

Abstract
The metabolism of poly(ADP-ribose) (PAR) in response to DNA strand breaks, which involves the concerted activities of poly(ADP-ribose) polymerases (PARPs) and poly(ADP-ribose) glycohydrolase (PARG), modulates cell recovery or cell death depending upon the level of DNA damage. While PARP inhibitors show high promise in clinical trials due to their low toxicity and selectivity for BRCA related cancers, evaluation of the therapeutic potential of PARG is limited by the lack of well-validated cell permeable inhibitors. In this study, Target-related Affinity Profiling (TRAP), an alternative to high-throughput screening, was used to identify a number of drug-like compounds from several chemical classes that demonstrated PARG inhibition in the low-micromolar range. A number of analogs of one of the most active chemotypes were synthesized to explore structure-activity relationship (SAR) for that series. This led to the discovery of a putative pharmacophore for PARG inhibition that contains a modified salicylanilide structure. Interestingly, these compounds also inhibit PARP-1, indicating strong homology in the active sites of PARG and PARP-1, and raising a new challenge for development of PARG specific inhibitors. The cellular activity of a lead inhibitor was demonstrated by the inhibition of both PARP and PARG activity in squamous cell carcinoma cells, although preferential inhibition of PARG relative to PARP was observed. The ability of inhibitors to modulate PAR metabolism via simultaneous effects on PARPs and PARG may represent a new approach for therapeutic development.
Keywords: poly(ADP-ribose), poly(ADP-ribose) glycohydrolase, poly(ADP-ribose) polymerase, PARG, PARP, PAR, salicylanilide, structure-activity relationship, dual inhibition, NAD, TRAP
Introduction
Activation of poly(ADP-ribose) (PAR) metabolism is an immediate response to genotoxic stress that promotes cell recovery at low levels of damage and cell death at high levels.1, 2 In response to DNA strand breaks, PAR synthesis is catalyzed from NAD+ by poly(ADP-ribose) polymerases 1 and 2 (PARP-1/2) and PAR degradation is catalyzed by poly(ADP-ribose) glycohydrolase (PARG).3–5 Activation of PARPs-1/2 results in the rapid accumulation of large, multi-branched PAR6 that covalently modifies PARP-1, histones, p53, and nuclear proteins involved in DNA repair.7 At higher levels of DNA damage, the coordinate activities of PARPs-1/2 and PARG can rapidly deplete the pool of cellular NAD(H), facilitating the release of mitochondrial proteins through signaling pathways that promote cell death.8, 9
Enzymes involved in PAR synthesis are promising therapeutic targets with development focused on PARP-1, the main synthesizing enzyme of PAR with a well defined role in cell fate determination,10–12 although PARP-2 selective inhibitors have been reported.13 PARP inhibitors are very well tolerated and have shown much promise in late stage clinical trials adjunctive with chemotherapy for triple negative breast cancers and as monotherapy for BRCA1/2 cancers.14–16 Much less is known concerning the therapeutic potential of PARG.17 The lack of high-throughput screening methods, lack of a crystal structure, and lack of bioavailable inhibitors has limited the evaluation of PARG. Complete PARG knockouts in mice are embryonic lethal,18 but deletion of the nuclear isoform of PARG results in hypersensitivity to DNA alkylating agents and radiation, demonstrating the requirement of a close coordination between PARP and PARG activities for responses to DNA damage.19, 20 RNA interference approaches have aimed to overcome some of the limitations associated with the gene manipulation, but have been limited by the inability to completely suppress PARG activity.21–23 However, PARG knockdown in combination with disruption of base excision repair has been shown to enhance the cytotoxicity of temozolomide.24 There are several reports of PARG inhibitors consisting of hydrolysable tannins,25–27 ethacridine,28 the ADP-ribose analogue ADP-(hydroxymethyl)-pyrrolidinediol (ADP-HPD),29–31 and N-bis-(3-phenyl-propyl)9-oxofluorene-2,7-diamide (GPI 16552).32 While these inhibitors have shown utility in-vitro, questions pertaining to potency, selectivity, and cell permeability remain.33–35
As an alternative to a high-throughput screening, we applied Target-related Affinity Profiling (TRAP) to identify the initial PARG inhibitors described in this report.36 TRAP technology characterizes a chemical library of small molecules by their binding affinities to a panel of proteins, which defines an “affinity fingerprint”. Activity information for a set of compounds (e.g., IC50 values) is combined with the affinity fingerprints of the compound to construct a fingerprint-based model for bioactivity. This model is used to select additional compounds for testing. Screening proceeds in an iterative manner, with batches of ~70 compounds tested followed by computational model refinement at each iteration. Such iterative cycles of compound selection, experimental assay, and model refinement have shown success in identifying bioactive molecules across a number of therapeutic targets.36
Chemistry
The simplest route to obtain salicylanilides involves amide coupling between commercially available salicylic acids and anilines (Scheme 1). The substituted amines 3a-g were created by cross coupling selected nitrobenzenes with various phenols using Cs2CO3 as the base in excess (3 equiv.) at 90°C, to obtain the substituted nitrobenzenes 2a-g. These nitrated compounds were selectively reduced to amines 3a-g in the presence of excess stannous chloride (SnCl2) in ethanol. Benzoic acids were converted to acid chlorides 5a-f by addition of excess thionyl chloride (SOCl2) under reflux. Hydroxyl methylation of 3,5-dichlorosalicylic acid was carried out prior to thionyl chloride treatment, by refluxing with potassium carbonate and iodomethane in acetone to give 4. The acid chlorides were coupled to corresponding amines in CH2Cl2 to generate 6a-m, 7, 8a-g.
