

Abstract
Neurodegeneration can be triggered by genetic or environmental factors. Although the precise cause is often unknown, many neurodegenerative diseases share common features such as protein aggregation and age dependence. Recent studies in Drosophila have uncovered protective effects of NAD synthase nicotinamide mononucleotide adenylyltransferase (NMNAT) against activity-induced neurodegeneration and injury-induced axonal degeneration1,2. Here we show that NMNAT overexpression can also protect against spinocerebellar ataxia 1 (SCA1)-induced neurodegeneration, suggesting a general neuroprotective function of NMNAT. It protects against neurodegeneration partly through a proteasome-mediated pathway in a manner similar to heat-shock protein 70 (Hsp70). NMNAT displays chaperone function both in biochemical assays and cultured cells, and it shares significant structural similarity with known chaperones. Furthermore, it is upregulated in the brain upon overexpression of poly-glutamine expanded protein and recruited with the chaperone Hsp70 into protein aggregates. Our results implicate NMNAT as a stress-response protein that acts as a chaperone for neuronal maintenance and protection. Our studies provide an entry point for understanding how normal neurons maintain activity, and offer clues for the common mechanisms underlying different neurodegenerative conditions.
Injury-induced axonal degeneration is dramatically delayed in wallerian degeneration slow (WldS) mice, a mutant strain that over-expresses a chimaeric protein containing the NAD synthase NMNAT3,4. The WldS chimaeric protein offers neuroprotection against axonal degeneration2,5–7 as well as a variety of neurodegenerative conditions8–11. WldS protein contains the amino (N)-terminal 70-amino-acid fragment of ubiquitination factor E4B (Ube4b), a unique 18-amino-acid linking region translated from the 5′ untranslated region (UTR) of Nmnat1, and the entire coding sequence of Nmnat14,12. Conflicting results exist in mammalian systems as to whether Nmnat1 exerts protective effects13–15 and whether NAD is required4,14,15. Drosophila contains only one NMNAT gene, whose overexpression delays axonal degeneration2. This study and our finding that NMNAT functions as a maintenance factor to protect against activity-induced neurodegeneration1 suggest that NMNAT alone can protect against multiple neurodegenerative insults. Our recent finding that enzymatically inactive NMNAT retains neuroprotective capabilities also exposed a hitherto unknown molecular function1.
To test if NMNAT is a general factor required for neuronal maintenance and protection, we first examined the effects of NMNAT overexpression in a Drosophila model for SCA1. Overexpression of wild-type NMNAT or enzyme-inactive NMNAT (NMNAT-WR)1 suppresses the degenerative phenotypes induced by overexpression of Drosophila ataxin-1 (dAtx-1). It also offers moderate protection against the severe phenotypes caused by overexpression of human ataxin-1 with an expanded (82) poly-glutamine tract (hAtx-1[82Q])16 (Fig. 1). These findings, and the observations that NMNAT protects from axonal injury2, from photoreceptor injury caused by intense light, and that its loss causes massive neurodegeneration1, indicate that NMNAT is a versatile neuroprotective agent.
Figure 1. NMNAT suppresses toxicity induced by ataxin-1.
