

Abstract
Germline mutations in SH3BP2 gene have been identified in patients with cherubism, a skeletal disorder characterized by excessive osteoclastic bone resorption that is limited to the mandible and maxilla. We previously demonstrated that SH3BP2 overexpression in Raw264.7 cells increased RANKL-induced osteoclastogenesis. Here, we examine the effect of decreased SH3BP2 on osteoclastogenesis. shRNAknockdown of SH3BP2 decreased PLCγ2 phosphorylation and NFATc1 expression, and reduced the expression of osteoclast-specific genes. In BMMs knockdown of SH3BP2 led to reductions in both the number and the surface area of TRAP positive and multinucleated osteoclasts. Bone resorptive activity was also dramatically blocked by shRNAknockdown of SH3BP2. Similarly Sh3bp2(−/−) deficient mice BMMs formed smaller osteoclasts that stained less with TRAP than wild-type mice. Taken together, this study demonstrates that SH3BP2 knockdown significantly decreases osteoclast differentiation and function. These results suggest that SH3BP2 plays a critical role in osteoclastogenesis and is a potential target for suppression of pathologic bone resorption.
Keywords: SH3BP2, Osteoclasts, RANKL, NFATc1, PLCγ2
Introduction
Although RANKL and M-CSF are recognized as the key signals for osteoclastogenesis (1–3), the critical downstream effector pathways that induce osteoclastogenesis have not been fully characterized. RANKL induces osteoclast formation via activation of nuclear factor of activated T cells c1 (NFATc1), a transcriptional factor that is necessary and sufficient to induce osteoclast differentiation and which is considered the master regulator of osteoclastogenesis (1, 4). NFATc1 increases transcription of osteoclast-specific genes such as tartrate resistant acid phosphatase (TRAP) (4), β3 integrin (5) and Cathepsin K (6). PLCγ stimulates NFATc1 activation and PLCγ2−/− mice have decreased numbers of osteoclasts and an osteopetrotic phenotype also (4, 7–9).
Cherubism is an autosomal dominant syndrome characterized by giant-cell bone resorptive tumors of the mandible and maxilla, substantial facial swelling and cervical lymphadenopathy (10, 11). The disorder is caused by heterozygous missense mutations of the Src homology 3 binding protein 2 (SH3BP2) gene, which encodes an adaptor protein (11). Recent work from our laboratory suggests that these mutations lead to a gain-of-function rather than a loss of activity (12). The basis for the unusual skeletal phenotype in cherubism patients with SH3BP2 mutations is not completely known, but previous overexpression studies indicate that SH3BP2 plays an important role in the activation of cells within the osteoimmune system (12–21). Sh3bp2(3bp2)(−/−) mice had increased accumulation of pre-B cells in the bone marrow and a block in the progression of transitional B cells in the spleen from the T1 to the T2 stage, but normal numbers of mature B cells (14). B-cell proliferation, cell cycle progression, PLCγ2 phosphorylation, calcium mobilization, NFATc1 dephosphorylation, and Erk and Jnk activation in response to B-cell receptor (BCR) ligation were all impaired (14). However osteoclastogenesis in particular has not examined in the Sh3bp2 (−/−) deficient mouse.
In the present study, we define the effect of decreased SH3BP2 on osteoclastogenesis. We used shRNA to knockdown SH3BP2 in osteoclastic cells and found significantly decreased RANKL-induced phosphorylation of PLCγ2 and reduced expression of NFATc1. Moreover, osteoclasts with decreased SH3BP2 and SH3BP2 (−/−) osteoclasts had less TRAP activity and less bone resorptive activity.
Methods
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic Foundation.