Scheme 1.

Synthesis of A-ring, B-ring, and C-ring substituted analogues.a
aReagents and Conditions: (i) HO-R, Cs2CO3, 90°C, 24 hrs; (ii) SnCl2, EtOH, 70°C, 3 hrs; (iii) CH3I, acetone, K2CO3, reflux, 3 hrs; (iv) SOCl2, 100°C, 30 min; (v) CH2Cl2, RT; (vi) NH4OH, CH2Cl2, RT; (vii) H2N-R, ether, RT.
The Schiff base 9 was readily formed by the mixture of 3,5-dichloro-salicylaldehyde and substituted amine 3a in ethanol (Scheme 2). When this product was reacted with an excess of sodium cyanoborohydride (5 equiv.) in ethanol the Schiff base was selectively reduced to the amine 10. Mono-N-alkylation of 3a was achieved by refluxing the amine and potassium carbonate in acetone in the presence of iodomethane (5 equiv.). This reaction gave both the mono-N-methyl 11 and di-N-methyl products, which were separated by column chromatography. Compound 11 was coupled to 5a in CH2Cl2 to generate 12.
Scheme 2.

Synthesis of linker modified analogues.a
aReagents and Conditions: (viii) 3,5-dichloro-salicylaldehyde, EtOH; (ix) NaCNBH3, EtOH; (x) CH3I, acetone, K2CO3, reflux 3 hrs; (xi) 5a, CH2Cl2, RT.
Results and Discussion
A novel screening approach has led to the discovery of lead PARG inhibitors
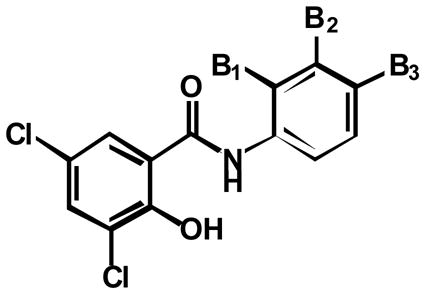
Target-related Affinity Profiling (TRAP)36 was used to search for lead inhibitors of PARG. For each round of screening, a hit was defined as a compound showing 70% inhibition of PARG at a defined concentration of test compound. In most cases, hits were subjected to dose-response evaluation to establish an approximate IC50 value. Four iterations of screening were completed and ~70 compounds were tested in each round. In total, 296 compounds were evaluated and ~30 compounds were identified with IC50 values below 100 μM. Of these, the 24 best hits were screened a second time at 10 μM and the 12 compounds with the highest degree of inhibition at this concentration were selected as the lead hits. The chemotype of the vast majority of these compounds was a substituted salicylanilide motif (Table 1). Common substitutions included the presence of a halide at two positions on the A ring of the salicylanilide (labeled A1 and A2) in 11 of the 12 hits, a chloro substitution on the B ring and an aromatic moiety attached to the B ring.
Table 1.
