

Abstract
Angiosarcoma (AS) is a distinct group of sarcomas characterized by up-regulation of vascular-specific receptor tyrosine kinases, including TIE1, KDR, TEK, and FLT1. In keeping with the clinical heterogeneity, gene expression profiling distinguishes two AS genomic clusters, which correlate with anatomic location and prior exposure to radiation. Furthermore, a high percentage of secondary AS, but not primary AS, show distinct 8q24 chromosomal gains, due to MYC amplification. In this study we mined the transcriptional output of 10 secondary and 11 primary AS to better define the dichotomy in the pathogenesis of these two clinical subsets. The oncogenic role of MYC was investigated further in secondary AS, as well as in radiation-induced atypical vascular lesions (AVL) and other radiation-associated sarcomas. High level MYC amplification was found in 100% of secondary AS, but in none of the AVL or other radiation-associated sarcomas. Co-amplification of FLT4 (encoding VEGFR3) was identified in 25% of secondary AS, but not in other types. Our findings reinforce the distinct pathogenesis of AS subtypes, with MYC amplification being an early, but necessary event in secondary AS. Secondary genetic hits, such as FLT4 gene co-amplification or KDR mutations, may play a role in tumor progression, as well as potential therapeutic targeting.
Keywords: angiosarcoma, radiation, MYC, FLT4, amplification, atypical vascular proliferation
Introduction
Angiosarcoma (AS) is an aggressive malignancy with a 5-year overall survival of ∼30%, characterized by biological heterogeneity based on anatomic primary site. Typical clinical characteristics include multifocal spread, local recurrence, and early hematogenous dissemination. Even with wide excision and irradiation, both locoregional recurrence and metastatic disease are common. Although post-radiation sarcomas are uncommon, ∼40% of all radiation induced sarcomas develop after radiotherapy for breast cancer, most of them being angiosarcoma subtype (Lagrange et al., 2000; Sheppard and Libshitz, 2001).
By array-comparative genomic hybridization, recurrent genetic abnormalities were identified recently in secondary but not primary AS (Manner et al., 2010), with frequent high-level amplifications in chromosome bands 8q24.21 (50%), 10p12.33 (33%) and 5q35.3 (11%). The hot spot in 8q24.21 was defined as being secondary to high-level MYC amplifications, found in more than half of AS associated with radiation and chronic lymphedema, but not in primary tumors (Manner et al., 2010). Amplification of MYC in these tumors did not predispose to higher-grade morphology or increased proliferation. These findings suggest that MYC amplification might be implicated in the pathogenesis of secondary AS. In this study we investigate further the oncogenic role of MYC in secondary AS, radiation-induced pre-malignant/atypical vascular lesions and other radiation-associated sarcomas. The transcriptional output of 21 AS was also mined to better define the dichotomy in the pathogenesis of primary versus secondary AS clinical subsets.
Material and Methods
Histopathology of Experimental and Control Groups
The pathology files were searched for diagnosis of radiation-induced angiosarcoma (AS) and atypical vascular lesions (AVL) arising in the skin or breast from patients treated with radiation for breast cancer. A total number of 12 AVL and 22 secondary AS occurring in the setting of radiation for breast cancer were retrieved with available tissue for further immunohistochemical work-up and molecular analysis. In 2 patients the AVL co-existed with an AS in the mastectomy specimen. The clinical follow-up in the remaining 10 AVL patients ranged from 1-7 years (median of 3 years); none of them progressed to an AS. The secondary AS patients were subdivided in two groups: based on prior radiation therapy for breast cancer (n=20) or the associated chronic lymphedema after breast cancer treatment (n=2). For comparison were also included 18 primary AS (breast, 6; head and neck, 6; bone and soft tissue, 5; visceral, 1), 2 secondary AS after radiation applied for other conditions than breast cancer, and 8 radiation-associated high grade sarcomas, with morphology other than AS (i.e. malignant fibrous histiocytoma). The latter group of tumors developed after radiation for breast, lymphoma or soft tissue tumors (i.e. desmoid tumor). The diagnosis of AS was re-confirmed in all cases based on the vasoformative features on light microcopy and immunoreactivity for CD31. The diagnosis of AVL was rendered in superficial dermal vascular lesions, with a distinct growth pattern parallel to the epidermis and lined by a single layer of hobnail endothelial cells, showing hyperchromatic nuclei with mild atypia (Fig 2A). Overt features of malignancy, such as high nuclear grade, prominent nucleoli or a more solid growth pattern were not associated with a diagnosis of AVL. An immunohistochemical stain for D2-40 (Ventana Medical Systems, Tucson, AZ, pre-diluted) was applied in all the AVL as well as on a TMA containing a primary and secondary AS tumors from 47 patients. The diagnosis of RT-induced sarcoma, NOS, was based on a high grade spindle and pleomorphic sarcoma, lacking vasoformative features and CD31 immunoreactivity.
Fig. 2.