Lentiviral vectors, pLKO.1 with short hairpin RNAs (shRNAs), containing SH3BP2-specific sequences were purchased from Open Biosystems (Huntsville, AL, USA). Three non-overlapping shRNA sequences targeting the murine SH3BP2 gene (GenBank accession No. NM_011893) were used: 5′-GCCTCTCAATAAATCAGAGAT-3′, (corresponding to the coding region positions 2145–2165), 5′-CCCATTCAAGATCATTCACAT-3 ′ (342–362) and 5 ′-AGTGAGGAATTATCGCATCTT-3′ (1575–1595). pLKO.1-empty and pLKO.1-scramble shRNA, which has a nontargeting scramble sequence, were used as control vectors (Addgene, Cambridge, MA, USA). Three independent SH3BP2 targeting shRNA-expressing lentiviruses were pooled to increase the knockdown efficiency of SH3BP2 expression. Infection of Raw264.7 cells or BMMs with recombinant lentivirus was conducted in the presence of 8 μg/ml polybrene (Sigma-Aldrich) for 16 hours, infected cells were selected in 3.5 μg/ml puromycin for 3 to 5 days.
In vitro osteoclast differentiation
For in vitro osteoclast differentiation, Raw264.7 cells infected with recombinant lentivirus (pLKO.1-SH3BP2 shRNA or pLKO.1-empty or pLKO.1-scramble shRNA) were cultured in αMEM supplemented with 10% FBS and 1% P/S containing 100 ng/ml RANKL. BMMs infected with recombinant lentivirus were cultured in the same medium but with 30 ng/ml M-CSF.
Immunoblot analysis
Whole cell lysates were collected from the cultured cells and samples containing equal amounts of protein were electrophoresed through 4–12% bis-tris polyacrylamide gels (Invitrogen) in MOPS buffer (Invitrogen) and electrophoretically transferred to nitrocellulose membranes, exposed to antibodies and incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer’s instructions, and antibody binding was detected by exposure to HyBlot CL Autoradiography film (Denville Scientific Inc., Metuchen, NJ, USA).
Quantitative real time PCR
The expression of osteoclast-specific genes was analyzed by quantitative real time PCR. We isolated total RNA from infected Raw264.7 cells cultured in αMEM containing 100 ng/ml RANKL using RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) at 24 hours intervals for up to 72 hours. First strand cDNA was transcribed from 5 μg of RNA using the SuperScript III First-Strand Synthesis System according to the manufacturer’s instructions (Invitrogen). PCR was performed using specific primers for murine TRAP, osteoclast-associated receptor (OSCAR), β3 integrin, Cathepsin K and β-actin. Primers used were: TRAP, 5′-GATGACTTTGCCAGTCAGCA-3′ and 5′-AACTGCTTTTTGAGCCAGGA-3′; OSCAR, 5′-GAGCTCTGCCTTTGATGGTC-3′ and 5′-CAAGGATCCCAGCTTCTCTG-3′; β3 integrin, 5′-GAAAGGCCAGTCAGAACTGC-3′and5′-TGTGGCCTCCCAGATTAAAG-3′; Cathepsin K, 5′-TTCTCCTCTCGTTGGTGCTT-3′ and 5′-AAAAATGCCCTGTTGTGTCC-3′; β-actin, 5′-GATCATTGCTCCTCCTGAGC-3′ and 5′-ACATCTGCTGGAAGGTGGAC-3′.
Real time PCR was performed in a 20 μl reaction mixture using the Maxima SYBR Green qPCR Master Mix (Fermentas, Glen Burnie, MD, USA) on the Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Standard curves were generated for all reactions. The PCR conditions were 1 cycle at 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. PCR was performed in duplicate for each template, and the no-template control contained water instead of the template. The values were normalized with those for β-actin and were analyzed using relative standard curve method (22).
TRAP and Bone resorption assay using dentine discs
TRAP activity of infected Raw264.7 cells was determined using cells that had been incubated with or without RANKL to induce osteoclast differentiation, after 5 days. After 5 or 7 days, TRAP staining was performed using the Leucocyte Acid Phosphatase Stain Kit (Sigma-Aldrich) with sodium tartrate according to the manufacturer’s instructions. The number of TRAP+, multinucleated osteoclasts containing three or more nuclei were counted under a light microscope, and the surface area that was occupied by TRAP+, multinucleated osteoclasts was measured using Image J software (NIH). BMMs infected with recombinant lentivirus were plated on dentine discs (Osteosite Dentine Discs; Immunodiagnostic Systems Inc, Fountain Hills, AR) and resorption pits were counted using a light microscope.