Active salicylanilides identified from TRAP screening
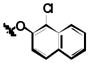
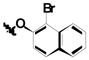
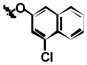
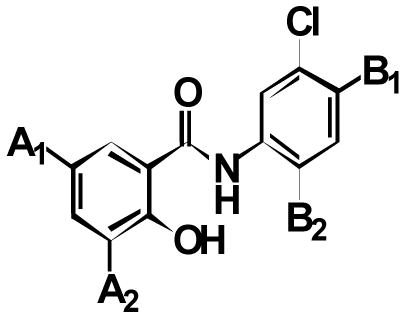 | ||||
|---|---|---|---|---|
| Cmpd | A1 | A2 | B1 | B2 |
| 1a | Cl | Cl | H |
|
| 1b | Cl | Cl | H |
|
| 1c | Cl | Cl | H |
|
| 1d | Cl | Cl | H |
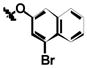
|
| 1e | Cl | Cl |
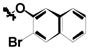
|
H |
| 1f | Cl | Br | H |
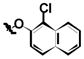
|
| 1g | Cl | Br | H |
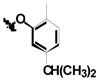
|
| 1h | Cl | I | H |
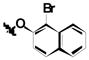
|
| 1i | Cl | I | H |
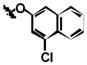
|
| 1j | Cl | H |
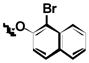
|
H |
| 1k | Br | Br | H |
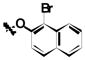
|
| 1l | Br | Br | H |
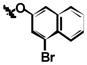
|
SAR studies have defined a new pharmacophore for PARG inhibition
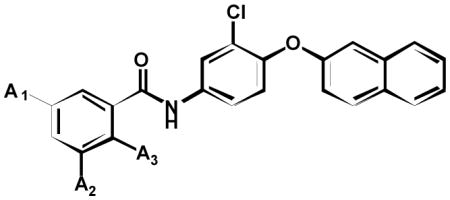
To further explore the SAR relationships for PARG inhibition, we synthesized a series of additional salicylanilide analogs. Compound 1e was taken forward as the lead hit since it contained the greatest selectivity towards PARG over PARP-1. Initially 1e was modified by removal of the C-ring bromo-substitution 6a to remove a significant amount of molecular weight without losing significant activity. To determine the importance of the A ring hydroxyl and halo substitutions, a number of analogs were synthesized and tested for PARG inhibition (Table 2). Removal of the hydroxyl group at position A3 6b resulted in loss of detectable inhibition of PARG, establishing this group as an important feature of the pharmacophore. Removal of both chloro substitutions 6c resulted in more than a 50-fold decrease in potency while removal of single chloro substitution 6d, 6e decreased potency 5–10 fold. The requirement of a chloro substitution for inhibition led us to hypothesize that an acidic hydroxyl was a component of the pharmacophore rather than a hydrogen bond donor (HBD) hydroxyl. This hypothesis was supported by the observation that methylation of the hydroxyl group 6f resulted in the loss of detectable PARG inhibition. In addition, PARG inhibition was retained when the acidic hydroxyl was replaced with a carboxylic acid 6g. In total, these experiments demonstrate an acidic substitution at the position of the A ring hydroxyl as a key component of the PARG inhibition pharmacophore.
Table 2.
SAR of A-ring substituted analogues
 | ||||
|---|---|---|---|---|
| Cmpd | A1 | A2 | A3 | PARG IC50 (μM) |
| 6a | Cl | Cl | OH | 12 ± 2 |
| 6b | Cl | Cl | H | NI |
| 6c | H | H | OH | ~500 |
| 6d | Cl | H | OH | 117 ± 60 |
| 6e | H | Cl | OH | 61 ± 12 |
| 6f | Cl | Cl | OMe | NI |
| 6g | H | H | COOH | 72 ± 7 |
NI (Non inhibitory): no detectable inhibition of PARG up to 500μM
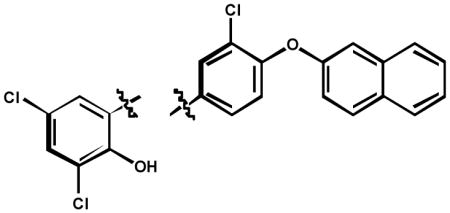
The next approach in defining the pharmacophore involved characterizing the involvement of the amide linker in PARG inhibition. Toward this end, a number of analogs were synthesized and tested for PARG inhibition properties (Table 3). The absence of the carbonyl group and amide hydrogen 9 resulted in loss of PARG inhibition. Removal of just the carbonyl group 10 also resulted in a loss of PARG inhibition. Methylation of the amide nitrogen 12 resulted in a 10-fold loss of PARG inhibition. These results argue that the pharmacophore involves the presence of a hydrogen bond acceptor (HBA), while the HBD is dispensable, but favorable for PARG inhibition.
Table 3.