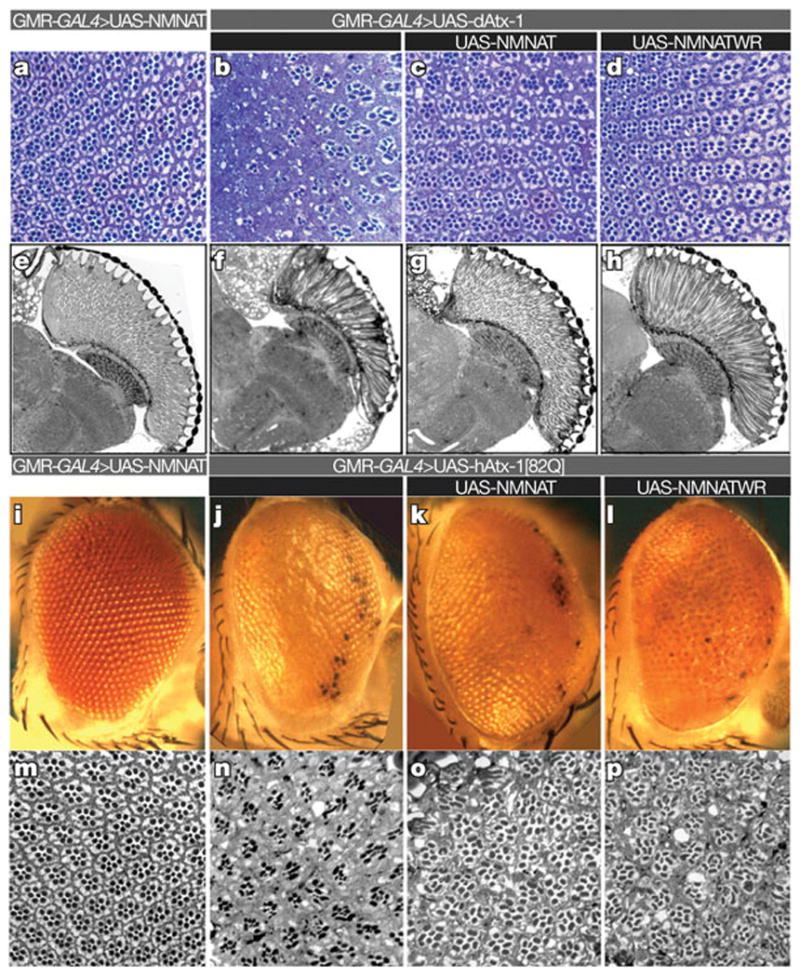
a–h, Ommatidial morphology of cross section (a–d) and horizontal section (e–h) of 15-day-old control fly (a, e), and flies overexpressing dAtx-1 (b–d, f–h). Overexpression of wild-type NMNAT (c, g) or enzyme-inactive NMNAT-WR (d, h) restores the rhabdomere degeneration phenotype. i–p, Eye exterior morphology and ommatidial morphology of two-day-old control fly (i, m), and flies overexpressing hAtx-1[82Q] (j–l, n–p). Overexpression of hAtx-1[82Q] leads to rough eye (j) and reduced rhabdomere size (n) phenotypes that are partly suppressed by co-expression of NMNAT (k, o) or NMNAT-WR (l, p).
To define the mechanisms of NMNAT function, we turned to a mammalian cell-culture-based assay to evaluate the effect of NMNAT on hAtx-1[82Q] aggregation. When transfected into Cos7 cells, hAtx-1[82Q] forms numerous and often large protein aggregates in both nucleus and cytoplasm17 (Fig. 2a, b), which are similar to the pathological aggregates found in the neurons of SCA1 mice or SCA1 patients17. hAtx-1[82Q] appears to be distributed diffusely and/or in aggregated forms in cells (Supplementary Fig. 1A–F). When NMNAT and hAtx-1[82Q] are co-expressed in cells, the percentage of cells with hAtx-1[82Q] aggregates is reduced and more cells contain diffuse hAtx-1[82Q] (Supplementary Fig. 1I). The fluorescence intensity of total hAtx-1[82Q] and its aggregates is significantly reduced (Fig. 2d). This reduction mimics the effect of Hsp7017 (Fig. 2d), a chaperone known to suppress the pathology associated with hAtx-1[82Q] expression in SCA1 mice18. This similarity between NMNAT and Hsp70 for aggregate formation was further supported when we analysed the solubility of the hAtx-1[82Q] protein by detergent fractionation and high-speed centrifugation. Co-expression of NMNAT or Hsp70 reduced the level in the detergent-insoluble fraction (Fig. 2e, g), arguing that NMNAT, like Hsp70, participates in aggregate reduction. To test whether NMNAT promotes protein degradation through the proteasome-mediated pathway17, we impaired the pathway with an inhibitor (MG-132) and evaluated its impact on the protein level of hAtx-1[82Q]. As shown in Fig. 2d, treatment with MG132 increased the total level of hAtx-1[82Q] and aggregates in all cells, including the cells that are co-transfected with NMNAT or Hsp70. Similarly, the level of detergent-insoluble hAtx-1[82Q] is increased in all cells (Fig. 2e, lanes 4–6 compared with lanes 1–3), suggesting that the proteasome is important in controlling aggregate formation. However, the insoluble hAtx-1[82Q] in Hsp70 or NMNAT co-transfected cells is lower than in vector-transfected cells (Fig. 2e, g). This suggests that NMNAT and Hsp70 act only partly through the proteasome to reduce aggregate levels, and that both Hsp70 and NMNAT might also function independently from the proteasome-mediated pathway. Human ataxin-1 without poly-Q expansion (hAtx-1[2Q]) also forms aggregates when expressed in these cells (Supplementary Fig. 1H). This is not surprising, as it has been shown that ataxin proteins without the poly-glutamine expansion also induce neurodegeneration when overexpressed16, and overexpression of Drosophila ataxin-1 with no poly-glutamine domain induces neurodegeneration (Fig. 1b, f). Importantly, NMNAT is also able to reduce the level of aggregation and the detergent-insoluble fraction of hAtx-1[2Q] (Fig. 2f, h and Supplementary Fig. 1J). These observations suggest that NMNAT reduces the aggregation and promotes the degradation of ataxin proteins, partly through the proteasome-mediated pathway.