Statistical analysis
Each experiment was performed at least three times independently and data are presented as means ± SD. Statistical significance between means was evaluated using the unpaired student’s t-test and all tests were two-tailed with differences considered significant at p<0.05.
Results
shRNA knockdown of SH3BP2 inhibits osteoclastogenesis in Raw264.7 cells
In control cells, both PLCγ2 phosphorylation and NFATc1 expression were significantly increased by RANKL stimulation in a time-dependent manner. By contrast, lentiviral shRNA knockdown of SH3BP2 inhibited the RANKL-induced increase in PLCγ2 phosphorylation and NFATc1 expression compared with cells infected with lentiviral control vector after 24 hours RANKL stimulation (Fig. 1).
Fig. 1.
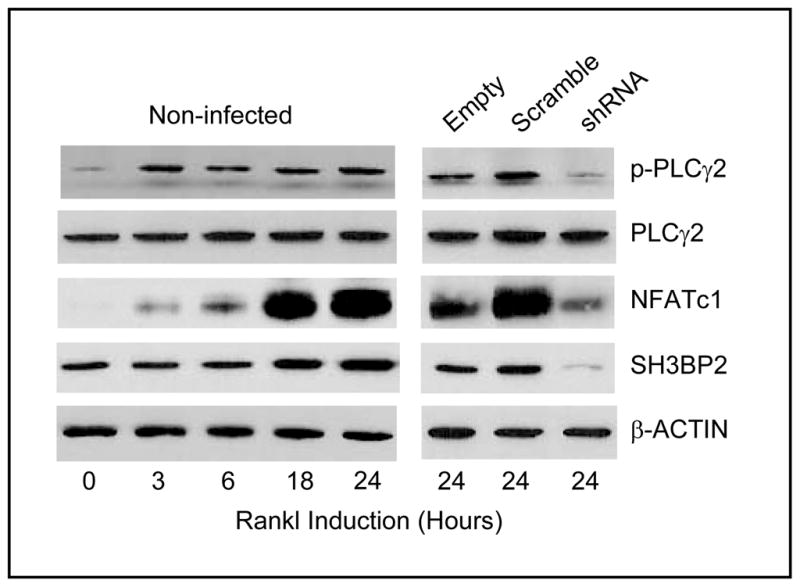
BMMs from Sh3bp2 homozygous knockout mice form large cells and are slower to become -TRAP positive in response to M-CSF and RANKL. SH3BP2 knockdown decreases osteoclastogenesis in Raw264.7 cells. Non-infected Raw264.7 cells and cells infected with lentivirus (control vectors or SH3BP2-shRNA) were cultured with RANKL 100 ng/ml for 24 hours. In non-infected cells, both PLCγ2 phosphorylation and NFATc1 expression were increased by RANKL in a time-dependent manner. Conversely, lentiviral shRNA knockdown of SH3BP2 dramatically decreased SH3BP2 protein expression and decreased both PLCγ2 phosphorylation and NFATc1 expression compared with control cells after 24 hours of RANKL stimulation.
The expression of osteoclast-specific genes, TRAP, OSCAR, β3 integrin and Cathepsin K, also was increased after 72 hours of RANKL stimulation of control cells. However, knockdown of SH3BP2 resulted in significantly less induction of osteoclast-specific genes by RANKL. Specifically, mRNA expression of TRAP was deceased to 32% of that with empty vector (*p<0.0001) and 34% of that with scramble shRNA (*p<0.05) at 72 hours (Fig. 2A), OSCAR was decreased to 28% of that with both empty vector and scramble shRNA (*p<0.05 compared with both controls) at 72 hours (Fig. 2B) and β3 integrin was decreased to 42% of that with empty vector (**p<0.005) and 47% of that with scramble shRNA (**p<0.01) at 72 hours (Fig. 2C). mRNA expression of Cathepsin K was decreased to 56% of that with empty vector and to 60% of that with scramble shRNA (these differences were not significant, p>0.05) (Fig. 2D). We used β-actin as a control and did not find a difference in β-actin levels between groups. These results suggest that reduced expression of SH3BP2 can inhibit RANKL-induced osteoclastogenesis, and are consistent with an important role for SH3BP2 in RANKL signaling.
