ. CC chemokine receptor 5 gene promoter activation by the cyclic AMP response element binding transcription factor. Blood. 2008;112:1610–9. doi: 10.1182/blood-2008-01-135111. [DOI] [PubMed] [Google Scholar]
- [12].Ruppert S, Cole TJ, Boshart M, Schmid E, Schutz G. Multiple mRNA isoforms of the transcription activator protein CREB: generation by alternative splicing and specific expression in primary spermatocytes. EMBO J. 1992;11:1503–12. doi: 10.1002/j.1460-2075.1992.tb05195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell. 1991;64:739–49. doi: 10.1016/0092-8674(91)90503-q. [DOI] [PubMed] [Google Scholar]
- [14].Baker JG, Hall IP, Hill SJ. Temporal characteristics of cAMP response element-mediated gene transcription: requirement for sustained cAMP production. Mol Pharmacol. 2004;65:986–98. doi: 10.1124/mol.65.4.986. [DOI] [PubMed] [Google Scholar]
- [15].Pati D, Meistrich ML, Plon SE. Human Cdc34 and Rad6B ubiquitin-conjugating enzymes target repressors of cyclic AMP-induced transcription for proteolysis. Mol Cell Biol. 1999;19:5001–13. doi: 10.1128/mcb.19.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Verma IM, Sassone-Corsi P. Proto-oncogene fos: complex but versatile regulation. Cell. 1987;51:513–4. doi: 10.1016/0092-8674(87)90115-2. [DOI] [PubMed] [Google Scholar]
- [17].Laoide BM, Foulkes NS, Schlotter F, Sassone-Corsi P. The functional versatility of CREM is determined by its modular structure. EMBO J. 1993;12:1179–91. doi: 10.1002/j.1460-2075.1993.tb05759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005;17:1343–51. doi: 10.1016/j.cellsig.2005.02.003. [DOI] [PubMed] [Google Scholar]
- [19].Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–80. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- [20].Goichberg P, Kalinkovich A, Borodovsky N, Tesio M, Petit I, Nagler A, et al. cAMP-induced PKCzeta activation increases functional CXCR4 expression on human CD34+ hematopoietic progenitors. Blood. 2006;107:870–9. doi: 10.1182/blood-2005-03-0941. [DOI] [PubMed] [Google Scholar]
- [21].Barcellos LF, Schito AM, Rimmler JB, Vittinghoff E, Shih A, Lincoln R, et al. CC-chemokine receptor 5 polymorphism and age of onset in familial multiple sclerosis. Multiple Sclerosis Genetics Group. Immunogenetics. 2000;51:281–8. doi: 10.1007/s002510050621. [DOI] [PubMed] [Google Scholar]
- [22].Sheikine YA, Hansson GK. Chemokines as potential therapeutic targets in atherosclerosis. Curr Drug Targets. 2006;7:13–27. doi: 10.2174/138945006775270240. [DOI] [PubMed] [Google Scholar]
- [23].Apostolakis S, Papadakis EG, Krambovitis E, Spandidos DA. Chemokines in vascular pathology (review) Int J Mol Med. 2006;17:691–701. [PubMed] [Google Scholar]
- [24].Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- [25].Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J Leukoc Biol. doi: 10.1189/jlb.1009671. [DOI] [PubMed] [Google Scholar]
- [26].Balistreri CR, Caruso C, Grimaldi MP, Listi F, Vasto S, Orlando V, et al. CCR5 receptor: biologic and genetic implications in age-related diseases. Ann N Y Acad Sci. 2007;1100:162–72. doi: 10.1196/annals.1395.014. [DOI] [PubMed] [Google Scholar]
- [27].Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996;35:3362–7. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- [28].Le Y, Zhou Y, Iribarren P, Wang J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol. 2004;1:95–104. [PubMed] [Google Scholar]
- [29].Robertson P, Means TK, Luster AD, Scadden DT. CXCR4 and CCR5 mediate homing of primitive bone marrow-derived hematopoietic cells to the postnatal thymus. Exp Hematol. 2006;34:308–19. doi: 10.1016/j.exphem.2005.11.017. [DOI] [PubMed] [Google Scholar]
- [30].Moriuchi M, Moriuchi H, Fauci AS. GATA-1 transcription factor transactivates the promoter for CCR5, a coreceptor for human immunodeficiency virus type 1 entry. Blood. 1999;93:1433–5. [PubMed] [Google Scholar]
- [31].Sundrud MS, Vancompernolle SE, Eger KA, Bruno TC, Subramaniam A, Mummidi S, et al. Transcription factor GATA-1 potently represses the expression of the HIV-1 coreceptor CCR5 in human T cells and dendritic cells. Blood. 2005;106:3440–8. doi: 10.1182/blood-2005-03-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moriuchi M, Moriuchi H. YY1 transcription factor down-regulates expression of CCR5, a major coreceptor for HIV-1. J Biol Chem. 2003;278:13003–7. doi: 10.1074/jbc.M204980200. [DOI] [PubMed] [Google Scholar]
- [33].Craig JC, Schumacher MA, Mansoor SE, Farrens DL, Brennan RG, Goodman RH. Consensus and variant cAMP-regulated enhancers have distinct CREB-binding properties. J Biol Chem. 2001;276:11719–28. doi: 10.1074/jbc.M010263200. [DOI] [PubMed] [Google Scholar]
- [34].Tinti C, Yang C, Seo H, Conti B, Kim C, Joh TH, et al. Structure/function relationship of the cAMP response element in tyrosine hydroxylase gene transcription. J Biol Chem. 1997;272:19158–64. doi: 10.1074/jbc.272.31.19158. [DOI] [PubMed] [Google Scholar]
- [35].Don J, Stelzer G. The expanding family of CREB/CREM transcription factors that are involved with spermatogenesis. Mol Cell Endocrinol. 2002;187:115–24. doi: 10.1016/s0303-7207(01)00696-7. [DOI] [PubMed] [Google Scholar]
- [36].Alexaki A, Quiterio SJ, Liu Y, Irish B, Kilareski E, Nonnemacher MR, et al. PMA-induced differentiation of a bone marrow progenitor cell line activates HIV-1 LTR-driven transcription. DNA Cell Biol. 2007;26:387–94. doi: 10.1089/dna.2006.0542. [DOI] [PubMed] [Google Scholar]
- [37].Wu L, Paxton WA, Kassam N, Ruffing N, Rottman JB, Sullivan N, et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–91. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deichmann M, Kronenwett R, Haas R. Expression of the human immunodeficiency virus type-1 coreceptors CXCR-4 (fusin, LESTR) and CKR-5 in CD34+ hematopoietic progenitor cells. Blood. 1997;89:3522–8. [PubMed] [Google Scholar]