Figure 2. NMNAT acts as a chaperone in cultured cells.
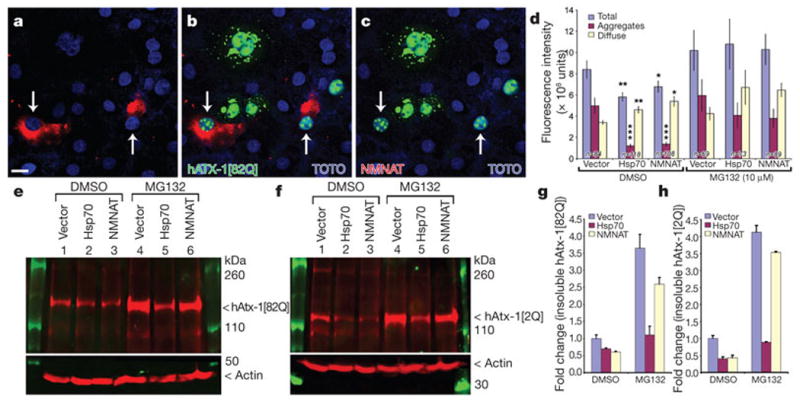
a–c, Cos7 cells are co-transfected with hAtx-1[82Q]-GFP (green) and NMNAT (red). TOTO3 staining (blue) marks nuclei. Scale bar in a for a–c, 20 μm. d, Quantification of GFP fluorescence. Aggregates are defined as objects at least 0.4 μm2 and 3,600 intensity units. Error bars, s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005 (analysis of variance post-hoc test). e–h, Western analysis (e, f) and quantification (g, h) of detergent-insoluble fractions of cells transfected with hAtx-1[82Q]-GFP (e) or hAtx-1[2Q]-GFP (f) and vector, NMNAT or Hsp70, and treated with either DMSO or 10 μM MG132. The level of insoluble hAtx-1[82Q] (g) or hAtx-1[2Q] (h) in the DMSO-treated cells is equal to 1 (fold). Error bars, s.e.m.; n = 4.
We next examined the in vivo response of NMNAT and Hsp70 upon the induction of aggregation of hAtx-1[82Q]. Endogenous NMNAT is located primarily in the cell bodies of most central nervous system neurons1, including those of the optic lobes (Fig. 3a). When hAtx-1[82Q] is overexpressed using the pan-neuronal driver nervana-GAL419, numerous hAtx-1[82Q] aggregates form in the cell bodies (Fig. 3b, l), similar to cultured cells (Fig. 2). However, unlike in cultured cells, we do not observe diffuse hAtx-1[82Q] localization. Interestingly, endogenous NMNAT is induced and recruited into these aggregates (compare Fig. 3 a, b, f, g). This upregulation and recruitment is very similar to the Hsp70 response when hAtx-1[82Q] is overexpressed (Fig. 3p, q), and expression of dAtx-1 or hAtx-1[82Q] increases the protein level of endogenous NMNAT (Supplementary Fig. 2) and Hsp70.
Figure 3. Endogenous and overexpressed NMNAT proteins are recruited with Hsp70 into the hAtx-1[82Q] aggregates.