Fig. 2.

mRNA expression of osteoclast specific genes is suppressed by SH3BP2 knockdown. Relative mRNA expression of osteoclast specific genes in infected Raw264.7 cells after 72 hours of RANKL stimulation was examined by quantitative real time PCR. SH3BP2 knockdown significantly suppressed the expression of TRAP (A), OSCAR (B) and β3 integrin (C) compared with controls. The differences in the expression of Cathepsin K (D) were not significant. (A) 32% of that with empty vector (*p<0.0001) and 34% of that with scramble shRNA (*p<0.05) at 72 hours, (B) *p<0.05 compared with both control vectors at 48 and 72 hours (28% of that with both controls at 72 hours), (C) *p<0.01 compared with empty vector at 48 hours and 42% of that with empty vector (**p<0.005) and 47% of that with scramble shRNA (**p<0.01) at 72 hours, (D) 56% of that with empty vector and 60% of that with scramble shRNA (no significant difference between the empty vector, scramble shRNA and SH3BP2 shRNA).
Impaired TRAP+, multinucleated osteoclast formation in SH3BP2 knockdown Raw264.7 cells
To determine whether SH3BP2 is essential in RANKL-induced osteoclast differentiation, Raw264.7 cells infected with either lentiviral control vector or SH3BP2 shRNA were incubated with or without RANKL to induce osteoclast differentiation. TRAP activity in non-infected Raw264.7 cells was increased by RANKL stimulation in a time-dependent manner. By contrast, TRAP activity in cells SH3BP2 knockdown cells was significantly decreased compared with control cells; TRAP activity was decreased to 29% of that with empty vector and 27% of that with scramble shRNA, *p<0.005 compared with both controls (Fig. 3A). In addition, the number of TRAP+, multinucleated osteoclasts derived from SH3BP2 knockdown Raw264.7 cells was significantly reduced compared with controls; the number of TRAP+, multinucleated osteoclasts derived from Raw264.7 cells infected with SH3BP2 shRNA was 42% of that with empty vector and 44% of that with scramble shRNA, (p<0.001 compared with both controls, Fig. 3B). These results implicate SH3BP2 in RANKL-induced osteoclast differentiation.
Fig. 3.

Osteoclast formation is impaired in Raw264.7 cells with decreased SH3BP2 expression. (A) In non-infected cells, TRAP activity was increased by RANKL in a time-dependent manner. shRNA knockdown of SH3BP2 significantly decreased TRAP activity compared with control cells; TRAP activity was decreased to 29% of that with empty vector and 27% of that with scramble shRNA, *p<0.005 compared with both controls. (B) The number of TRAP+, multinucleated (MN) osteoclasts (OCs) derived from the cells infected with SH3BP2 shRNA was significantly reduced compared with controls; the number of TRAP+, multinucleated osteoclasts derived from Raw264.7 cells infected with SH3BP2 shRNA was 42% of that with empty vector and 44% of that with scramble shRNA, **p<0.001 compared with both controls.
RANKL-induced NFATc1 and phospho-PLCγ2 is inhibited in SH3BP2 knockdown BMMs
We further examined the effect of SH3BP2 knockdown in mouse BMMs by immunoblot analysis. RANKL stimulation of BMMs with knockdown of SH3BP2 showed little or no induction of SH3BP2, NFATc1 and phospho-PLCγ2 compared with control BMMs. By contrast, total PLCγ2 expression in BMMs with SH3BP2 shRNA was not decreased compared with control vectors (Fig. 4).
