

Abstract
Background
We recently showed that PARP-1 may play a role in allergen (ovalbumin)-induced airway eosinophilia, potentially through a specific effect on IL-5 production. We also reported that while IL-5 replenishment promotes reversal of eosinophilia in lungs of PARP-1−/− mice, IL-4 or IgE replenishment do not, suggesting a potentially significant regulatory relationship between PARP-1 and IL-5.
Objective
To explore the mechanism by which PARP-1 regulates IL-5 production and to determine how PARP-1 inhibition blocks allergen-induced eosinophilia.
Methods
This study was conducted using a murine model of allergic airway inflammation and primary splenocytes.
Results
PARP-1 knockout-associated reduction in IL-5 upon allergen exposure occurs at the mRNA level. Such an effect appears to take place after IL-4 receptor activation as PARP-1 inhibition exerted no effect on JAK1/JAK3 activation. STAT-6 protein was severely downregulated in spleens of PARP-1−/− mice without any effect on mRNA levels, suggesting an effect on protein integrity rather than gene transcription. Interestingly, the degradation of STAT-6 in PARP-1−/− mice required allergen stimulation. Additionally, PARP-1 enzymatic activity appears to be required for STAT-6 integrity. The dowregulation of STAT-6 coincided with mRNA and protein reduction of GATA-3 and occupancy of its binding site on the IL-5 gene-promoter. IL-4 was sufficient to induce STAT-6 downregulation in both PARP-1−/− mice and isolated splenocytes. Such degradation may be mediated by calpain, but not by proteasomes.
Conclusion
These results demonstrate a novel function of PARP-1 in regulating IL-5 expression during allergen-induced inflammation and explain the underlying mechanism by which PARP-1 inhibition results in IL-5 reduction.
Keywords: Allergen-induced eosinophilia, IL-4, Transgenic/Knockout Mice, Lung
Introduction
The control of inflammation has long been one of the major therapeutic goals of medicine, as inflammatory processes are involved in the pathogenesis of diseases that affect all physiological systems, including cardiac, pulmonary, and neurological. Over the last decades, the family of poly(ADP-ribose)polymerases (PARP) has emerged as an important player in the development and progression of inflammatory disease. The involvement of the primary member of the family, PARP-1, in the inflammatory process is not entirely clear. This enzyme is believed to mediate inflammation through the promotion of cell death via ATP depletion as well as the transcription of inflammatory factors (for review (1–3). We have demonstrated the involvement of PARP-1 in the pathogenesis of allergen-induced inflammation (4–6) and airway hyperresponsiveness (AHR) (6) upon allergen (ovalbumin, OVA) exposure in a mouse model of asthma. PARP-1 activity appears to be critical for allergen-induced inflammation and airway AHR as poly(ADP-ribosyl)ation is evident in lungs of OVA-exposed animals (4, 7). Furthermore, inhibition of PARP-1 pharmacologically with old generation as well as novel drugs confers a marked protection against the manifestation of airway inflammation and AHR upon allergen exposure (4, 7, 8). PARP-1 inhibition was also shown to reduce the severity of cough and the occurrence of dyspnea in a guinea pig asthma model (7). In a number of studies, our laboratory has shown that inhibition of PARP-1, either pharmacologically or genetically, markedly attenuated OVA-induced eosinophilic infiltration as well as reduced the expression of Th2 cytokines, particularly those downstream of IL-4. A primary target of PARP-1 inhibition is cytokine IL-5, which is known to be crucial for eosinophilia (9). In a phenotype-reversal experiment, we were able to reestablish eosinophilia in OVA-challenged PARP-1−/− mice. However, intranasal administration of either IL-4 or IgE completely failed to reverse eosinophilia in OVA-challenged PARP-1−/− mice (5). These results clearly establish a role for PARP-1 in the pathogenesis of OVA-induced lung inflammation in our murine model of allergic airway inflammation and also describe a potentially important regulatory relationship between PARP-1 and IL-5. More importantly, these results suggest that the role PARP-1 may be upstream of IL-5, but downstream of IL-4.
Upon ligand binding, IL-4 receptor (IL-4R) heterodimerization promotes the activation of members of the Janus family of protein kinases (JAK1 and JAK3) (reviewed (10, 11). While JAK1 is constitutively associated with the α chain of the IL-4R (IL-4Rα), JAK3 is constitutively associated with the γ chain of the receptor. The two JAK proteins are subsequently activated by trans-phosphorylation of the specific and conserved tyrosine residues located in their activation loops. The activated JAK kinases initiate several intracellular signaling cascades by phosphorylating multiple tyrosine residues in the cytoplasmic domain of IL-4Rα, which facilitates the recruitment of the IL-4Rα-specific transcription factor STAT-6 via its SH2 domain. STAT-6 then homodimerizes upon phosphorylation by JAK kinases, which ultimately promotes its translocation to nuclei of stimulated cells. Nuclear STAT-6 then binds to the promoters of target genes such as GATA-3 and, thus, participating in their expression (10, 11). The goal of the present study was to explore the mechanism by which PARP-1 regulates IL-5 production and to determine how PARP-1 inhibition blocks allergen-induced eosinophilia by focusing primarily on the IL-4 signaling pathway. Given that stimulation of immune cells with IL-4 results in IL-5 production (9), we examined the various steps of the signaling cascade to determine whether there are interruptions in the pathway. Interference with any of the latter processes would explain an attenuation of Th2 inflammation, which can be bypassed using IL-5 but not IL-4.
Materials and Methods
Animals
C57BL/6J wild type and C57BL/6J PARP-1−/− mice were bred in a specific-pathogen free facility at LSUHSC and allowed unlimited access to sterilized chow and water. Maintenance and experimental protocols were approved by the LSUHSC Animal Care and Use Committee. The generation of PARP-1−/− mice on a C57BL/6J genetic background was described (5).