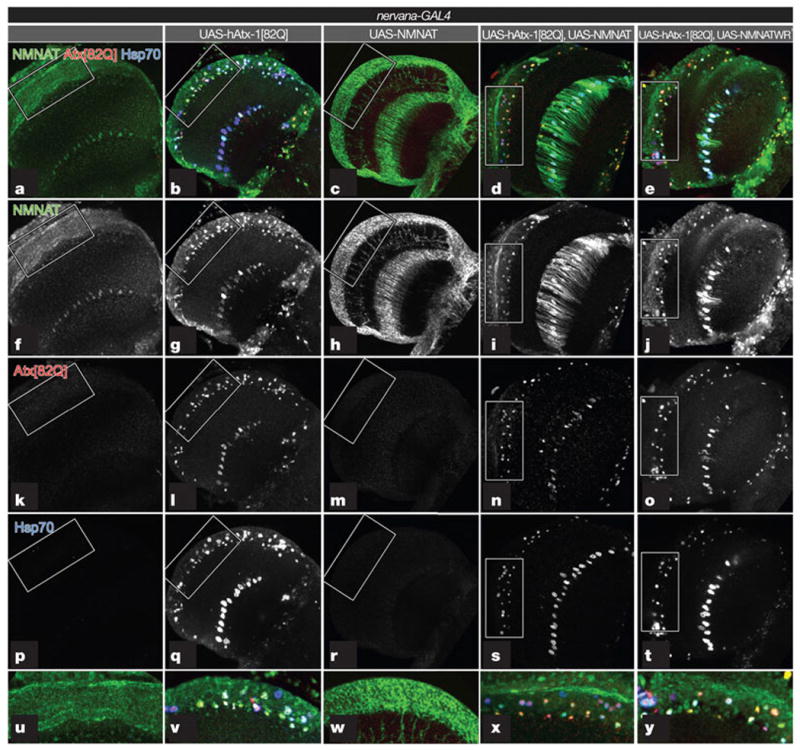
a–e, Seven-day-old adult fly brains were stained for NMNAT (green; f–j), hAtx-1[82Q] (red; k–o) and Hsp70 (blue; p–t). Higher magnifications of boxed areas are shown in u–y. Endogenous NMNAT is primarily nuclear (f). hAtx-1[82Q] forms nuclear aggregates (l). Endogenous NMNAT is upregulated and recruited to the aggregates (g). NMNAT is distributed to the cytoplasm when singly overexpressed (h), but is recruited to ataxin aggregates when co-expressed with hAtx-1[82Q] (i, n). Enzymatically inactive NMNAT-WR is also recruited into ataxin aggregates when co-overexpressed with hAtx-1[82Q] (j, o). Hsp70 is upregulated and recruited into the aggregates (q, s, t).
When NMNAT is overexpressed with nervana-GAL4, the protein is not only present in cell bodies, but also in axons (Fig. 3c, h, w). This is consistent with its protective effects in axonal degeneration when overexpressed2. To test the effects of hAtx-1[82Q] on the localization of overexpressed NMNAT, we overexpressed both proteins together. NMNAT is recruited into the hAtx-1[82Q] aggregates (see also Fig. 3d, i, n, x) and so is the enzymatically inactive NMNAT-WR (Fig. 3e, j, o, y). The recruitment of NMNAT into hAtx-1[82Q]-induced aggregates is specific, because green fluorescent protein (GFP) is not incorporated into the aggregates when co-expressed (Supplementary Fig. 3). The addition of NMNAT into hAtx-1[82Q] aggregates likely reduces the toxicity associated with hAtx-1[82Q] because the total level of hAtx-1[82Q] is reduced when co-expressing NMNAT or NMNAT-WR (Supplementary Fig. 3E). Taken together, our studies suggest that, in cultured cells, NMNAT functions with the proteasome to reduce the level of aggregation; in the fly brain, NMNAT is recruited into the hAtx-1[82Q] aggregates to reduce the neuronal toxicity induced by hAtx-1[82Q].
In wild-type brains, the level of Hsp70 was below our detection limit (Fig. 3p). However, when hAtx-1[82Q] is overexpressed, Hsp70 expression is strongly induced (compare Fig. 3p, q) and it co-localizes with endogenous NMNAT and hAtx-1[82Q] aggregates (Fig.3b, q, v). When overexpressed, both enzymatically active and inactive forms of NMNAT are recruited with Hsp70 into hAtx-1[82Q] aggregates (Fig. 3i, j, s, t). These data indicate that NMNAT is upregulated and recruited into the hAtx-1[82Q] aggregates in a manner similar to Hsp70, drawing yet another parallel between the function of these two proteins.