Fig. 4.
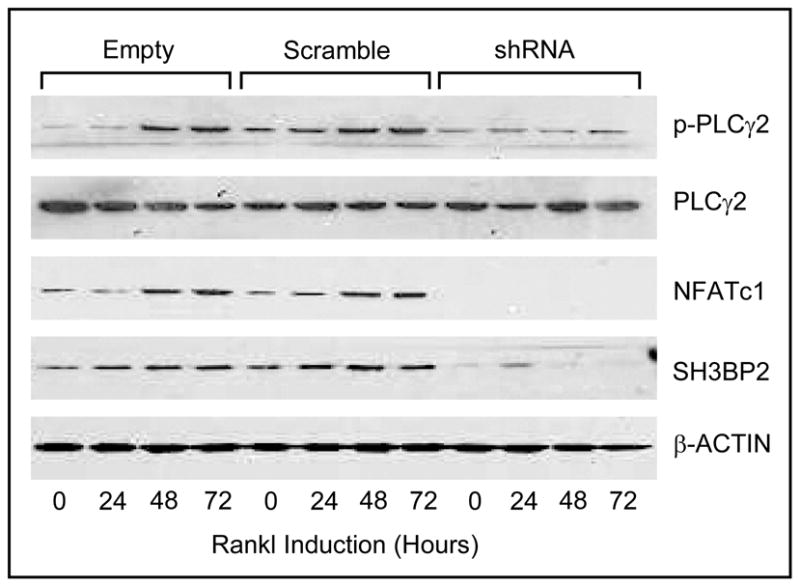
shRNA knockdown of SH3BP2 prevents osteoclastogenesis in BMMs. Expression of NFATc1 and PLCγ2 phosphorylation were increased by RANKL in control BMMs. In contrast, in RANKL stimulated BMMs infected with SH3BP2 shRNA, SH3BP2 expression was strongly silenced, NFATc1 was diminished, and PLCγ2 phosphorylation was not detected.
shRNA knockdown of SH3BP2 decreases osteoclastogenesis
To evaluate whether knockdown of SH3BP2 in BMMs affects their differentiation into osteoclasts, we performed TRAP staining of infected BMMs after culture in the presence of RANKL for 7 days. The number of TRAP+, multinucleated osteoclasts derived from BMMs infected with SH3BP2 shRNA was significantly decreased compared with controls (42% of empty vector and 44% of scramble shRNA, *p<0.0001 compared with both controls). We also measured the surface area occupied by TRAP+, multinucleated osteoclasts derived from infected BMMs, and it was significantly reduced by knockdown of SH3BP2 compared with controls (24% of both empty vector and scramble shRNA, **p<0.0001 compared with both controls) (Fig. 5). We did not observe an increase in cell death in any group of cells. These results suggest that loss of SH3BP2 expression can suppress both RANKL-induced osteoclast differentiation and mature osteoclast formation.
Fig. 5.

BMMs with decreased SH3BP2 expression fail to differentiate into mature osteoclasts. The number of TRAP+, multinucleated (MN) osteoclasts (OCs) derived from BMMs with decreased SH3BP2 expression was significantly fewer than controls (42% of that with empty vector and 44% of that with scramble shRNA, *p<0.0001 compared with both controls). The surface area occupied by the TRAP+, multinucleated (MN) osteoclasts (OCs) was reduced in SH3BP2 shRNA infected BMMs compared to control infections (24% of that with both empty and scramble shRNA, **p<0.0001 compared with both controls).
SH3BP2 knockdown significantly suppressed osteoclast bone resorptive function
Since bone resorption is a characteristic feature of osteoclasts, we evaluated whether SH3BP2 knockdown in BMMs affects osteoclastic resorptive function on dentine discs(22–25). BMMs infected with lentiviral control vectors exhibited numerous resorption pits on the discs, however, BMMs infected with SH3BP2 shRNA showed less resorptive capacity compared with controls (19% of empty vector, *p<0.005 and 21% of scramble shRNA, *p<0.05) (Fig. 6). The decrease in resorption pits indicates that SH3BP2 can mediate the bone resorptive function of osteoclasts.