SAR of linker modified analogues
 | ||
|---|---|---|
| Cmpd | PARG IC50 (μM) | |
| 5a |
|
12 ± 2 |
| 9 |
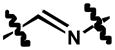
|
NI |
| 10 |
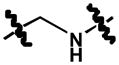
|
NI |
| 12 |
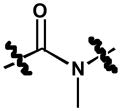
|
140 ± 20 |
NI (Non inhibitory): no detectable inhibition of PARG up to 500μM
SAR experiments also examined the involvement of the B ring and C ring in PARG inhibition. A number of analogs were prepared that either eliminated or modified the B ring and/or C ring (Table 4). Complete elimination of the C-ring 8a resulted in a complete loss of PARG inhibition. When the hydroxyl was removed from the B-ring 8c, PARG inhibition returned although at low potency. It is likely the acidic nature of the hydroxyl on the B-ring in compound 8a is very unfavorable. When the Cring was deconstructed to a 4-methylphenyl 6l and phenyl substitution 6m, potency was decreased 2-fold and 5-fold respectively. Collectively these compounds indicate a large, hydrophobic substitution is favorable at the C-ring position
Table 4.
SAR of B-ring and C-ring substituted analogues
 7 |
 6a, 6h-m 6a, 6h-m8a-g |
|||
|---|---|---|---|---|
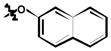
| Cmpd | B1 | B2 | B3 | PARG IC50 (μM) |
| 6a | H | Cl |
|
12 ± 2 |
| 6h | H | H |
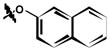
|
42 ± 9 |
| 6i | H | F |
|
27 ± 2 |
| 6j | H | CF3 |
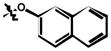
|
21 ± 3 |
| 6k | H | CH3 |
|
25 ± 7 |
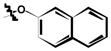
| 6l | H | Cl |
|
26 ± 2 |
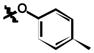
| 6m | H | Cl |
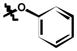
|
61 ± 10 |
| 7 | - | - | - | NI |
| 8a | H | Cl | OH | NI |
| 8b | Cl | H | H | PI |
| 8c | H | Cl | H | 261 ± 75 |
| 8d | H | H | Cl | ~500 |
| 8e | CH3 | H | H | PI |
| 8f | H | CH3 | H | PI |
| 8g | H | H | CH3 | PI |
| 8h | H | H | H | NI |
NI (Non inhibitory): inhibition of PARG does not exceed 10% at 500μM PI (Partially inhibitory): inhibition of PARG is between 10–30% at 500μM
Elimination of the C-ring and B-ring 7 resulted in loss of detectable PARG inhibition. Replacement of the B-ring in the absence of any B-ring substitution 8h was still not enough to detect PARG inhibition. Only once the B-ring chloro-substitution 8c was replaced, was PARG inhibition detected again. Displacement of the chloro-substitution to the ortho- and para-positions of the B-ring 8b, 8d resulted in a slight loss of potency. Switching out the chloro-substitution for a methyl substitution at all three positions 8e-g on the B-ring also resulted in a slight loss of potency. This indicated the B-ring substitution may serve to provide hydrophobicity rather than electron-withdrawing effects important for potency. To test this observation, substitutions were made at the B-ring in the presence of the C-ring. Replacing the chloro-substitution of the B-ring with substitutions of greater electronegativity 6i, 6j resulted in nearly a 2-fold loss in potency. When a methyl group was added instead 8k, again only a slight loss of potency was observed. However, removal of a substitution at this position 8h resulted in a 3-fold loss in potency. Taken together it is indicated that this B-ring substitution slightly contributes to PARG inhibition due to its hydrophobicity rather than electron-withdrawing effects. These experiments define the B-ring and B-ring substitution as a necessary component of the pharmacophore, but potency is greatly enhanced by the presence of the C-ring. Figure 1 represents the pharmacophore for PARG inhibition derived from the SAR studies shown in Tables 2, 3, and 4.
Figure 1.
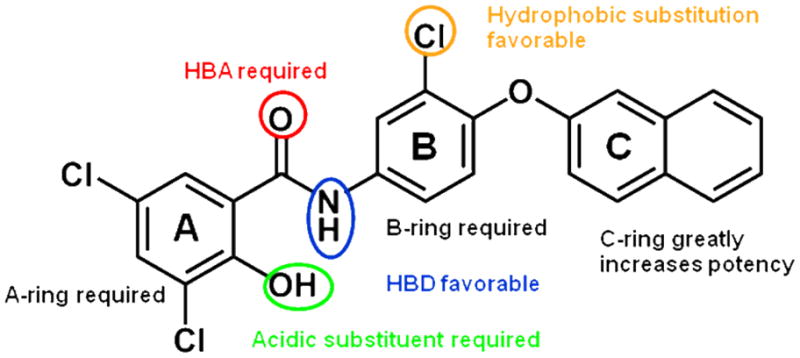
Key pharmacophoric elements related to PARG inhibition
It has been shown that salicylanilides form stable intermolecular hydrogen bonds, resulting in rigid, planar conformations.37 This is likely the case for compounds 6c and 6f. However, in all other compounds that contain electron withdrawing groups at the 3 and 5 position of the A-ring, we predict the hydroxyl is predominantly ionized at physiological pH. This ionized form may form an electron delocalized “pseudo-ring” with the amide nitrogen proton, or may simply be absent of intramolecular hydrogen bonding. While the exact conformation that effectively inhibits PARG is not currently known, it is likely that the negative charge plays a key role in inhibition, since the PAR substrate of PARG is a large, highly negatively charged polymer. It is possible the anionic hydroxyl (or carboxylic acid 6g) mimics the negatively charged characteristic of PAR.