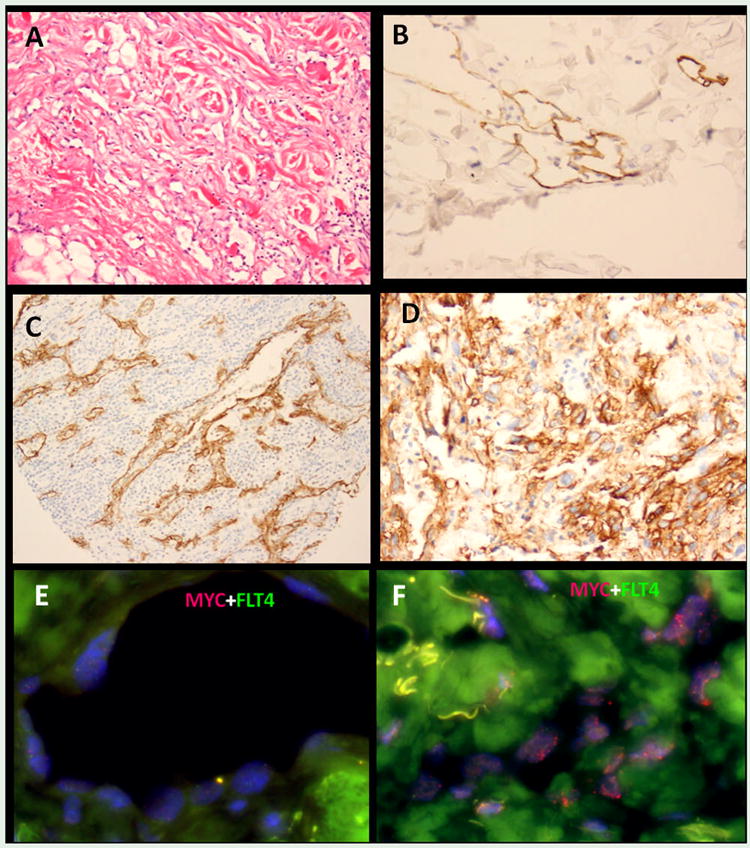
Atypical vascular lesion (AVL) microscopic appearance with dilated channels lined by hobnailed endothelium infiltrating dermis (A) and showing immunoreactivity for D2-40 (B). D2-40 expression is also seen in a subset of AS, occasionally with a strong pattern staining, as seen in this scalp AS, associated with brisk lymphoid infiltrate (C) or in a spleen AS (D). FISH analysis showed no abnormalities of MYC or FLT4 by FISH in the AVL (E), however the adjacent RT-induced AS diagnosed synchronously showed evidence of MYC amplification (F).
Gene Expression Profiling of Angiosarcoma
The transcriptional profiling data from 21 AS samples previously studied on the U133A Affymetrix platform was re-analyzed (Antonescu et al., 2009) for differentially expressed genes in primary versus secondary AS. The expression intensities were normalized using the robust multiarray average method, which includes background adjustment, quantile normalization across arrays, and probe-level expression measure summarization using median polish on the log2 scale, for each probe set. A list of differentially expressed genes between primary versus secondary AS was obtained based on fold changes (FC) > 2 and a P value cutoff of <0.001.
Fluorescence In Situ Hybridization (FISH)
FISH on interphase nuclei from paraffin embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BACs), covering MYC (RP11-440N18), TPD52 (RP11-668P13; RP11-880M10), and FLT4 (RP11-586L9). BAC clones were chosen according to USCS genome browser (http://genome.uscs.edu). The BAC clones were obtained from BACPAC sources of Children's Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer's instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described (Antonescu et al., 2009). Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). The genomic location of each BACs set was verified by hybridizing them to normal metaphase chromosomes.
MYC overexpressing cell lines, Western Blotting and Co-Immunoprecipitation
Two cell lines with oncogenic MYC activation were used for studying MYC isoforms expression and MYC/MAX interaction. First, a MYC amplified human breast adenocarcinoma cell line, SKBR3, was obtained from ATCC, which was grown in modified McCoy's 5A medium + 10% fetal bovine serum. Second, NIH-3T3 cells were transfected with a pMSCV-Myc-IRES-GFP (Addgene Inc, Cambridge, MA), containing a wild type human MYC cDNA cloned into a mouse stem cell retroviral vector. The transfection was performed using GenJet DNA lipofectamine reagent Ver.II (Signagen Laboratories, Gaithersburg MD), according to the manufacturer's protocol. Transfected cells were selected by flow cytometry for GFP expression.
Western Blotting (WB) was performed to assess the expression of MYC protein and investigate the preferential expression of MYC isoforms within different AS subtypes. In addition, co-immunoprecipitation (IP) was used to identify the MYC-MAX interaction and heterodimerization in both human AS tumor samples and MYC-expressing cell lines. Frozen tissue from primary and secondary AS samples and cells from MYC-transfected NIH-3T3 and SKBR3 were homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors as previously described (Antonescu et al., 2005; Guo et al., 2007). Blots were probed with anti-MYC monoclonal antibody, 9E10 (Abcam, Cambridge, MA), anti-MAX monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-VEGFR2 antibody (Cell Signaling Technology, Danvers, MA). MAX protein was co-IP with MYC protein using mouse monoclonal anti-MYC antibody and protein A coated agarose beads. Immunoprecipitates were subjected to Western Blotting, both MYC and MAX being detected with rabbit polyclonal antibodies against MYC and MAX (Cell Signaling Technology, Danvers, MA).