Protocol for OVA challenge model, tissue processing, and cytokine assessment
Six- to 8-wk-old male mice were sensitized and challenged essentially as described (5). Briefly, mice received i.p. injections of 100 µg of grade V chicken OVA (Sigma-Aldrich, St Louis, MO) mixed with 2 mg of aluminum hydroxide in saline once a week for 2 consecutive weeks, followed by a challenge with aerosolized OVA 1 week after the second sensitization. The OVA aerosol was generated by a Bennett nebulizer (DeVilbiss, PA). Control groups were not sensitized or challenged. The mice used in each experiment were of the same litter or the same family. Mice were then left to recover and sacrificed 24 h later for bronchoalveolar lavage (BAL) for cytokine determination. Spleens were also removed to be subjected to homogenization followed by protein or RNA extraction.
BAL fluids were assessed for IL-5 using a single-plex assay kit (Bio-Rad Laboratories, Hercules, CA) as per instructions of the manufacturer and as described (6). In some experiments, naïve mice received a single i.p. injection of 100 ng recombinant mouse IL-4 (R&D Systems, Minneapolis, MN; cat# 404-ML) in120 µl saline. Mice were sacrificed 6 or 12 h after injection. Spleens were then collected and processed for protein or RNA extraction.
Isolation of splenocytes, immunoblot analysis, nuclear protein extraction, electrophoretic mobility shift assay (EMSA), and conventional and real-time RT-PCR
Splenocytes were isolated essentially as described (5). The cells were counted and cultured in complete medium and treated with 10 ng/ml recombinant mouse IL-4 for the time intervals indicated in the figures. In some experiments, splenocytes were treated with IL-4 in the presence or absence of the proteasome inhibitor MG132 (Sigma Aldrich, cat# c2211) or the calpain and proteasome inhibitor Acetyl-L-Leucyl-L-Leucyl-L-Norleucinal (ALLN) (Santa Cruz Biotech, sc-29119).
Whole protein extracts were subjected to immunoblot analysis with antibodies to STAT-6 (sc-621), JAK1 (sc-1677), JAK3 (sc-513), actin (sc-1615), or GATA-3 (sc-22206) all of which were purchased from Santa Cruz Biotechnology or to antibodies to phospho-JAK1 (ab5493), phospho-JAK3 (ab61102), phospho-STAT-6 (ab54461), or ubiquitin (ab7780) which were purchased from Abcam Inc (Cambridge, MA).
RNA was extracted from either whole spleens or isolated splenocytes using standard methods and cDNA was generated using reverse transcriptase III (Invitrogen, Carlsbad, CA). The primers (Integrated DNA technologies, Coralville, IA) used for the PCR reactions were as follows: STAT-6: forward, 5’-CTTCAACGACAACAGCTT-3’; reversed, 5’-CTGGCTCATTGAGGAGAAGG-3’; GATA3: forward: 5’-AGCCGCGCTGGGTGAGCCA-3’; reversed: 5’-CGTGGTGGATGGACGTCTTG-3’; IL-5: forward, 5’-GAAAGAGACCTTGACACAGCTG-3’; reversed, 5’-GAACTCTTGCAGGTAATCCAGG-3’; and β-actin: forward, 5'-ACC GTG AAA AGA TGA CCC AGA TC -3'; reverse, 5'-TAG TTT CAT GGA TGC CAC AGG -3'. The resulting PCR products were subjected to agarose electrophoresis. For real-time PCR, the specific primers were as follows: IL-5: forward, 5'- GGG CTT CCT GCT CCT ATC TA -3'; reverse, 5'- CAG TCA TGG CAC AGT CTG AT -3'; STAT-6: forward, 5'- CTC TGT GGG GCC TAA TTT CCA -3'; reverse, 5'- CAT CTG AAC CGA CCA GGA ACT -3'; GATA-3: forward, 5'- CTC GGC CAT TCG TAC ATG GAA -3'; reverse, 5'- GGA TAC CTC TGC ACC GTA GC -3'; and β-actin: forward primer, 5’- TACAGCTTCACCACCACAGC-3’, reverse primer, 5’- TCTCCAGGG AGGAAGAGGAT-3’. Quantitative determination of gene expression levels using a 2-step cycling protocol was performed on a MyIQ Cycler (Bio-Rad, Hercules CA). Relative expression levels were calculated using the 2[-Delta Delta C(T)] method (13). Quantities of all targets from the test samples were normalized to the mouse β-actin housekeeping gene.
Nuclear extracts were prepared from spleens collected from the different experimental groups using an extraction kit (Active Motif, Carlsbad, CA) according to the manufacturer’s instructions. Nuclear extracts were then subjected to EMSA as described (14) using a radiolabeled oligonucleotide containing the STAT-6 consensus sequence 5'–GTA TTT CCC AGA AAA GGA AC–3’ (Santa Cruz Biotech).
Chromatin immunoprecipitation assay (ChIP)
Splenocytes were treated with 10 ng/ml IL-4 for 14 h and fixed with formaldehyde to cross-link the chromatin and nuclear proteins. The ChIP assay was conducted using a kit from Active Motif following to the manufacturer's instructions. Briefly, after enzymatic shearing, antibodies against mouse GATA-3 were added to precipitate the sheared chromatin. After cross-linking reversal, DNA was isolated and one-tenth of the final precipitated DNA was used in each reaction. The following primers were used to target the GATA-3-binding site on the mouse IL-5 promoter: 5'-TCGCCTTTATTAGGTGTCCTC-3' and 5'-GGCCTTCAGCAAAGGAAGAG-3' to target the −70 to −59 region.
Data analysis
All data are expressed as means ± SEM of values from at least six mice per group or triplicate cell preparations. PRISM software (GraphPad, San Diego, CA) was used to analyze the differences between experimental groups by Two Way ANOVA followed by Bonferroni post-tests to compare replicate means by row.