Fig. 6.

BMMs with decreased SH3BP2 expression exhibit decreased bone resorptive capacity. Numerous resorption pits were detected on the dentine discs of controls and almost no resorption pit was detected from BMMs infected with SH3BP2 shRNA (19% of that with empty vector and 21% of that with scramble shRNA, *p<0.005 compared with empty vector and p<0.05 compared with scramble shRNA).
Sh3bp2−/− analysis
BMMs from Sh3bp2 deficient (−/−) mice were noted to form smaller osteoclasts than wild-type mice (Fig. 7) and the osteoclasts stained less avidly with TRAP than their wild-type counterparts (Fig. 7).
Fig. 7.

Sh3bp2(−/−) BMMs formed smaller osteoclasts with less TRAP staining and novel external cell membrane protrusions than wild-type BMMs. BMMs were treated with RANKL (100ng/ml) and m-CSF (30ng/ml) and osteoclast size, density of TRAP staining and osteoclast area (by F-actin staining) were analyzed and revealed significant differences.
Discussion
In this study we found that shRNA knockdown of SH3BP2 decreases RANKL-induced phosphorylation of PLCγ2 and NFATc1 expression and inhibited mRNA expression of osteoclast-specific genes. We also found that NFATc1 level as well as PLCγ2 phosphorylation were increased by RANKL stimulation in BMMs and that these increases were not present in SH3BP2 knockdown BMMs. These results suggest that SH3BP2 may play a role in RANKL-induced PLCγ2 phosphorylation and NFATc1 expression and that SH3BP2 could regulate the osteoclastogenic response to RANKL. Previous studies support this hypothesis (26, 27). Ueki et al created a P416R Sh3bp2 knock-in mouse to replicate the most common human activating SH3BP2 mutation (P418R) in patients with cherubism(26, 27). Myeloid cells from this “cherubism” mouse have increased response to M-CSF and RANKL stimulation (26, 27). Aliprantis et al reported that mutant Sh3bp2 bone marrow cells from this same “cherubism” mouse displayed increased induction of the Nfatc1/A isoform in response to RANKL stimulation and that SH3BP2 is upstream of NFATc1 in RANKL-induced osteoclastogenesis (26).
In this study, we also found that shRNA knockdown of SH3BP2 significantly decreased RANKL-induced TRAP activity and reduced the number of TRAP+, multinucleated osteoclasts derived from Raw264.7 cells. Using BMMs, we showed that SH3BP2 knockdown leads to significant decreases both in the number and the surface area of TRAP+, multinucleated osteoclasts. These results suggest that SH3BP2 may be required for osteoclast differentiation. Indeed, myeloid cells from the “cherubism” mouse (P416R Sh3bp2 knock-in mouse) form increased numbers of abnormally enlarged osteoclasts (26, 27).
We also demonstrate for the first time that resorption pits on discs with SH3BP2 knockdown BMMs were significantly decreased compared with resorption pits on discs with BMMs with no SH3BP2 knockdown indicating a decrease in osteoclast function. Taken together, SH3BP2 knockdown inhibits not only osteoclastogenesis but also actual bone resorptive function. A separate study has examined SH3BP2 knockdown in RAW 264.7 cells (28). These investigators found similarly an important role for SH3BP2 in osteoclastogenesis (28). Our study is the first however to look at SH3BP2 knockdown in BMMs and the first to examine osteoclastogenesis in the Sh3bp2(−/−) mouse.
The SH3BP2 knockout mouse does not have osteopetrosis (14). However, this lack of obvious bone resorptive abnormality could be due to compensation by other proteins in the SH3BP2 knockout mice. GNAS provides an example of another gene which is important for bone remodeling in humans but the knockout GNAS mouse does not have an obvious bone phenotype (29, 30).