Results
D2-40 immunostaining is consistently seen in AVL and a subset of AS
All except one AVL showed reactivity for D2-40, and the reactivity varied from strong and diffuse (Fig. 2B), to often more weak and patchy pattern of staining. In contrast D2-40 immunostaining was negative in 74% of AS tested. D2-40 was focal and weak in 11% of cases, while in 7 (15%) it showed a diffuse and strong expression, with no apparent correlation with the anatomic location of AS (RT-breast, 3; head and neck, 2; soft tissue, 1; visceral, 1) (Fig 2 B,C). CD31 immunoreactivity was uniformly positive in both AVL and AS.
MYC overexpression is a hallmark of secondary AS
As prior genomic studies suggest a divergence in the pathogenesis of AS based on the prior exposure to radiation, we hypothesized that transcriptional output may provide further insight in defining these 2 clinical subsets. Thus we compared the gene expression profile of 10 secondary AS, including 8 RT-induced and 2 post-lymphedema (PL) AS to a group of 11 primary AS. Using a cutoff of p<0.001 and fold change (FC) >2, there were 526 genes differentially expressed between the 2 groups. The top 6 genes upregulated in secondary AS were: SCN3A (sodium channel, voltage-gated, type III, alpha subunit; FC, 10.1), PRAME (preferentially expressed antigen in melanoma; FC, 8.4), COL9A3 (collagen, type IX, alpha 3; FC, 8.0), RELN (reelin; FC, 7.6), CETP (cholesteryl ester transfer protein, plasma; FC, 6.0) and MYC (v-myc myelocytomatosis viral oncogene homolog (avian); FC, 5.9)(Fig. 1 - bar chart, Table 1).
Fig. 1.
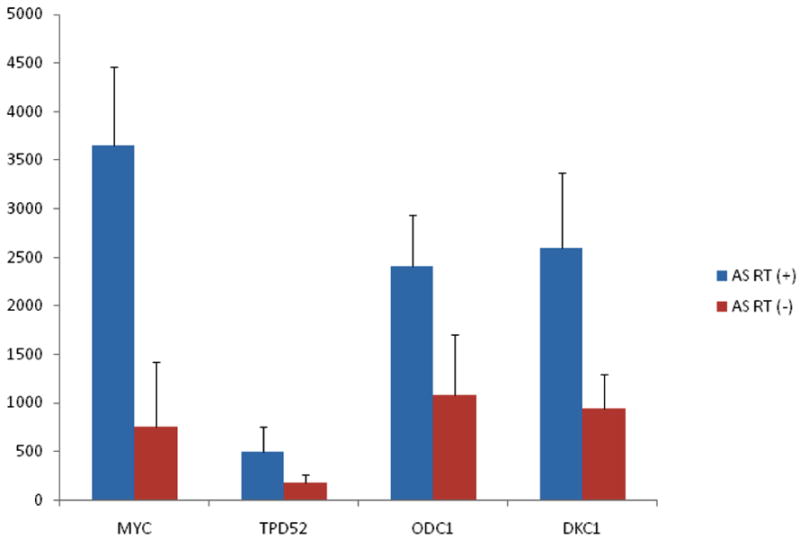
MYC and TPD52 mRNA expression levels were upregulated in secondary compared to primary AS tumors. MYC downstream genes, ODC1 and DKC1 were also upregulated in secondary AS.
Table 1. Differentially expressed genes in secondary AS in comparison with primary AS.
| Gene Symbol | Gene title | Fold change | Chromosomal location | Gene ontology biological process |
|---|---|---|---|---|
| SCN3A | Sodium channel, voltage-gated, type III, alpha subunit | 10.1 | 2q24 | Transport |
| PRAME | Preferentially expressed antigen in melanoma | 8.4 | 22q11.22 | -- |
| COL9A3 | Collagen, type IX, alpha 3 | 8.0 | 20q13.3 | -- |
| RELN | Reelin | 7.6 | 7q22 | Cell morphogenesis involved in differentiation |
| CETP | Cholesteryl ester transfer protein, plasma | 6.0 | 16q21 | Lipid metabolic process |
| MYC | Myc myelocytomatosis viral oncogene homolog (avian) | 5.9 | 8q24.21 | B cell apoptosis |
| FLT4 | FMS-like tyrosine kinase 4 | 5.3 | 5q35.3 | Protein amino acid phosphorylation |
| TPD52 | Tumor protein D52 | 2.8 | 8q21 | Anatomical structure morphogenesis |
| DKC1 | Dyskeratosis congenita 1, dyskerin | 2.8 | Xq28 | Pseudouridine synthesis |
| ODC1 | Ornithine decarboxylase 1 | 2.6 | 2p25 | Kidney development |
As MYC up-regulation appears to play an important role in secondary AS, we then investigated the expression levels of other MYC target genes. Two direct transcriptional targets of MYC, ODC1 (ornithine decarboxylase 1; FC, 2.2) and DKC1 (dyskeratosis congenita 1; FC, 2.8) were overexpressed in secondary AS group, with P <0.001. In addition, TPD52 (tumor protein D52; FC, 2.8) located in 8q21, a gene that is amplified in a subset of breast cancers in association with or independent of MYC gene amplification, was found to be upregulated in secondary AS. In contrast, MAX, the heterodimerization partner of MYC, showed low level expression in all AS tumors and did not parallel the high expression of MYC in secondary AS. The FLT4 (FMS-like tyrosine kinase 4, FC, 5.3), encoding the vascular endothelial growth factor receptor 3 (VEGFR3), also showed high level of expression in secondary AS.