Results and Discussion
PARP-1 gene knockout reduces allergen-induced IL-5 production at the mRNA level without affecting expression or activation of IL-4 receptor-associated kinases
The mechanism by which PARP-1 inhibition results in a reduction in eosinophilic infiltration in a mouse model of asthma remains unclear. The ability of IL-5 replenishment, but not of either IL-4 or IgE, to restore eosinophilia in OVA-treated PARP-1−/− mice suggested initially that PARP-1 may play a role upstream of IL-5 but downstream of IL-4. We postulated that PARP-1 gene deletion may be associated with a disconnect between IL-4 and IL-5, potentially through dysfunctional IL-4 receptor (IL-4R)-associated signal transduction. Figure 1A confirms the significant reduction in IL-5 production in BAL fluids of OVA-challenged PARP-1−/− mice compared with their wild-type (WT) counterparts (5, 6, 15). An important question to address was whether the effect of PARP-1 gene deletion on IL-5 occurred at the transcriptional level. Fig. 1B (upper panels) shows that PARP-1 gene knockout was associated with a severe reduction in IL-5 mRNA upon OVA challenge compared with their WT counterparts as assessed by RT-PCR analysis of RNA isolated from spleens of the different experimental groups and confirmed by real-time PCR (Fig. 1B, bottom panel). These results suggest that PARP-1 may regulate IL-5 at the mRNA level. We and others have shown the involvement of PARP-1 in the transcription of many inflammatory genes, namely those dependent on NF-κB (16, 17), through a number of processes including direct transcription factor binding (reviewed (18).
Figure 1. PARP-1 gene deletion-associated inhibition of IL-5 occurs at the mRNA level without an effect on IL-4R activation.
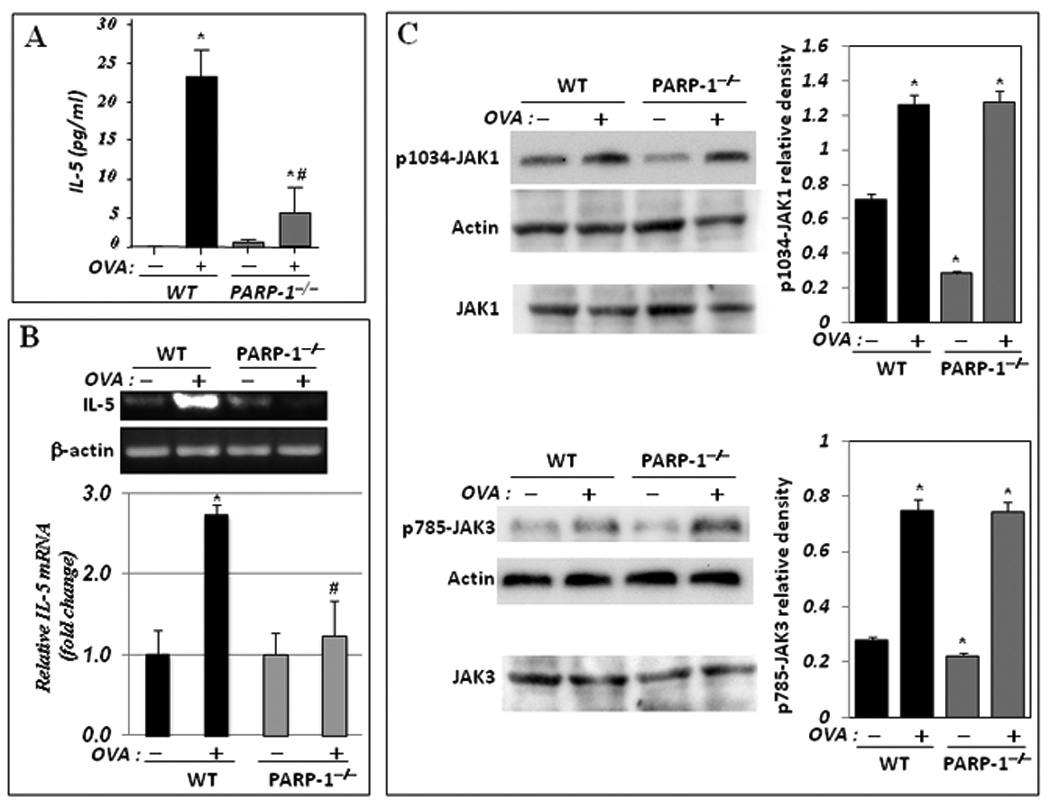
(A) WT and PARP-1−/− mice were sensitized to and challenged with OVA. Mice were then sacrificed and lungs were subjected to BAL and spleens were collected for RNA or protein extraction. (A) BAL fluids were assessed for IL-5 using a BioRad single-plex system. Data are given as means ± SEM of values obtained from at least six mice per group. *, difference from unchallenged mice, p < 0.01; #, difference from WT mice subjected to the OVA challenge, p < 0.01. (B) Total RNA, extracted from portions of the collected spleens, was subjected to cDNA generation followed by conventional (upper panels) or real-time (bottom panel) PCR with primers specific to murine IL-5 or β-actin. (C) Protein extracts were prepared from the remaining portions of the collected spleens and subjected to immunoblot analysis with antibodies to JAK1, JAK3, the phosphorylated form of JAK1 at tyrosine residue 1034 (p1034-JAK1), the phosphorylated form of JAK3 at tyrosine residue 785 (p785-JAK3), or actin. Note that JAK1 and JAK3 blots (C, bottom panels) are of the same samples used for p1034-JAK1 and p785-JAK3, respectively but were generated using a different gel. The immunoblots were quantified using Adobe Photoshop CS and data is expressed as relative density; *, Difference from untreated WT control, p< 0.01.