In conclusion, our results indicate that reducing the expression of SH3BP2 decreases RANKL-dependent osteoclastogenesis. These findings are consistent with previous models of increased expression or function of SH3BP2 showing increased osteoclastogenesis (20) and the Sh3bp2 “cherubism” knockin mouse with increased numbers of osteoclasts (27). Although further studies are needed to explore the mechanism of SH3BP2 action in osteoclastogenesis, our findings provide important new insights into the molecular understanding of osteoclastogenesis and identify SH3BP2 as a potential target for novel therapies aimed at reducing osteoclastic bone resorption.
Acknowledgments
This work was supported in part by US Public Health Service Research grants K08-AR47661 (SAL), R01-DE018237 (SAL) and the OREF+ (SAL). Dr Raif Geha generously provided the SH3BP2 knockout(−/−) mouse.
Footnotes
All authors have no conflicts of interest
Contributor Information
Teruya Kawamoto, Email: trykwmt@med.kobe-u.ac.jp.
Chun Fan, Email: chunf@ccf.org.
Robert J Gaivin, Email: gaivinr@ccf.org.
Michael A Levine, Email: levinem@email.chop.edu.
Steven A Lietman, Email: lietmas@ccf.org.
References
- 1.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102:1130–1139. doi: 10.1002/jcb.21553. [DOI] [PubMed] [Google Scholar]
- 3.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 4.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 5.Crotti TN, Flannery M, Walsh NC, Fleming JD, Goldring SR, McHugh KP. NFATc1 regulation of the human beta3 integrin promoter in osteoclast differentiation. Gene. 2006;372:92–102. doi: 10.1016/j.gene.2005.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, Hisatake K, Nogi Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279:45969–45979. doi: 10.1074/jbc.M408795200. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Wang X, Di L, Fu G, Chen Y, Bai L, Liu J, Feng X, McDonald JM, Michalek S, et al. Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis. J Biol Chem. 2008;283:29593–29601. doi: 10.1074/jbc.M802493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epple H, Cremasco V, Zhang K, Mao D, Longmore GD, Faccio R. Phospholipase Cgamma2 modulates integrin signaling in the osteoclast by affecting the localization and activation of Src kinase. Mol Cell Biol. 2008;28:3610–3622. doi: 10.1128/MCB.00259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takayanagi H. The role of NFAT in osteoclast formation. Ann N Y Acad Sci. 2007;1116:227–237. doi: 10.1196/annals.1402.071. [DOI] [PubMed] [Google Scholar]
- 10.Novack DV, Faccio R. Jawing about TNF: new hope for cherubism. Cell. 2007;128:15–17. doi: 10.1016/j.cell.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 11.Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, Ninomiya C, doAmaral C, Peters H, Habal M, et al. Mutations in the gene encoding c-Abl- binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125–126. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 12.Lietman SA, Kalinchinko N, Deng X, Kohanski R, Levine MA. Identification of a novel mutation of SH3BP2 in cherubism and demonstration that SH3BP2 mutations lead to increased NFAT activation. Hum Mutat. 2006;27:717–718. doi: 10.1002/humu.9433. [DOI] [PubMed] [Google Scholar]
- 13.Chen G, Dimiriou I, La Rose J, Ilangumaran S, Yeh WC, Turner M, Gommerman J, Rottapel R. The 3BP2 adapter protein is required for optimal B Cell Activation and T-independent type 2 humoral response. Mol Cell Biol. 2007 doi: 10.1128/MCB.01014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de la Fuente MA, Kumar L, Lu B, Geha RS. 3BP2 deficiency impairs the response of B cells, but not T cells, to antigen receptor ligation. Mol Cell Biol. 2006;26:5214–5225. doi: 10.1128/MCB.00087-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deckert M, Rottapel R. The adapter 3BP2: how it plugs into leukocyte signaling. Adv Exp Med Biol. 2006;584:107–114. doi: 10.1007/0-387-34132-3_8. [DOI] [PubMed] [Google Scholar]