Consistent MYC gene amplification is detected in all radiation and chronic lymphedema associated AS, but not in radiation-induced AVL or other AS subgroups
High level MYC amplification by FISH was detected in all 21 samples from 20 patients with radiation-induced AS (Fig. 3A). Both patients with AS developed in the setting of chronic lymphedema secondary to breast surgery and radiation also showed similar pattern of MYC amplification. However, no gene copy abnormalities were detected in any of the 12 AVL tested. Furthermore, no MYC abnormality was detected in the AVL lesions seen adjacent to the radiation-induced AS present within the same mastectomy resection in 2 patients (Fig. 2D). In both cases, MYC amplification was detected only in the AS cells but not in the synchronous AVL (Fig. 2E). No MYC amplification was detected in the original breast carcinoma available for FISH analysis in four patients who subsequently developed AS or sarcoma, NOS, after radiation therapy. No MYC amplifications were detected in any of the 18 primary AS, regardless of anatomic location (Fig. 3C). Furthermore, there was no MYC amplification in any of the radiation-induced sarcoma with a non-AS morphology (Fig. 3D). MYC was also found to be amplified in SKBR3, confirming previous results based on Southern blotting analysis (Kozbor and Croce, 1984). Only one of the 2 AS developed in the setting of radiation for other conditions than breast cancer showed evidence of MYC amplification. This patient received radiation therapy for a low grade fibromyxoid sarcoma of the thigh and subsequently developed a high grade angiosarcoma 12 years later.
Fig 3.
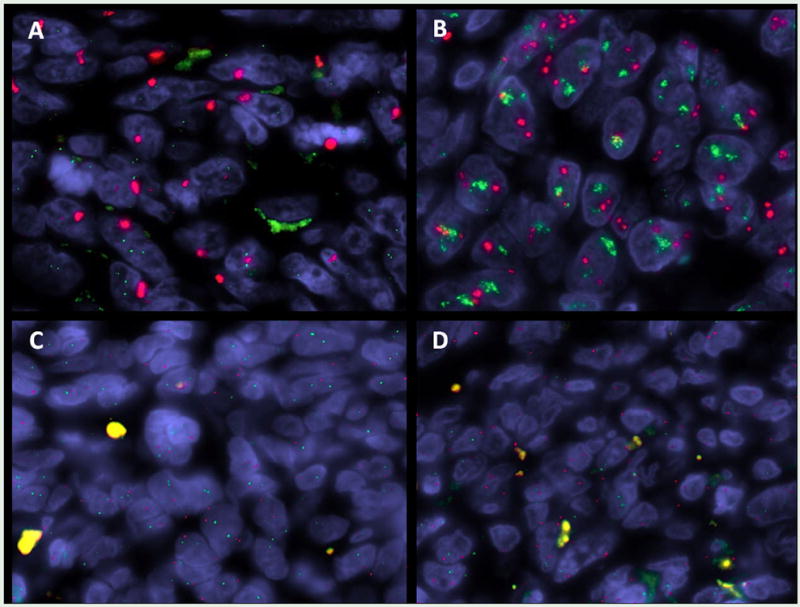
FISH analysis showing evidence of high level MYC amplification in an RT-associated AS carrying a KDR exon 15 mutation (A); co-amplification of MYC and FLT4 in a RT-associated AS; in contrast no evidence of copy number changes of MYC or FLT4 were noted in primary AS (C) or in RT-induced sarcoma (without an angiosarcoma morphology)(D).
As a subset of breast cancer with MYC abnormalities show co-amplification with other genes near the 8q24 region and occasionally in conjunction with TPD52 on 8q21 (Choschzick et al., 2010), we investigated 9 secondary AS for TPD52 gene copy number alterations by FISH, also because TPD52 mRNA levels were overexpressed in this group compared to the primary AS. None of the cases were associated with TPD52 gene amplification (data not shown).