Signal transduction through the IL-4 receptor is a complex and a crucial pathway that promotes the effects of the T cell-mediated pathogenesis of asthma (12). To determine whether the decrease in IL-5 mRNA expression is linked to a defect in IL-4R-associated signal transduction, we examined the expression levels and activation states of JAK1 or JAK3 upon OVA challenge. Figure 1C shows that PARP-1 gene deletion affected neither the integrity of JAK1 and JAK3 expression nor their activation as assessed by phosphorylation on tyrosines 1034 and 785, respectively. These results clearly suggest that the effect of PARP-1 gene deletion on IL-5 mRNA expression may occur after receptor activation through JAK1 and JAK3 phosphorylation
PARP-1 inhibition is associated with STAT-6 degradation in spleens in an allergen-dependent manner and linked to a severe reduction in GATA-3 expression
The phosphorylated residues on JAK1 and JAK3 serve as docking sites for STAT-6 (10, 11). (12, 19). Subsequently, STAT-6 binds to the phosphorylated cytoplasmic sequences, becomes phosphorylated, and disengages from the receptor. Phosphorylated STAT-6 then homodimerizes and translocates into the nucleus, where it serves as a transcription factor for various genes including GATA-3, which in turn drives the expression of IL-5 (10, 11). Accordingly, we next examined the fate of STAT-6 in WT or PARP-1−/− mice upon OVA challenge. The expression levels of STAT-6 appeared to be comparable between naïve WT and PARP-1−/− mice (Fig. 2A). It is important to note that OVA challenge culminated in STAT-6 phosphorylation in spleens of WT mice and that such event was largely absent in OVA-challenged PARP-1−/− mice (Fig. 2B). Surprisingly, while the levels of STAT-6 remained largely unchanged in OVA-challenged WT mice, its levels were severely reduced in spleens of PARP-1−/− mice upon OVA challenge (Fig. 2A), suggesting involvement of an allergen-induced phenomenon. Fig. 2C shows that STAT-6 DNA binding activity, as assessed by EMSA, was almost completely absent in nuclear extracts isolated from spleens of OVA-challenged PARP-1−/− mice as compared to their WT counterparts confirming the results attained using immunoblot analysis.
Figure 2. PARP-1 gene deletion promotes STAT-6 protein degradation in spleens in an allergen-dependent manner with a consequent reduction in GATA-3 expression.
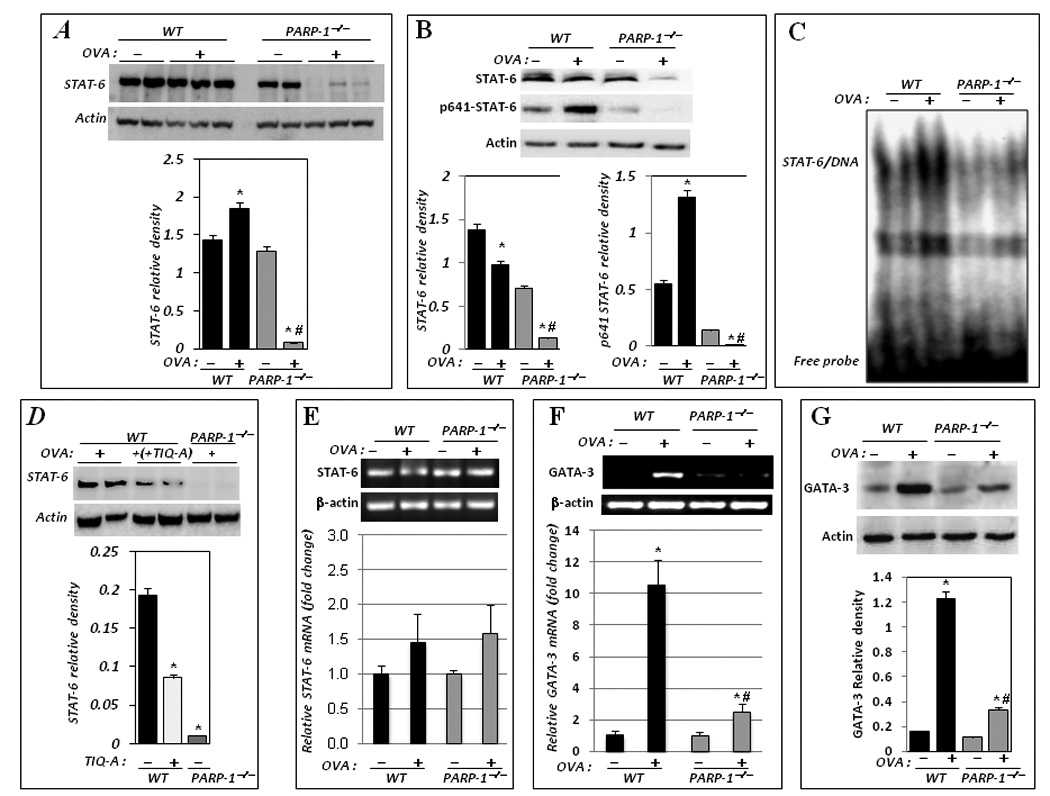
Spleens from the different experimental groups were subjected to total or nuclear protein or RNA extraction. Total protein extracts were then subjected to immunoblot analysis with antibodies to STAT-6 or actin (A) or to its phosphorylated form on tyrosine residue 641 (p641-STAT-6) (B); the immunoblots were quantified using Adobe Photoshop CS and data is expressed as relative density; *, difference from untreated control, p< 0.05; #, difference from OVA-treated WT mice, p< 0.05. (C) Nuclear extracts were subjected to EMSA using a radiolabeled oligonucleotide containing the STAT-6 consensus sequence. (D) Mice received an i.p. injection of TIQ-A (6 mg/kg) or vehicle alone 1 h prior OVA challenge. Mice were sacrificed 24 h later. Spleens were removed for protein extraction after which proteins were subjected to immunoblot analysis with antibodies to STAT-6 or actin; extracts from spleens of OVA-challenged PARP-1−/− mice were used as a control for STAT-6 degradation. The immunoblots were quantified as above and data is expressed as relative density; *, difference from OVA-treated WT mice, p< 0.05. Total RNA was subjected to cDNA generation followed by conventional (upper panels) or real-time (lower panels) PCR with primers specific to STAT-6 (E) or GATA-3 (F); β-actin was used as an internal control. *, difference from OVA-treated WT mice, p< 0.01. (G) Protein extracts were subjected to immunoblot analysis with antibodies to murine GATA-3 or actin.