Next, we assayed whether NMNAT has chaperone activity. To measure the in vivo chaperone activity of NMNAT, we used the assay in which refolding of heat-denatured luciferase is monitored as a measure for chaperone activity20. In this assay, heat denaturation, combined with the protein-synthesis inhibitor cycloheximide, renders the endogenous levels of molecular chaperones insufficient to recover luciferase activity fully (Fig. 4a). When we transfect NMNAT as well as its adenylyltransferase-inactive forms (NMNAT-WR and -H30), they all protect luciferase from unfolding during heat shock (red bars in Fig. 4b) and enhance refolding after heat shock (yellow bars in Fig. 4b). In this assay, the chaperone activities of all forms of NMNAT are similar to that of mammalian Hsp70 or Drosophila Hsp83, the homologue of mammalian Hsp9021. These data indicate that NMNAT protects proteins from unfolding and promotes refolding, either by acting as a chaperone or by regulating the activity of other chaperones.
Figure 4. In vivo and in vitro chaperone activity assays.
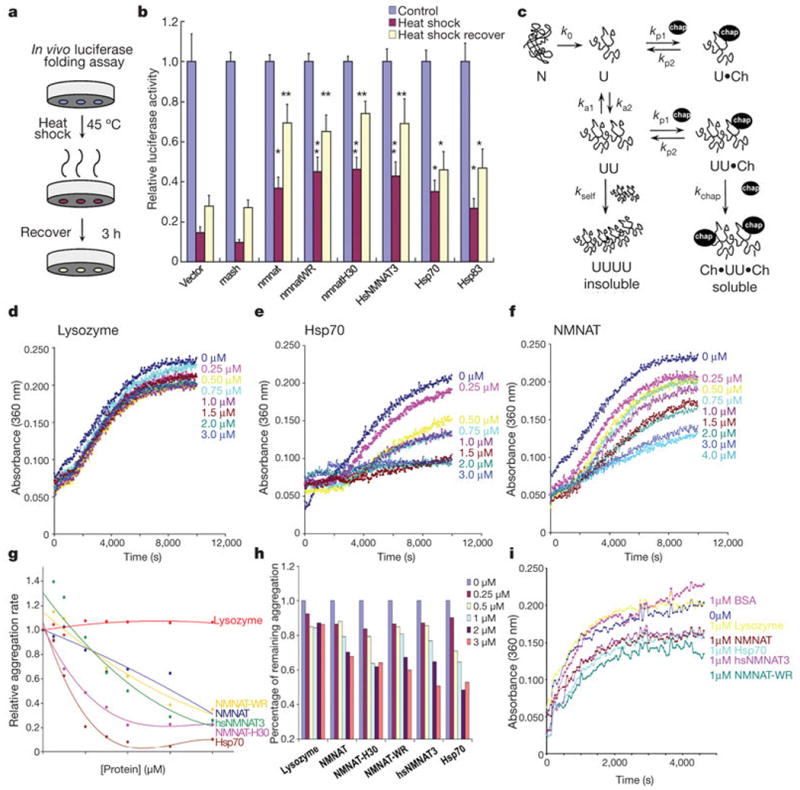
a, b, Luciferase activity was measured without heat shock as controls (blue columns), after 15 min heat shock at 45 °C (red columns) or after 3 h recovery at room temperature (yellow columns). The luciferase activities relative to the controls are displayed. Error bars, s.e.m. *P <0.05, **P <0.01; (analysis of variance post-hoc test); n = 4, triplicate sampling. c, Protein aggregation and its inhibition by chaperones. d–h, Aggregation of citrate synthase22 is prevented by addition of Hsp70 (e, g), NMNAT (f, g) or human hsNMNAT3 (g) but not by lysozyme (d). g, The relative aggregation rate is calculated by the change in absorbance per minute and the aggregation rate of CS alone is set to 1. (h) The percentage of remaining aggregation is calculated at the saturation point (8000 s). i, Insulin aggregation is inhibited by Hsp70, NMNAT, NMNAT-WR or hsNMNAT3 but not BSA or lysozyme.