Secondary AS show high level expression of MYC protein with the predominance of MYC-II isoform
WB was performed to investigate the MYC protein expression pattern in different AS subsets and the potential preferential expression of various isoforms [MYC-I (66 kDa), MYC-II (62kDa) and MYC-S (42 kDa)](Ramsay et al., 1984; Spotts et al., 1997). Eight radiation-induced AS and 12 primary AS (soft tissue and bone, 6, breast, 3, visceral, 3) were studied showing strong MYC protein expression in secondary AS, with MYC-II as the predominant isoform. In fact, MYC-II was preferentially expressed in secondary AS, while being undetectable in primary AS, regardless of anatomic location. However, MYC-I was variably expressed in both secondary and primary AS tumors (Fig. 4). MYC-amplified SKBR3 and MYC-transfected NIH-3T3 cells showed abundant MYC-II expression without detectable levels of MYC-I. MYC-S was expressed in all types of AS tumors and tested cell lines (data not shown).
Fig. 4.
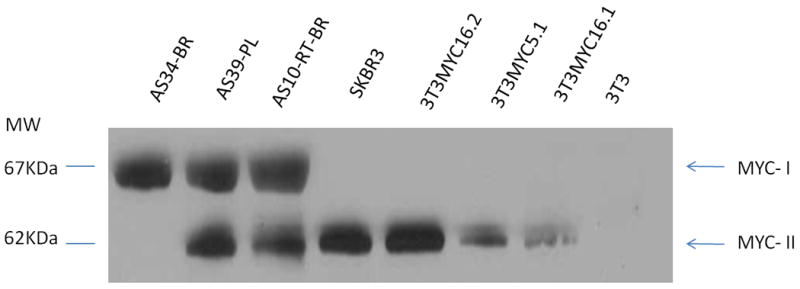
MYC isoforms in AS tumors. Western Blotting with anti-MYC antibody demonstrated that MYC-II (62KDa) expression was abundant in secondary AS tumors (RT-BR, post-radiation breast; PL, post-lymphedema), SKBR3 and MYC-transfected NIH3T3 cells, while not detected in primary AS (BR, primary breast AS). MYC-I (67KDa) was expressed in AS tumors, regardless to RT-exposure, but not in the tested cell lines (SKBR3 and MYC-transfected NIH3T3).
To investigate the MYC/MAX heterodimerization we examined the expression of MAX by WB and subjected tumor tissue lysates to co-IP. MAX protein was detected in all AS tumors by WB. Potential MYC/MAX dimers were precipitated with an anti-MAX antibody and the immunocomplex was subjected to WB to detect MYC and MAX proteins at molecular weights of 62-67 KDa and 22 KDa, respectively, using corresponding antibodies. Results showed that only MYC-II (62KDa) co-precipitated with MAX (22KDa) (Fig.5), and the interaction was only detected in secondary AS tumors. Results were then confirmed by another round of reversed co-IP, by precipitating MAX protein with an anti-MYC antibody and subsequent WB. In addition, co-IP of MYC/MAX was performed on SKBR3 and MYC transfected NIH-3T3 cell lysates, resulting in similar MYC/MAX heterodimers formation in the cells with MYC amplification/overexpression. We also examined whether MYC/MAX interaction can be identified in other tumors lacking oncogenic MYC activation, however no heterodimers were detected using a KIT-mutated gastrointestinal stromal tumor (GIST) and a GIST cell line as controls.
Fig. 5.
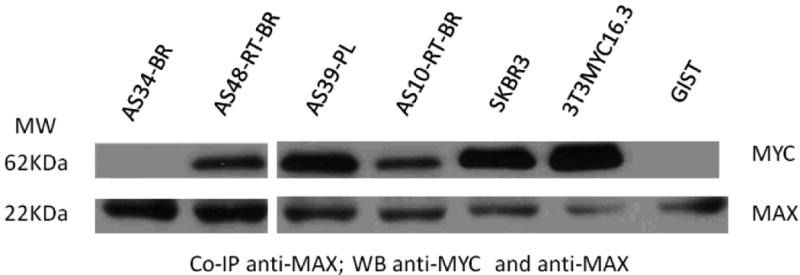
MYC/MAX heterodimers were detected in secondary AS tumors (RT-BR, post-radiation breast; PL, post-lymphedema), MYC amplified SKBR3 and MYC-transfected NIH3T3 cells, but not in primary AS (BR, primary breast AS) and GIST cell line. AS tumor lysates were co-IP with anti-MAX antibody and the immunoprecipitates were detected with anti-MYC and anti-MAX antibodies separately. MAX was detected in all tested samples.
High level FLT4 amplification is detected in a subset of secondary AS in conjunction with MYC amplification
As FLT4 mRNA expression was significantly upregulated in secondary AS and one of the three amplified hot spots in this AS subset was previously described to be located in 5q35 (Manner et al., 2010), we hypothesized that FLT4 may be a potential candidate for gene amplification. As such we designed FISH BAC probes spanning the FLT4 gene in 5q35 (Table 1) to test this possibility, and indeed, a high level amplification pattern was detected in 5 of the 20 (25%) radiation induced AS and in one of the two post-lymphedema induced AS. FLT4 copy number alterations were always found to be co-amplified with MYC (Fig. 3B). In contrast none of the 12 AVL or the 18 primary AS cases tested showed FLT4 gene copy number abnormalities by FISH (Figs. 2E, 3C).