The involvement of PARP-1 in the fate of STAT-6 upon OVA challenge was confirmed using the specific and potent inhibitor of the enzyme TIQ-A (Fig. 2D). The latter results also indicate that the actual activity of the PARP-1 is necessary for STAT-6 protein integrity upon allergen exposure. Interestingly, STAT-6 mRNA levels were not altered in spleens of either WT or PARP-1−/− mice subjected to OVA challenge or left untreated as assessed by conventional and confirmed by real-time PCR (Fig. 2E); a slight increase in STAT-6 message was observed in spleens of both WT and PARP-1−/− upon OVA exposure. These results suggest that the effect of PARP-1 gene knockout in reducing STAT-6 levels was not due to a change in transcription. Rather, PARP-1 gene knockout may promote conditions conducive to the degradation of the signal transducer. As stated above, GATA-3 is a key transcription factor necessary for driving IL-5 expression and directly dependent on STAT-6 activation (20). GATA-3 gene expression requires binding of STAT-6 to its promoter. Accordingly, we surmised that the loss of STAT-6 would lead to a reduction in GATA-3 expression. Indeed, STAT-6-reduced expression in spleens of OVA-challenged PARP-1−/− mice coincided with a severe reduction in GATA-3 mRNA as assessed by conventional and confirmed by real-time PCR (Fig. 2F), which culminated in a drastic reduction in protein levels as assessed by immunoblot analysis (Fig. 2G). Together, these results suggest that PARP-1 may regulate IL-5 production by influencing the integrity of STAT-6, affecting, as a result, the downstream expression levels of GATA-3 mRNA and protein.
The reduction of STAT-6 levels associated with PARP-1 gene knockout upon allergen exposure may occur in response to IL-4 stimulation
Given that PARP-1 gene deletion may be associated with a disconnect between IL-4-mediated signal transduction and IL-5 expression, we wished to determine whether OVA-induced STAT-6 degradation in PARP-1−/− mice may be achieved by exposure to IL-4 alone. Intraperitoneal administration of IL-4 induced little to no change in the expression levels of STAT-6 in the spleens of treated WT mice (Fig. 3A). However, administration of IL-4 induced a marked and significant reduction in the expression levels of STAT-6 in the spleens of treated PARP-1−/− mice (Fig. 3B). To expand our efforts in understanding the mechanism of the association between PARP-1 gene deletion and STAT-6 degradation, we examined whether direct exposure to cytokines would induce degradation of STAT-6 in an in vitro splenocyte system. Figure 3C shows that STAT-6 protein expression was markedly reduced in IL-4-treated splenocytes derived from PARP-1−/− mice; such an effect was not observed in IL-4-treated splenocytes derived from WT mice. The reduction in STAT-6 levels in IL-4-treated PARP-1−/− splenocytes culminated in a severe reduction in IL-5 mRNA expression as compared to the WT counterparts (Fig. 3D) as assessed by RT-PCR. To fully confirm that such event took place at the promoter level, we examined the effect of PARP-1 gene deletion on GATA-3 occupancy of its binding site on the IL-5 promoter in IL-4 simulated splenocytes. Fig. 3E shows that in WT splenocytes, IL-4 treatment induced a robust binding of GATA-3 to its site on the −70 to −59 region of the mouse IL-5 gene promoter using a ChIP assay with antibodies against the transcription factor. The GATA-3 binding to the IL-5 gene promoter was largely absent in IL-4-treated PARP-1−/− splenocytes (Fig. 3E) strongly supporting the notion that PARP-1 is required for IL-5 gene expression by affecting the fate of STAT-6 and the consequent expression of GATA-3 in response to IL-4 exposure. These results confirm our hypothesis regarding the stage at which PARP-1 inhibition may affect IL-5 expression and ultimately eosinophilia. A number of reports have demonstrated the effect of decreased STAT-6 expression through knockout studies; indeed, Stat-6−/− mice are unable to mount a Th2 response (for review (19). In addition to STAT-6, NF-κB has been shown to play a role in regulating GATA-3 expression (20). Given the intimate relationship between PARP-1 and NF-κB, it is conceivable that PARP-1 may regulate GATA-3 through both STAT-6 and NF-κB. Unraveling such a possibility certainly requires additional experimentation.
Figure 3. IL-4 promotes a severe reduction in STAT-6 protein levels spleens of treated PARP-1−/− mice and in vitro in treated PARP-1−/− splenocytes.
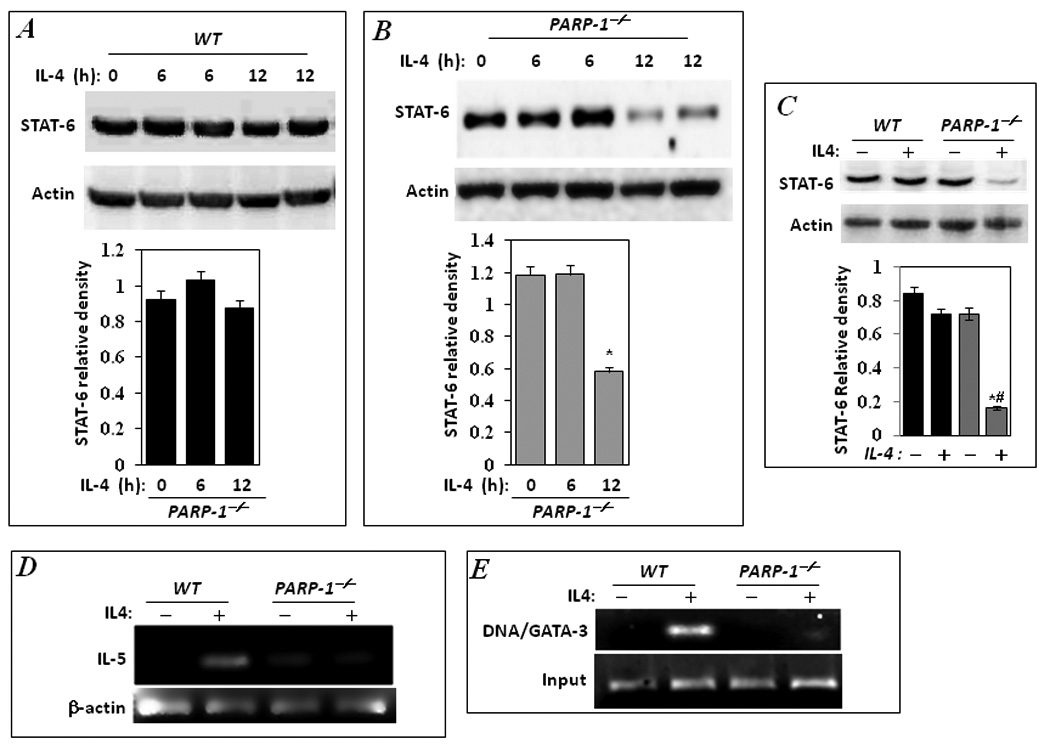
WT (A) and PARP-1−/− (B) mice received a single i.p. injection of IL-4 (100ng/mouse). Mice were sacrificed 6 or 12 h later and spleens were removed and processed for protein isolation. Proteins were then subjected to immunoblot analysis with antibodies to STAT-6 or actin; the immunoblots were quantified using Adobe Photoshop CS and data is expressed as relative density; *, difference from untreated control, p< 0.05. Splenocytes were isolated from naive WT or PARP-1−/− mice and stimulated with IL-4 (10ng/ml) for 12 hours. Cells were lysed then subjected to total protein or RNA extraction. A portion of the cells was also subjected to cross-linking with formaldehyde as described in the Methods. (C) Protein extracts were subjected to immunoblot analysis with antibodies to STAT-6 or actin. The immunoblots were quantified and data is expressed as relative density; *, difference from untreated control, p< 0.05; #, difference from IL-4-treated WT mice, p< 0.05. (D) Total RNA was subjected to cDNA generation followed by conventional PCR with primers specific to mouse IL-5; β-actin was used as an internal control. (E) Formaldehyde-fixed cells were subjected to ChIP assay using antibodies to GATA-3. Levels of immunoprecipitated chromatin fragments (−70 to −59 region) of the mouse IL-5 gene promoter input were examined by PCR.