PARG inhibitors also show inhibition of PARP-1
We next examined the selectivity of compounds containing the PARG pharmacophore. Recent secondary structure predictions suggest that the active site domain of PARG has a high degree of homology to the ADP-ribosyl transferase domain of PARPs.38 Accordingly, we examined a number of the PARG inhibitors described in Tables 1 to 4 for effects on PARP-1. Each of these compounds also showed significant inhibition of PARP-1 (Table 5), although one compound 1e had a PARG selectivity of 15-fold. Also included in Table 5 are values taken from previous studies showing that the PARG inhibitor ADP-HPD does not inhibit PARP-130 and conversely that the PARP inhibitor benzamide does not inhibit PARG,39 demonstrating that selective inhibition of these two enzymes can be achieved. It should be noted that the PARG inhibitors did not inhibit the activity of other NAD+ binding enzymes that bind via other NAD+ binding motifs, SIRT1 and alcohol dehydrogenase (data not shown). The results shown in Table 5 point out a significant future challenge for the development of PARG specific inhibitors. On the other hand, they describe the discovery of a novel class of inhibitors with the potential to disrupt the coordination of PARP and PARG activities needed for cell responses to DNA damage.
Table 5.
Cross inhibition with PARP-1
PARG inhibitors inhibit both PARP and PARG in intact cells but show a preference for PARG inhibition
The efficacy of lead compounds was assessed by examining effects on PARP and PARG activity in a squamous cell carcinoma cell line (SCC-25). Effects on PARP activity in cells were examined by determination of NAD(H) depletion following treatment with the DNA damaging agent N-methyl-N-nitro-N-nitroso-guanidine (MNNG) as shown in Figure 2. When cells were treated for 30 min with MNNG, a dose-dependent decrease in NAD(H) content was observed (Figure 2A). The depletion of NAD(H) after 5 μg/mL MNNG treatment occurred for about 30 minutes, at which point consumption began to level off (Figure 2B). The complete inhibition of NAD(H) depletion by pre-incubation with the PARP inhibitor benzamide (Figure 2C) demonstrates that the depletion represents PARP activity in intact cells. Treatment of cells with active compounds 6a and 8d resulted in partial inhibition of NAD(H) consumption while treatment with inactive compounds 6b and 7 had minimal effect (Figure 2C). For all compounds, a concentration of 1mM was utilized for proof of principle of cell permeability. These results of Figure 2 demonstrate that 6a and 8d are causing PARP inhibition in intact cells. However, it is not possible to distinguish between direct inhibition of PARP activity and indirect inhibition due to increased automodification secondary to PARG inhibition.
Figure 2.

Depletion of NAD(H) in SCC-25 cells following genotoxic stress: (A) Treatment with varying concentrations of MNNG for 30 minutes; (B) A time course of NAD(H) levels following treatment with MNNG (5μg/mL); (C) Effects on NAD(H) depletion in cells treated with MNNG (5μg/mL) for 30 minutes, pretreated with 1mM of benzamide (Bz), 6a, 8d, 6b, or 7.
To examine for effects on PARG activity in intact cells, PAR turnover experiments were completed. SCC-25 cells were first treated with MNNG to induce PAR formation. The addition of 1mM benzamide at 20 minutes inhibits further PARP activity, thus decreases in PAR content are due exclusively to PARG activity (Figure 3A). When 1mM 6a was added along with benzamide, PAR turnover was strongly inhibited following a short lag period (Figure 3A). These data further demonstrate that 6a effectively inhibits PARG in intact cells.
Figure 3.

PAR formation in SCC-25 cells following genotoxic stress: (A) Time course of PAR formation after treatment of MNNG (5μg/mL). Cells were treated with 1mM benzamide with compound 6a (□) and without compound 6a (△) at 20 minutes as indicated by the arrow; (B) Time course of PAR formation following treatment of MNNG (5μg/mL), pretreated with 1mM compound 6a (□) and without pretreatment (■).