In addition to FISH analysis for copy number alterations, we performed full length cDNA sequencing of FLT4 of 11 AS, as previously described (Antonescu et al., 2009). Tested tumors were selected based on highest FLT4 mRNA expression and included two primary and nine secondary AS (six post-RT and three post-lymphedema). Among the secondary AS group, 7 tumors lacked FLT4 gene amplification. However no FLT4 mutations were detected.
As FLT4 encodes VEGFR3, with a role in lymphatic specification (Karkkainen et al., 2000), we questioned if FLT4-amplified AS may induce lymphatic differentiation. However there was no correlation between the FLT4 amplification pattern and the D2-40 expression. None of the D2-40 positive AS tumors showed FLT 4 abnormalities; instead, D2-40 was present in two MYC amplified post-RT breast AS and one tumor with both KDR mutation and MYC amplification. The time period from radiation exposure to AS development ranged from 5.6-11 years (median 8.2) in the FLT4 and MYC co-amplified group, with no difference in morphologic appearance, as compared to MYC amplified alone tumors.
Discussion
Angiosarcomas (AS) represent a heterogeneous group of malignant vascular tumors, occurring not only in different anatomic locations, but also in distinct clinical settings, such as after radiation therapy or associated chronic lymphedema. While representing only 2% of soft tissue sarcomas, vascular sarcomas provide unique insight into the general process of tumor angiogenesis. The clinical features of AS vary by anatomic site in terms of patient age, gender, median survival, and response to chemotherapy. By expression profiling, we have previously shown that AS have distinct up-regulation of vascular-specific receptor tyrosine kinases, including TIE1, KDR, SNRK, TEK, and FLT1 (Antonescu et al., 2009). Full-sequencing of these five candidate genes identified 10% of patients harboring KDR mutations. A KDR-positive genotype was associated with strong KDR protein expression and was restricted to the breast anatomic site, with or without prior exposure to radiation. Transient transfection of KDR mutants into COS-7 cells demonstrated ligand-independent activation of the kinase, which was inhibited by sunitinib and sorafenib (Antonescu et al., 2009).
In our initial study, the unsupervised clustering of gene expression profiling identified two distinct AS genomic clusters, which correlated with anatomic location and prior exposure to radiation (Antonescu et al., 2009). The first group included radiation-induced breast AS and post-lymphedema AS, while primary breast and bone and soft tissue AS clustered in a second group. In keeping with our observations, Manner et al identified that a high percentage of secondary AS, but not primary AS, show distinct chromosomal gain patterns by CGH, including MYC amplification at 8q24 region (Manner et al., 2010), suggesting a split in the pathogenesis of the two sarcoma types. As such we sought to use our expression data in order to further investigate this dichotomy and study the oncogenic role of MYC in other radiation-associated neoplasia, as well as to mine additional novel candidate targets involved in their pathogenesis. Thus MYC overexpression secondary to high level gene amplifications was identified preferentially in radiation-induced and lymphedema-associated AS, but not in other clinical settings. In contrast with the previous study where MYC amplification was found in only 55% of secondary AS (Manner et al., 2010), our results show that high level MYC amplification was identified in 100% of tumors that developed in the setting of radiation therapy after breast cancer, while it was absent in all primary AS, of any anatomic location. The high predilection for MYC amplification present only in secondary AS tumors, particularly after radiation therapy for breast cancer, questions if this represents a true driver oncogenic event or a site-specific epi-phenomenon. Since a subset of breast cancers show evidence of MYC amplification, we hypothesized that radiation therapy in these patients may predispose to MYC-amplified angiosarcomas. However, that was not the case, since the initial breast cancer studied in 4 patients with tissue available did not show evidence of MYC abnormality. Furthermore, none of the synchronous AVL lesions detected in the mastectomy specimens adjacent to the radiation induced AS showed evidence of MYC amplification. Both results reinforce that MYC abnormalities represent a primary event in the angiosarcoma-genesis. It is however possible that the anatomic location, i.e. breast, may influence the endothelial susceptibility in the setting of radiation; as endothelial cell diversity is a well recognized phenomenon, with heterogenous expression signatures depending on tissue of origin (Chi et al., 2003).
MYC plays a critical role in tumorigenesis. The two common mechanisms of MYC oncogenic activation include either gene amplification or gene rearrangement, as seen in a subset of breast carcinomas and most of Burkitt lymphomas, respectively ( Taub et al., 1982; Hamlyn and Rabbitts, 1983). Dysregulated MYC expression promotes cell proliferation through inappropriate entry to S phase from G1 phase. Interestingly, over-expression of MYC in vitro induced alterations of G1/S arrest secondary to ionizing radiation (Sheen and Dickson, 2002). As MYC overexpression was implicated as an early oncogenic event, being amplified in both in situ and invasive breast carcinoma (Watson et al., 1993), we hypothesized that if radiation associated-AVL represent a precursor lesion of secondary AS, then MYC amplification might be detected at this stage. However none of the 12 AVL tested, even if adjacent to an AS, showed any evidence of MYC abnormality, in keeping with a distinct pathogenesis.