Regulation by protein degradation has been reported for STAT-6 (21, 22). However, such regulation was strictly associated with the phosphorylation status of STAT-6, as the fate of its unphosphorylated form remains largely unaltered (21, 23, 24). Interestingly, however, Andrews et al. (25) reported that STAT-6 could be continuously activated upon IL-4 exposure through a constant cycle of activation, deactivation, nuclear export, and reactivation, implying that protein degradation does not play a major role in the downregulation of activated STAT-6. Accordingly, the results of our study uncovered a novel mechanism by which STAT-6 is regulated, shedding important light on the underlying mechanism by which PARP-1 inhibition blocks IL-5 production and subsequent eosinophilia upon allergen exposure.
PARP-1 knockout-associated STAT-6 degradation upon IL-4 stimulation is mediated by calpain but not by proteasome-associated proteolytic activity
As stated above, a number of reports suggested the involvement of proteases in the regulation of STAT-6 (primarily that of the phosphorylated form) upon IL-4 exposure (21, 22). The two major proteases suggested to play a role in the degradation of the phospho-STAT-6 are proteasomes and calpains (22). Accordingly, we surmised that PARP-1 gene deletion may promote STAT-6 degradation by enhancing the activity of either proteasomes or calpains. We therefore examined the effect of the proteasome inhibitor, MG132, and the calpain inhibitor, ALLN, on the integrity of STAT-6 in PARP-1−/− splenocytes upon IL-4 stimulation. Figure 4A and B shows, contrary to our prediction, that inhibition of proteosomal activity by MG132 did not block STAT-6 degradation in PARP-1−/− splenocytes upon IL-4 treatment despite a clear accumulation of ubiquitinated proteins, indicative of proteasomal inhibition (Fig. 4C). Interestingly, treatment with the calpain inhibitor markedly blocked STAT-6 degradation in IL-4-treated PARP-1−/− splenocytes (Fig. 4A and B). Treatment with ALLN also moderately increased the level of STAT-6 in IL-4-treated WT cells (data not shown). It is important to note that ALLN exhibits some inhibitory activity on proteasomes as shown by numerous reports; however, it is considered a better calpain inhibitor. Since MG132 did not block STAT-6 degradation, the blockage of STAT-6 degradation by ALLN may be strictly related to the inhibition of calpains.
Figure 4.
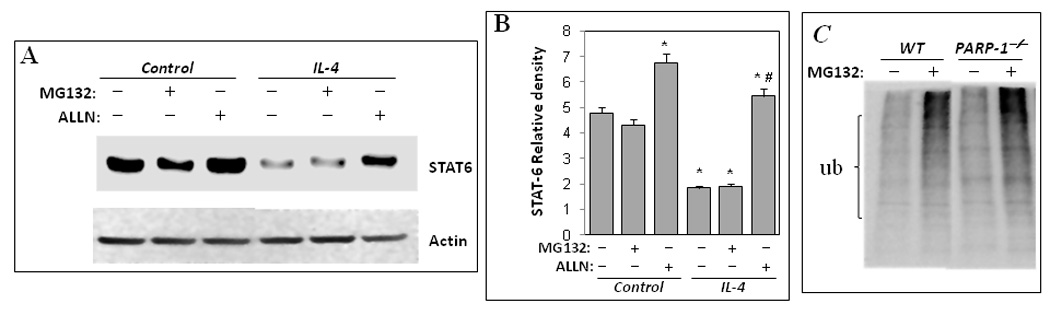
Calpain inhibition blocks degradation of STAT-6 in PARP-1−/− splenocytes upon IL-4 stimulation. Splenocytes isolated from WT or PARP-1−/− mice were treated with IL-4 in the absence or presence of 20 µM of the proteasome inhibitor MG132 or the calpain inhibitor ALLN. Cells were collected 12 h after treatment and subjected to protein extraction. Proteins were then subjected to immunoblot analysis with antibodies to STAT-6 or actin. (B) The immunoblot results in A were quantified and expressed as STAT-6 relative (over actin). *Difference from the control that received neither IL-4 nor protease inhibitors; #, Difference from IL-4-treated cells without protease inhibitors, p< 0.01. (C) Lysates from control cells that were treated with MG132 or left untreated were subjected to immunoblot analysis with antibodies to ubiquitin.