To examine the relative inhibition of 6a on PARP and PARG in SCC-25 cells, PAR content following MNNG treatment was examined in the presence and absence of 6a. The activities of PARP and PARG are closely coordinated, thus the levels of PAR over time represent the relative activities of PARP and PARG. At the dose of MNNG used, PAR levels rapidly rise and reach a plateau at approximately 20 minutes and then slowly decline as PAR turnover continues but NAD(H) substrate is depleted (Figure 3B). When cells were pre-incubated with 6a, PAR accumulation at 10 min was increased approximately 3-fold and PAR levels continued to increase gradually over the course of the experiment (Figure 3B). These results, demonstrate a preferential inhibition of PARG over PARP by 6a. The same conclusion can be drawn from the results observed on PARP and PARG activity separately where 6a showed approximately 40% inhibition of PARP activity (Figure 2C) but nearly complete inhibition of PARG (Figure 3B) under the conditions of the experiments.
Implications for therapeutic development
The close coordination of PARP and PARG activities required for cellular responses to DNA damage and the results of clinical trials of PARP inhibitors for cancer treatment that report both low toxicity and promising efficacy14 dictate an evaluation of the therapeutic potential of PARG for cancer therapy. Additionally, the therapeutic potential of targeting PAR metabolism extends beyond cancer therapy to other pathological conditions that result from acute or chronic genotoxic stress. Upon DNA strand break recognition, PARP-1 activity is elevated nearly 500-fold.40 High levels of DNA damage lead to concerted PARP and PARG activity that in turn results in rapid cellular depletion of NAD(H) and release of mitochondrial components that promote cell death.41, 42 Protective effects seen with PARP inhibition have therapeutic implications that include chronic heart disease, diabetic cardiovascular complications, circulatory shock, neurological diseases, ischemia/reperfusion injury and Parkinson’s disease.43–45 PARP-1 activity is negatively regulated by PAR automodification and PARG is required for removing auto-modification.17 Thus, inhibition of PARG potentially represents an approach to prevent cell death by indirect inhibition of PARP activity.
Previously, a number of studies have reported positive effects in model systems of 13 (GPI 16552), a member of a group of symmetrically di-substituted aromatic PARG inhibitors developed by Guilford Pharmaceuticals, Inc.32, 46, 47 However, the cell permeability and selectivity of this class of inhibitors has not been reported. While we have demonstrated cellular activity of the lead PARG inhibitors described here, the observation that these inhibitors also inhibit PARP-1 in vitro and PARP activity in intact cells raises a new challenge for the development of PARG selective inhibitors. While the lead PARG inhibitors described here are not completely selective for PARG, the high degree of selectivity of inhibitors such as ADP-HPD for PARG30 provides evidence that future SAR studies have the potential to lead to highly selective and cell permeable PARG inhibitors. While more research is needed, the current PARG inhibitors show a more potent inhibition of PARG than PARP in intact cells (Figures 2 and 3). Indeed, while it is possible to determine effects on PARG activity in the absence of PARP activity (Figure 3A), inhibition of PARP activity in intact cells can be due to both direct inhibition of PARP and indirect inhibition of PARP as a result of increased automodification that inhibits PARP activity secondary to PARG inhibition.17
Our results, together with other work from our laboratory,38 make it likely that the active sites of PARG and PARP-1 (and likely other PARPs) share a high degree of similarity. Inhibition of PARG is not observed with PARP inhibitors such as benzamide that target the nicotinamide binding pocket of the NAD+ binding site of PARPs,39 as PARG most likely does not contain such a site. However, current highly potent PARP inhibitors are designed to reach outside of the nicotinamide binding pocket into the ADP-ribose region of the NAD+ site, as to gain favorable ADME properties and selectivity among PARP’s.11 This added bulkiness of current PARP-1 inhibitors raises the possibility of PARG cross inhibition. PARG inhibition may not be a concern with highly potent PARP inhibitors specific for a particular cellular PARP but could possibly cause off target effects related to altered regulation of the coordinate function of a non-target PARP with its specific PARG partner. Consequently, the possible effect of current PARP clinical candidates on PARG would be of interest.
The molecules reported here represent only the initial steps in the development of molecules that will allow the evaluation of the therapeutic potential of targeting PARG. The current molecules provide proof of principle that cell permeable PARG inhibitors can be developed, but further studies will be needed to provide a definitive assessment of general toxicity and therapeutic potential of PARG inhibition.