High expression of MYC-II isoform in secondary but not primary AS suggests a distinct pathogenetic mechanism of AS developing in the setting of radiation or lymphedema. The proliferative and transformation role of MYC-II was previously demonstrated in models in vitro, whereas the role of MYC-I remains less clear (Blackwood, 1994; Hann et al., 1994). MYC exerts its transcriptional activation function through heterodimerization with MAX. The MYC/MAX complex binds to E-box element which contains a CAC(G/A)TG motif and initiates gene transcription (Kretzner et al., 1992). Our screening detected MYC/ MAX interaction only in secondary AS tumors, MYC-amplified SKBR3 cells and MYC-transfected NIH-3T3 cell. In addition, MAX mRNA expression did not parallel the high levels of MYC in secondary AS, but showed equally low expression at both mRNA and protein level in primary and secondary AS. In keeping with this observation, inhibition of the MYC/MAX heterodimerization using the10058F4 inhibitor (Sigma-Aldrich, St. Louis, MO) did not affect cell growth in SKBR3 (data not shown). This result suggests that the MYC transactivation function in MYC-amplified AS might require additional mechanisms outside the MYC/MAX interaction.
A novel finding was the high FLT4 mRNA expression in secondary AS, as a result of high level of gene amplification. Interestingly, the gene amplification of FLT4 was found in 25% of secondary AS and only in association with MYC amplification. This result suggests that FLT4 over-expression may represent a required second hit in the progression of secondary AS and raise the possibility of targeting FLT4 as a potential therapeutic option for secondary AS. Notably, of the six patients with both MYC and FLT4 amplification, three patients were treated with sorafenib and showed either a Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 complete response in one patient or a partial response in the remaining two. The patient with complete response is presently alive with no evidence of disease progression, being on sorafenib therapy for 26 months. No patient with MYC amplified only AS was treated with sorafenib to be able to compare clinical responses between the two groups. It also appears that FLT4 amplification is mutually exclusive with KDR gene abnormalities, since none of the FLT4 amplified tumors carried KDR mutations. In contrast, 13% of tumors showing MYC gene amplification without a FLT4 abnormality revealed KDR mutations, as previously reported (Antonescu et al., 2009).
A number of lymphatic-associated antibodies have been recently developed (D2-40, VEGFR3, Prox-1) and shown to be frequently expressed in AS, suggesting that lymphatic or mixed lymphatic and blood vascular differentiation is common in AS. In an immunohistochemical study of 49 AS, more than half of the tumors were positive for all three markers: D2-40 (53%), VEGFR3 (57%) and Prox-1 (76%) (Mankey et al., 2010). Although there was no correlation between expression of these lymphatic markers and the anatomic location or radiation/lymphedema exposure, the authors suggest that a subset of the immunopositive tumors show distinct morphologic features, in keeping with lymphatic differentiation (i.e. lymphangiosarcoma): well-differentiated vasoformative appearance with inter-anastomotic channels, prominent hobnailing, lymphoid aggregates and lack of red blood cells. Of interest 9 of these 10 tumors were located in the head and neck (scalp). In keeping with these observations, both head and neck AS tumors present on our TMA were strongly positive for D2-40 and had morphologic features of lymphatic lineage, however lacked FLT4, MYC or KDR abnormalities. D2-40 is a sensitive marker for benign lymphangioma and Kaposi sarcoma, while it is typically not expressed in hemangioma. The high incidence of D2-40 expression in AVL, together with their hobnailed morphology and lack of red blood cells is in keeping with differentiation toward lymphatic lineage. As previous studies have shown that most AVL follow a benign course and do not progress to AS (Gengler et al., 2007), alternative designations have been proposed to replace the ‘atypical’ terminology and reflect their lymphatic derivation, such as: acquired progressive lymphangioma or lymphangioma circumscriptum. However, D2-40 staining pattern alone cannot be used to distinguish a diagnosis of radiation-associated AVL over a radiation-induced AS.
In summary our results show that MYC amplification is one of the hallmarks of secondary AS and can potentially be used as a molecular diagnostic tool to distinguish from other atypical vascular lesions or sarcoma types in difficult cases or when biopsy material is limited. The FLT4 gene amplification present in a subset of RT-induced AS provides a basis for the activity of tyrosine kinase inhibitors in the treatment of these patients.
Acknowledgments
Milagros Soto for editorial assistance, Nicole Moracco for providing clinical information, and Maria Mariano for help with tumor procurement.