- 16.Foucault I, Le Bras S, Charvet C, Moon C, Altman A, Deckert M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood. 2005;105:1106–1113. doi: 10.1182/blood-2003-08-2965. [DOI] [PubMed] [Google Scholar]
- 17.Maeno K, Sada K, Kyo S, Miah SM, Kawauchi-Kamata K, Qu X, Shi Y, Yamamura H. Adaptor protein 3BP2 is a potential ligand of Src homology 2 and 3 domains of Lyn protein-tyrosine kinase. J Biol Chem. 2003;278:24912–24920. doi: 10.1074/jbc.M301201200. [DOI] [PubMed] [Google Scholar]
- 18.Qu X, Kawauchi-Kamata K, Miah SM, Hatani T, Yamamura H, Sada K. Tyrosine phosphorylation of adaptor protein 3BP2 induces T cell receptor-mediated activation of transcription factor. Biochemistry. 2005;44:3891–3898. doi: 10.1021/bi048353o. [DOI] [PubMed] [Google Scholar]
- 19.Saborit-Villarroya I, Del Valle JM, Romero X, Esplugues E, Lauzurica P, Engel P, Martin M. The adaptor protein 3BP2 binds human CD244 and links this receptor to Vav signaling, ERK activation, and NK cell killing. J Immunol. 2005;175:4226–4235. doi: 10.4049/jimmunol.175.7.4226. [DOI] [PubMed] [Google Scholar]
- 20.Lietman SA, Yin L, Levine MA. SH3BP2 is an activator of NFAT activity and osteoclastogenesis. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lietman SA, Yin L, Levine MA. SH3BP2 mutations potentiate osteoclastogenesis via PLCgamma. J Orthop Res. 28:1425–1430. doi: 10.1002/jor.21164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larionov A, Krause A, Miller W. A standard curve based method for relative real time PCR data processing. BMC Bioinformatics. 2005;6:62. doi: 10.1186/1471-2105-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA. The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem. 2004;279:13984–13992. doi: 10.1074/jbc.M213067200. [DOI] [PubMed] [Google Scholar]
- 24.Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169:1516–1523. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- 25.Zhu LL, Zaidi S, Moonga BS, Troen BR, Sun L. RANK-L induces the expression of NFATc1, but not of NFkappaB subunits during osteoclast formation. Biochem Biophys Res Commun. 2005;326:131–135. doi: 10.1016/j.bbrc.2004.10.212. [DOI] [PubMed] [Google Scholar]
- 26.Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, Ostrowski MC, Olsen BR, Glimcher LH. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–3789. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, Saheki Y, Kawai T, Mukherjee PM, Reichenberger E, et al. Increased Myeloid Cell Responses to M-CSF and RANKL Cause Bone Loss and Inflammation in SH3BP2 “Cherubism” Mice. Cell. 2007;128:71–83. doi: 10.1016/j.cell.2006.10.047. [DOI] [PubMed] [Google Scholar]
- 28.GuezGuez A, Prod’homme V, Mouska X, Baudot A, Blin-Wakkach C, Rottapel R, Deckert M. 3BP2 Adapter protein is required for receptor activator of NFkappaB ligand (RANKL)-induced osteoclast differentiation of RAW264.7 cells. J Biol Chem. 285:20952–20963. doi: 10.1074/jbc.M109.091124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Germain-Lee EL, Schwindinger W, Crane JL, Zewdu R, Zweifel LS, Wand G, Huso DL, Saji M, Ringel MD, Levine MA. A mouse model of albright hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology. 2005;146:4697–4709. doi: 10.1210/en.2005-0681. [DOI] [PubMed] [Google Scholar]
- 30.Yu S, Yu D, Lee E, Eckhaus M, Lee R, Corria Z, Accili D, Westphal H, Weinstein LS. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein alpha-subunit (Gsalpha) knockout mice is due to tissue-specific imprinting of the gsalpha gene. Proc Natl Acad Sci U S A. 1998;95:8715–8720. doi: 10.1073/pnas.95.15.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]