Conclusions
The identification of several small molecule inhibitors of PARG has been obtained using a novel screening technology. Analogues based on these screening hits were designed to determine SAR that led to a modified salicylanilide pharmacophore for PARG inhibition. Analysis of the A-ring reveals an acidic hydroxyl as a key feature necessary for PARG inhibition. Further analysis of the amide linker reveals that a HBD is essential for PARG inhibition and a HBA is favorable for PARG inhibition. A Bring is required for inhibition and a hydrophobic substituent on the B-ring is highly favorable. The Cring contributes greatly to potency, even though the extra size is not favorable in terms of obtaining desirable drug-like properties. The cross-inhibition observed with PARP-1 is most likely attributed to the similarities of substrates in the active sites of both enzymes. Furthermore, 5a demonstrated cell permeability for both PARP and PARG inhibition in intact cells. These studies point to future challenges for obtaining inhibitors highly selective for PARG. At the same time, inhibitors that disrupt PAR metabolism via effects on both cellular PARPs and PARG may represent a new approach for the therapeutic targeting of PAR metabolism.
Experimental section
Reagents
High specific activity (22,833 U/mg) PARP-1 was from Trevigen Inc. (Gaithersburg, MD). [32P]-NAD+ was from Perkin Elmer (Waltham, MA). All rPARG-CF was prepared as a 3.3 mg/mL stock as previously described.48
PARG assay
This assay utilizes the conditions previously reported.49 Reaction mixtures were prepared in a 1.5-mL microfuge tube by adding 10 μL of 3 × glyco assay buffer (150 mM potassium phosphate buffer, pH 7.5, 150 mM KCl, 0.3 mg/mL BSA and 30 mM 2-mercaptoethanol), 5 μL of 60 μM [α-32P]ADP-ribose polymers (approx. 5000 cpm/μL), 7 μL of deionized water, and 3 μL of test inhibitor in 50% DMSO. The reaction was initiated by the addition of 5 μL of PARG (0.3 μg/mL) and the reaction mixture was incubated at 37°C for 5 min. The reaction was then stopped by the addition of 3 μL of 1% SDS. Next, 3 μL of the reaction mixture was loaded on to a 2×10 cm poly(ethyleneimine)–cellulose F TLC plate, prespotted with 1 μL of 10 mM ADP-ribose. The spot was dried and the plate was placed into a TLC tank with 100 mL of methanol and developed until the solvent front reached the top of the plate. Next, the plate was transferred into a second tank containing 100 mL of a solution comprising 0.9 M acetic acid and 0.3 M LiCl. After developing the plate as before, it was air-dried and the ADP-ribose spot was visualized by using a hand-held short-wave UV lamp. The origin and ADP-ribose spot were excised, placed into 20-mL scintillation vials with 10 mL of EcoLume added, and the radioactivity was counted in a Beckman liquid scintillation counter.
PARP-1 assay
PARP-1 was diluted by PARP dilution buffer containing 50 mM Tris-HCl (pH 8.0), 1 mM DTT, 4 mM MgCl2, 1 mM PMSF, 2 μg/mL aprotinin, and 0.3 mg/mL BSA. The reaction was initiated by addition of PARP-1 to the PARP reaction mixture containing 100 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 2 mM DTT, 5 μg histone H1, 5 μg activated DNA, 0.1 mM NAD+, and 50,000 cpm [32P]-NAD+. The incubation time was 15 minutes. The reaction was terminated by addition of 12.5 Kl ice-cold 100% TCA, and 5 Kl of BSA (10 mg/mL) was added and the mixture was cooled at −20°C for 10 minutes, before a 15 minute centrifugation at 4°C and 14,000 rpm. The supernatant was collected and the pellet was washed with 100 Kl of 20% TCA and subjected to a second centrifugation for 15 minutes at 4°C at 14,000 rpm. The supernatant was collected as the wash and the pellet was suspended in 100 Kl 88% formic acid and collected as the pellet.
NAD assay
The NAD recycling assay as described by Jacobson and Jacobson was followed with minor adjustments.50 Briefly, 90% confluent SCC-25 cells in 35mm dishes were pretreated for 5 minutes with test compound before being treated with 5 μg/mL MNNG. After 30 minutes the media was removed and washed with 1mL PBS twice before being treated with 0.2 mL of ice-cold 0.5 M HClO4. Cells were scraped from the dish and washed with an additional 0.2 mL of HClO4. Cell extracts were then centrifuged at 14,000 rpm for 10 minutes at 4°C. Supernatants were neutralized with 135 μL 2 M KOH/0.66M KH2PO4 (to adjust pH to 7.0–7.5) and used for the measurement of NAD. The KClO4 precipitate was removed and 20 μL of the supernatant was incubated for 30 minutes at 37°C with 2 mM phenazine ethosulfate, 0.5 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide, 50 mM EDTA, 600 mM ethanol, and 120 mM Bicine, pH 7.8 and alcohol dehydrogenase (50 μg/mL, Sigma) in a 96-well plate. Absorbance was determined at 570 nm using a VERSAmax plate reader. Detected NAD content was normalized to cell protein content.