To distinguish between these possibilities, we used an in vitro biochemical assay to measure chaperone activity. This assay measures the chaperone's ability to reduce thermally or chemically induced aggregation of a model protein substrate such as citrate synthase or insulin22 (Fig. 4c). As shown in Fig. 4e, Hsp70 efficiently reduces thermally induced aggregation of citrate synthase at 43 °C in the absence of ATP, in a concentration-dependent manner, confirming that it acts as a chaperone23. Lysozyme, which has no known chaperone activity, does not effectively suppress thermally induced aggregation of citrate synthase (Fig. 4d). Incubation of citrate synthase with increasing amounts of NMNAT results in a concomitant decrease in citrate synthase aggregation (Fig. 4f). The chaperone activity is quantified by the relative aggregation rate (Fig. 4g), and by the percentage of aggregation reduction at the saturation point (Fig. 4h). The reduction in light scattering (absorbance at 360 nm) is not due to a loss of substrate proteins by degradation, as the levels of citrate synthase remain the same with or without added chaperones (Supplementary Fig. 4). Similar results were obtained with insulin as a model substrate (Fig. 4i), where aggregation of insulin is induced by adding the reducing agent DTT. As in the hAtx-1[82Q] aggregation experiments, enzymatically inactive NMNAT-WR and another inactive NMNAT, NMNAT-H30,1 are both able to suppress protein aggregation effectively (Fig. 4 g, h). This further suggests that the chaperone activity of NMNAT is independent of its NAD synthesis activity. Both wild-type and enzymatically inactive NMNAT efficiently inhibit aggregation in a similar manner to Hsp70, unlike bovine serum albumin (BSA) or lysozyme (Fig. 4i). Note that the human homologue hsNMNAT3 displays chaperone activity similar to the Drosophila proteins (Fig. 4b, g, h, i), suggesting that the NMNAT homologues function similarly1.
If NMNAT has chaperone activity independent of its NAD synthesis ability, there may be a specific domain associated with this function. We therefore created three truncated proteins: NMNAT-ΔC, deleting the carboxy (C)-terminal domain including the ATP binding motif; NMNAT-ΔN, deleting the N-terminal catalytic motif; and NMNAT-ΔCN, deleting both N and C termini (Supplementary Fig. 5A). The N-terminal catalytic motif is not essential for the chaperone activity (Supplementary Fig. 5C); however, the C-terminal domain containing the ATP-binding domain is required, as neither NMNAT-ΔC nor NMNAT-ΔCN have chaperone activity (Supplementary Fig. 5B, D). These data provide additional evidence that NMNAT's chaperone and NAD synthesis activities can be dissociated.
To explore further the structural nature of the chaperone function, we searched for primary sequence similarity between NMNAT and known chaperones, but did not find any sequence homology. However, a DALI search24 using the structural coordinates of NMNAT1 (Protein Data Bank identity code 1KKU) and NMNAT3 (identity code 1NUR)25 against the entire Protein Data Bank revealed that both NMNAT1 and NMNAT3 share similarity with chaperone universal stress protein A (UspA; identity code 1JMV)26, with a Z score of 5.1, and Hsp100 (identity code 1JBK)27, with a Z score of 2.8. A structural superposition of NMNAT1 and UspA showed 13% sequence identity and a root mean squared deviation of 3.0 Å over the entire length of the protein. Hence, NMNAT proteins display structural similarities with known chaperones.
The induction and recruitment of NMNAT upon hAtx-1[82Q] aggregate formation suggest that NMNAT is also a stress-response protein similar to heat-shock proteins. The redistribution of over-expressed NMNAT and co-localization with Hsp70 at intracellular aggregates suggest that NMNAT may act in concert or in parallel with Hsp70. Our data also indicate that NMNAT and Hsp70 act independently, as we observe additive effects when mixing the two proteins in our in vitro assay (Supplementary Fig. 6). The chaperone function of NMNAT is likely linked to the proteasome-mediated pathway, as NMNAT is able to reduce the aggregation and promote the degradation of misfolded proteins. The chaperone activity offers an explanation for the broad protective activity of NMNAT in different neurodegenerative conditions. Interestingly, a recent study on the protein–protein interaction network for human inherited ataxias has put NMNAT within the interactome of human ataxin-128.
In summary, our work in Drosophila indicates that NMNAT functions as a chaperone independently of its enzymatic activity. Several studies on wallerian degeneration have suggested that the NAD synthesis activity of NMNAT is required for protection of axon degeneration14,29. This difference might be partly due to the distinct mechanisms underlying the injury-induced degeneration and other types of neurodegeneration. Further characterization of the role of NMNAT in different degenerative processes will help reveal the mechanisms of neurodegeneration.