Supported by: NIH grants P01 CA47179 (CRA, SS, RGM), RC2 CA148260 (RGM), P50 CA140146 (CRA, RGM), NCI-ASCO Cancer Foundation Clinical Investigator Team Leadership Supplemental Award (RGM), Cycle for Survival (CRA, RGM), and the Shuman Fund for GIST research (CRA, RGM).
References
- Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- Antonescu CR, Yoshida A, Guo T, Chang NE, Zhang L, Agaram NP, Qin LX, Brennan MF, Singer S, Maki RG. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 2009;69:7175–7179. doi: 10.1158/0008-5472.CAN-09-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood MJ. Utilization management and data acquisition: a case study. Benefits Q. 1994;10:38–42. [PubMed] [Google Scholar]
- Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A. 2003;100:10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choschzick M, Lassen P, Lebeau A, Marx AH, Terracciano L, Heilenkotter U, Jaenicke F, Bokemeyer C, Izbicki J, Sauter G, Simon R. Amplification of 8q21 in breast cancer is independent of MYC and associated with poor patient outcome. Mod Pathol. 2010;23:603–610. doi: 10.1038/modpathol.2010.5. [DOI] [PubMed] [Google Scholar]
- Gengler C, Coindre JM, Leroux A, Trassard M, Ranchere-Vince D, Valo I, Michels JJ, Guillou L. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584–1598. doi: 10.1002/cncr.22586. [DOI] [PubMed] [Google Scholar]
- Guo T, Agaram NP, Wong GC, Hom G, D'Adamo D, Maki RG, Schwartz GK, Veach D, Clarkson BD, Singer S, DeMatteo RP, Besmer P, Antonescu CR. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13:4874–4881. doi: 10.1158/1078-0432.CCR-07-0484. [DOI] [PubMed] [Google Scholar]
- Hamlyn PH, Rabbitts TH. Translocation joins c-myc and immunoglobulin gamma 1 genes in a Burkitt lymphoma revealing a third exon in the c-myc oncogene. Nature. 1983;304:135–139. doi: 10.1038/304135a0. [DOI] [PubMed] [Google Scholar]
- Hann SR, Dixit M, Sears RC, Sealy L. The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 1994;8:2441–2452. doi: 10.1101/gad.8.20.2441. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- Kozbor D, Croce CM. Amplification of the c-myc oncogene in one of five human breast carcinoma cell lines. Cancer Res. 1984;44:438–441. [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, Eisenman RN. Myc and Max proteins possess distinct transcriptional activities. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- Lagrange JL, Ramaioli A, Chateau MC, Marchal C, Resbeut M, Richaud P, Lagarde P, Rambert P, Tortechaux J, Seng SH, de la Fontan B, Reme-Saumon M, Bof J, Ghnassia JP, Coindre JM. Sarcoma after radiation therapy: retrospective multiinstitutional study of 80 histologically confirmed cases. Radiation Therapist and Pathologist Groups of the Federation Nationale des Centres de Lutte Contre le Cancer. Radiology. 2000;216:197–205. doi: 10.1148/radiology.216.1.r00jl02197. [DOI] [PubMed] [Google Scholar]
- Mankey CC, McHugh JB, Thomas DG, Lucas DR. Can lymphangiosarcoma be resurrected? A clinicopathological and immunohistochemical study of lymphatic differentiation in 49 angiosarcomas. Histopathology. 2010;56:364–371. doi: 10.1111/j.1365-2559.2010.03484.x. [DOI] [PubMed] [Google Scholar]
- Manner J, Radlwimmer B, Hohenberger P, Mossinger K, Kuffer S, Sauer C, Belharazem D, Zettl A, Coindre JM, Hallermann C, Hartmann JT, Katenkamp D, Katenkamp K, Schoffski P, Sciot R, Wozniak A, Lichter P, Marx A, Strobel P. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34–39. doi: 10.2353/ajpath.2010.090637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay G, Evan GI, Bishop JM. The protein encoded by the human proto-oncogene c-myc. Proc Natl Acad Sci U S A. 1984;81:7742–7746. doi: 10.1073/pnas.81.24.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen JH, Dickson RB. Overexpression of c-Myc alters G(1)/S arrest following ionizing radiation. Mol Cell Biol. 2002;22:1819–1833. doi: 10.1128/MCB.22.6.1819-1833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DG, Libshitz HI. Post-radiation sarcomas: a review of the clinical and imaging features in 63 cases. Clin Radiol. 2001;56:22–29. doi: 10.1053/crad.2000.0599. [DOI] [PubMed] [Google Scholar]
- Spotts GD, Patel SV, Xiao Q, Hann SR. Identification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by fulllength c-Myc proteins. Mol Cell Biol. 1997;17:1459–1468. doi: 10.1128/mcb.17.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PH, Safneck JR, Le K, Dubik D, Shiu RP. Relationship of c-myc amplification to progression of breast cancer from in situ to invasive tumor and lymph node metastasis. J Natl Cancer Inst. 1993;85:902–907. doi: 10.1093/jnci/85.11.902. [DOI] [PubMed] [Google Scholar]