Together, these results show another important and novel function of PARP-1 in regulating the function of a transcription factor critical for mediating inflammation. A great deal of work has focused on the mechanism by which PARP-1 influences transcription factors by focusing primarily on promoter region binding (reviewed (26). The influence of PARP-1 on STAT-6 integrity is likely independent of ability of the transcription factor to bind target genes such as GATA-3; rather, PARP-1 may influence the targeting process via degradation by calpain but not by proteasomes. Interestingly, studies by Ullrich et al. (27, 28) and Ullrich & Grüne (29) reported that PARP-1 activity may influence 20S proteasome-mediated degradation of damaged histones in tumor cells upon exposure to oxidative stress. Proteasome and PARP-1 were shown to play closely interacting roles during the removal of protein carbonyls, single-strand breaks and 8-hydroxy-2'-deoxyguanosine enhancing, as a result, the selective degradation of oxidatively damaged histones (30). Although our results do not categorically rule out the potential involvement of proteasomes in STAT-6 degradation in PARP-1−/− cells upon allergen or IL-4 exposure, the data do suggest that calpains may be the primary culprits in this specific situation.
It is noteworthy that the involvement of PARP-1 enzymatic activity in regulating transcription factors remains controversial (18, 31). Our laboratory demonstrated that such enzymatic activity is crucial for the activation of at least NF-κB (4, 17). The transcriptional activity of a number of other factors including NFAT, HES1, Sp1, and Elk1 has also been shown to require the enzymatic activity of PARP-1 (reviewed (18). Virag et al (8) reported that inhibition of PARP-1 enzymatic activity with a drug termed PJ34 does not reduce IL-5 production upon OVA exposure leading to the conclusion that the blockade of allergen-induced airway inflammation by the drug may be attributable to a reduction in MIP-1α, IL-12, and TNF. The failure of PJ34 to reduce IL-5 production upon OVA-challenged mice may be attributable to the marginal elevation (1.5–2 folds) in the cytokine, which is largely different from the 20–25 folds increase observed in the model used in the current study (see Fig. 1). It is noteworthy that the model used in the latter study exhibited basal inflammatory markers in non-sensitized control mice including eosinophils, IL-5, and IL-13, which may have confounded the effect of PARP-1 inhibition on the cytokine. The additional effect of the PARP-1 inhibitor TIQ-A on STAT-6 expression reported in this study provides additional support for the role of PARP-1 in IL-5 gene expression and ultimate production of the cytokine. How PARP-1 influences the fate of STAT-6 upon allergen exposure or direct stimulation by IL4 will certainly require extensive and detailed studies. Such studies should be aimed at determining the potential post-translational changes to STAT-6 that render it more susceptible to degradation by calpain. It is important to mention that activation of STAT-6 may be required for its degradation by calpains, since this phenomenon was only observed upon allergen exposure in vivo or treatment with IL-4 both in vivo and in vitro. These results provide additional support for the potential of PARP-1 inhibition as a viable strategy to block allergen-induced inflammation. It noteworthy that the relationship between PARP-1 and human asthma has recently been exemplified by the report showing that the PARP-1 Val762Ala polymorphism that reduces its enzymatic activity by 40% was associated with a reduced risk of asthma in humans (32).
Acknowledgments
Funding Sources: This work was supported in part by grants HL072889 from the National Institute of Health and funds from the Louisiana Cancer Research Consortium to A. H. Boulares as well as by a National Institute of Health NRSA fellowship to Rahul Datta.
Abbreviations
- PARP-1
Poly(ADP-ribose)polymerase-1
- STAT-6
Signal Transducer and Activator of Transcription-6
- OVA
Ovalbumin
- JAK
Janus Kinase
- BAL
bronchoalveolar lavage
- ALLN
Acetyl-L-Leucyl-L-Leucyl-L-Norleucinal
- GATA-3
GATA binding protein-3
- IL-4R
IL-4 receptor
Footnotes
- Rahul Datta: The graduate student who led the study and conducted most of the experiments. No conflict to report.
- Amarjit S. Naura: Contributed to the training, experimental design, troubleshooting and conduction of some of the in vivo experiments. No conflict to report.
- Mourad Zerfaoui: Contributed to the training, troubleshooting and conduction of some of the in vitro experiments. No conflict to report.
- Youssef Errami: Conducted some the in vitro experiments and the statistical analyses. No conflict to report.
- Mustapha Oumouna: The original scientist who contributed to the original observation on IL-5 and the first to train the lead author. No conflict to report
- Hogyoung Kim: Conducted some of the in vitro experiments and help in the troubleshooting. No conflict to report.
- Jihang Ju: Conducted some of the in vitro experiments and help in the troubleshooting. No conflict to report.
- Virginia P Ronchi: Discussion and advice on the proteasome experiment; also provided reagents. No conflict to report.
- Arthur Haas: Discussion and advice on the proteasome experiment; also provided reagents. No conflict to report.
- Hamid Boulares: The principal investigator; contributed to the design of the experiment and the training of the first author as well as provided the financial support for the conducted work. No conflict to report.