Methods Summary
The ataxin-1 aggregation assay was performed as described17 with modifications. Retina sections and transmission electron microscopy were performed as described1. Western blot analysis was performed with infrared-dye-conjugated secondary antibodies and imaged on an Odyssey® system (LI-COR Biosciences). The luciferase folding assay was performed as described21,30. Confocal microscopy was performed with a Zeiss LSM 510 confocal Axiovert 200M microscope. Fluorescence analysis was performed with MetaMorph 5.07 (Molecular Devices/Universal Imaging Corp.), Amira 3.0 (TGS, Inc.) and Adobe Photoshop 7.0. The aggregation measurements were performed as described22. We searched three-dimensional structures on DALI sever24 (http://www.ebi.ac.uk/dali/); the structure superposition was done with PyMol structure analysis software.
Supplementary Material
Acknowledgments
We thank H. Zoghbi and R. Morimoto for reagents. We thank H. Zoghbi, H. Gilbert, H.-C. Lu, X. Zhou, P. Tsoulfas and A. Malhotra for technical suggestions and discussions. We also thank G. McNamara and the Analytical Imaging Core Facility at the University of Miami for imaging assistance. Some confocal imaging was supported by the BCM Mental Retardation and Developmental Disabilities Research Center. R.G.Z., P.R.H., C.M.H. were supported by the HHMI. H.J.B. is an HHMI investigator. R.G.Z. and F.Z. are also supported by the PhRMA Foundation and the Florida Department of Health, James and Esther King Biomedical Research Program.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions R.G.Z. and H.J.B. conceived the experiments. R.G.Z., P.R.H., Y.C. and C.M.H. performed the genetic and in vivo experiments. R.G.Z. and F.Z. performed the in vitro experiments. R.G.Z. and H.J.B. wrote the manuscript.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Zhai RG, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacDonald JM, et al. The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 3.Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the WldS mutant mouse. J Physiol (Lond) 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mack TG, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 5.Ribchester RR, et al. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS) Eur J Neurosci. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 6.Hoopfer ED, et al. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Ludwin SK, Bisby MA. Delayed wallerian degeneration in the central nervous system of Ola mice: an ultrastructural study. J Neurol Sci. 1992;109:140–147. doi: 10.1016/0022-510x(92)90160-m. [DOI] [PubMed] [Google Scholar]
- 8.Gillingwater TH, Haley JE, Ribchester RR, Horsburgh K. Neuroprotection after transient global cerebral ischemia in WldS mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- 9.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 10.Samsam M, et al. The WldS mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sajadi A, Schneider BL, Aebischer P. WldS-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 12.Conforti L, et al. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conforti L, et al. NAD + and axon degeneration revisited: Nmnat1 cannot substitute for WldS to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- 14.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, et al. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuda H, et al. The AXH domain of ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Cummings CJ, et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nature Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 18.Cummings CJ, et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 19.Sun B, Xu P, Salvaterra PM. Dynamic visualization of nervous system in live Drosophila. Proc Natl Acad Sci USA. 1999;96:10438–10443. doi: 10.1073/pnas.96.18.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 21.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 22.Chang Z, et al. Mycobacterium tuberculosis 16-kDa antigen (Hsp16.3) functions as an oligomeric structure in vitro to suppress thermal aggregation. J Biol Chem. 1996;271:7218–7223. [PubMed] [Google Scholar]
- 23.Kubo Y, et al. Two distinct mechanisms operate in the reactivation of heat-denatured proteins by the mitochondrial Hsp70/Mdj1p/Yge1p chaperone system. J Mol Biol. 1999;286:447–464. doi: 10.1006/jmbi.1998.2465. [DOI] [PubMed] [Google Scholar]
- 24.Holm L, Sander C. New structure–novel fold? Structure. 1997;5:165–171. doi: 10.1016/s0969-2126(97)00176-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, et al. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- 26.Sousa MC, McKay DB. Structure of the universal stress protein of Haemophilus influenzae. Structure. 2001;9:1135–1141. doi: 10.1016/s0969-2126(01)00680-3. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Sha B. Crystal structure of the E. coli Hsp100 ClpB N-terminal domain. Structure. 2003;11:323–328. doi: 10.1016/s0969-2126(03)00030-3. [DOI] [PubMed] [Google Scholar]
- 28.Lim J, et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 29.Jia H, et al. Identification of a critical site in Wld(s): essential for Nmnat enzyme activity and axon-protective function. Neurosci Lett. 2007;413:46–51. doi: 10.1016/j.neulet.2006.11.067. [DOI] [PubMed] [Google Scholar]
- 30.Michels AA, et al. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem. 1997;272:33283–33289. doi: 10.1074/jbc.272.52.33283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.