References
- 1.Pacher P, Szabo C. Role of Poly(ADP-ribose) polymerase 1 (PARP-1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors. Cardiovasc Drug Rev. 2007;25(3):235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virag L. Poly(ADP-ribosyl)ation in asthma and other lung diseases. Pharmacol Res. 2005;52(1):83–92. doi: 10.1016/j.phrs.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Gero D, Szabo C. Poly(ADP-ribose) polymerase: a new therapeutic target? Curr Opin Anaesthesiol. 2008;21(2):111–121. doi: 10.1097/ACO.0b013e3282f63c15. [DOI] [PubMed] [Google Scholar]
- 4.Boulares AH, Zoltoski AJ, Sherif ZA, Jolly P, Massaro D, Smulson ME. Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2003;28(3):322–329. doi: 10.1165/rcmb.2001-0015OC. [DOI] [PubMed] [Google Scholar]
- 5.Oumouna M, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177(9):6489–6496. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 6.Naura AS, Hans CP, Zerfaoui M, You D, Cormier SA, Oumouna M, et al. Post-allergen challenge inhibition of poly(ADP-ribose) polymerase harbors therapeutic potential for treatment of allergic airway inflammation. Clin Exp Allergy. 2008;38(5):839–846. doi: 10.1111/j.1365-2222.2008.02943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki Y, Masini E, Mazzocca C, Cuzzocrea S, Ciampa A, Suzuki H, et al. Inhibition of poly(ADP-ribose) polymerase prevents allergen-induced asthma-like reaction in sensitized Guinea pigs. J Pharmacol Exp Ther. 2004;311(3):1241–1248. doi: 10.1124/jpet.104.072546. [DOI] [PubMed] [Google Scholar]
- 8.Virag L, Bai P, Bak I, Pacher P, Mabley JG, Liaudet L, et al. Effects of poly(ADP-ribose) polymerase inhibition on inflammatory cell migration in a murine model of asthma. Med Sci Monit. 2004;10(3):BR77–BR83. [PubMed] [Google Scholar]
- 9.Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med. 2004;10(10):493–499. doi: 10.1016/j.molmed.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Haque SJ, Sharma P. Interleukins and STAT signaling. Vitam Horm. 2006;74:165–206. doi: 10.1016/S0083-6729(06)74007-9. [DOI] [PubMed] [Google Scholar]
- 12.Steinke JW, Borish L. Th2 cytokines and asthma. Interleukin-4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res. 2001;2(2):66–70. doi: 10.1186/rr40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Boulares HA, Giardina C, Navarro CL, Khairallah EA, Cohen SD. Modulation of serum growth factor signal transduction in Hepa 1–6 cells by acetaminophen: an inhibition of c-myc expression, NF-kappaB activation, and Raf-1 kinase activity. Toxicol Sci. 1999;48(2):264–274. doi: 10.1093/toxsci/48.2.264. [DOI] [PubMed] [Google Scholar]
- 15.Naura AS, Datta R, Hans CP, Zerfaoui M, Rezk BM, Errami Y, et al. Reciprocal regulation of iNOS and PARP-1 during allergen-induced eosinophilia. Eur Respir J. 2009;33(2):252–262. doi: 10.1183/09031936.00089008. [DOI] [PubMed] [Google Scholar]
- 16.Zerfaoui M, Suzuki Y, Naura AS, Hans CP, Nichols C, Boulares AH. Nuclear translocation of p65 NF-kappaB is sufficient for VCAM-1, but not ICAM-1, expression in TNF-stimulated smooth muscle cells: Differential requirement for PARP-1 expression and interaction. Cell Signal. 2008;20(1):186–194. doi: 10.1016/j.cellsig.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zerfaoui M, Errami Y, Suzuki Y, Naura AS, Kim H, Ju J, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export of p65 NF-kB and retention upon TLR4 stimulation. J Immunol. 2010;185:1894–1902. doi: 10.4049/jimmunol.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294–302. doi: 10.1016/j.ceb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006;17(3):173–188. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Barnes PJ. Role of GATA-3 in allergic diseases. Curr Mol Med. 2008;8(5):330–334. doi: 10.2174/156652408785160952. [DOI] [PubMed] [Google Scholar]
- 21.Hanson EM, Dickensheets H, Qu CK, Donnelly RP, Keegan AD. Regulation of the dephosphorylation of Stat6. Participation of Tyr-713 in the interleukin-4 receptor alpha, the tyrosine phosphatase SHP-1, and the proteasome. J Biol Chem. 2003;278(6):3903–3911. doi: 10.1074/jbc.M211747200. [DOI] [PubMed] [Google Scholar]
- 22.Hendry L, John S. Regulation of STAT signalling by proteolytic processing. Eur J Biochem. 2004;271(23–24):4613–4620. doi: 10.1111/j.1432-1033.2004.04424.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Dai X, Haas AL, Wen R, Wang D. Proteasome-dependent down-regulation of activated Stat5A in the nucleus. Blood. 2006;108(2):566–574. doi: 10.1182/blood-2005-12-4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Moriggl R, Stravopodis D, Carpino N, Marine JC, Teglund S, et al. A small amphipathic alpha-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. Embo J. 2000;19(3):392–399. doi: 10.1093/emboj/19.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrews RP, Ericksen MB, Cunningham CM, Daines MO, Hershey GK. Analysis of the life cycle of stat6. Continuous cycling of STAT6 is required for IL-4 signaling. J Biol Chem. 2002;277(39):36563–36569. doi: 10.1074/jbc.M200986200. [DOI] [PubMed] [Google Scholar]
- 26.Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol Life Sci. 2002;59(9):1534–1553. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullrich O, Reinheckel T, Sitte N, Hass R, Grune T, Davies KJ. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc Natl Acad Sci U S A. 1999;96(11):6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ullrich O, Ciftci O, Hass R. Proteasome activation by poly-ADP-ribose-polymerase in human myelomonocytic cells after oxidative stress. Free Radic Biol Med. 2000;29(10):995–1004. doi: 10.1016/s0891-5849(00)00399-3. [DOI] [PubMed] [Google Scholar]
- 29.Ullrich O, Grune T. Proteasomal degradation of oxidatively damaged endogenous histones in K562 human leukemic cells. Free Radic Biol Med. 2001;31(7):887–893. doi: 10.1016/s0891-5849(01)00672-4. [DOI] [PubMed] [Google Scholar]
- 30.Catalgol B, Wendt B, Grimm S, Breusing N, Ozer NK, Grune T. Chromatin repair after oxidative stress: role of PARP-mediated proteasome activation. Free Radic Biol Med. 2010;48(5):673–680. doi: 10.1016/j.freeradbiomed.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Altmeyer M, Hottiger MO. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging (Albany NY) 2009;1(5):458–469. doi: 10.18632/aging.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tezcan G, Gurel CB, Tutluoglu B, Onaran I, Kanigur-Sultuybek G. The Ala allele at Val762Ala polymorphism in poly(ADP-ribose) polymerase-1 (PARP-1) gene is associated with a decreased risk of asthma in a Turkish population. J Asthma. 2009;46(4):371–374. doi: 10.1080/02770900902777791. [DOI] [PubMed] [Google Scholar]