PAR assay
High throughput chemiluminescent ELISA (HT PARP in vivo pharmacodynamic assay II developed by Trevigen) was used to quantify ADP-ribose polymers in adherent cultured cells. The procedure followed was according to the supplied protocol, with minor adjustments. Briefly, 90% confluent SCC-25 cells in 35mm dishes were treated with 5 μg/mL MNNG. Cells were harvested by addition of lysis buffer containing 1% SDS. Detected PAR levels were normalized to cell count.
SAR studies
Inhibition assays were performed in duplicate with selected inhibitors in 5% DMSO. The catalytic activity of PARG and PARP-1 were measured in the presence of varying concentrations of inhibitors. Data were fit to the equation describing a sigmoidal dose response relation between % activity (100 to 0) and log[inhibitor] using GraphPad prism version 4 for Windows, GraphPad Software, San Diego, CA, www.graphpad.com. The procedure resulted in calculated values for logIC50 and standard error which are reported.
Chemistry
Starting materials were purchased from Aldrich and Alfa-Aesar and used without further purification unless otherwise specified. Flash column chromatography was performed on silica gel (200–400 mesh, 60 Å) eluting with CH2Cl2. Proton (1H NMR) nuclear magnetic resonance spectra were recorded on a Bruker Avance spectrometer at 300 MHz. Chemical shifts (δ) are in parts per million (ppm) relative to Si(CH3)4, and coupling constants (J) are in hertz. The NMR solvent used was DMSO-d6. For routine aqueous workup, the reaction mixture was extracted with CH2Cl2 or EtOAc. The organic layer was washed twice with Na2HCO3, once with brine, dried over anhydrous MgSO4, and concentrated with a Büchi rotary evaporator. All compounds tested in assays were >95% purity as determined by RP-HPLC, using 0.5% TFA (aq):MeOH (2:8) elution phase.
General procedure for the synthesis of compounds 2a-2g
To selected nitrobenzenes (4 mmol) in toluene were added substituted phenols (6 mmol, 1.5 equiv) and Cs2CO3 (12 mmol, 3 equiv) and left stirring under nitrogen. The reaction was kept at 100°C and left until completion (24–36 hours). The reaction progress was monitored by TLC (15% EtOAc/hexane), and once all nitro aryl halide was reacted, the solvent was removed in vacuo and redissolved in EtOAc. The mixture was then filtered, and routine aqueous workup was performed on the filtrate. The organic phase was concentrated and purified by column chromatography (5% EtOAc/hexane) to obtain compounds 2a-2g as red and yellow oils.
2-(2-chloro-4-nitrophenoxy)naphthalene (2a)
(57%); Rf = 0.60 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.48 (d, J = 2.7 Hz, 1H), 8.16 (dd, J = 9.1, 2.8 Hz, 1H), 8.07 (d, J = 8.9 Hz, 1H), 7.99 (d, J = 7.1 Hz, 1H), 7.90 (d, J = 7.0 Hz, 1H), 7.67 (d, J = 2.0 Hz, 1H), 7.54 (tt, J = 6.9, 5.3 Hz, 2H), 7.40 (dd, J = 8.9, 2.4 Hz, 1H), 7.12 (d, J = 9.1 Hz, 1H).
2-(4-nitrophenoxy)naphthalene (2b)
(52%); Rf = 0.67 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.27 (d, J = 9.2 Hz, 2H), 8.07 (d, J = 8.9 Hz, 1H), 8.00 (d, J = 7.0 Hz, 1H), 7.92 (d, J = 7.4 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.55 (m, 2H), 7.39 (dd, J = 8.9, 2.4 Hz, 1H), 7.21 (d, J = 9.2 Hz, 2H).
2-(2-fluoro-4-nitrophenoxy)naphthalene (2c)
(58%); Rf = 0.45 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.37 (dd, J = 10.8, 2.7 Hz, 1H), 8.10 (d, J = 2.7 Hz, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.98 (d, J = 7.3 Hz, 1H), 7.89 (d, J = 7.2 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.53 (m, 2H), 7.42 (dd, J = 8.9, 2.4 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H).
2-(4-nitro-2-(trifluoromethyl)phenoxy)naphthalene (2d)
(66%); Rf = 0.70 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.55 (d, J = 2.6 Hz, 1H), 8.45 (dd, J = 9.2, 2.7 Hz, 1H), 8.11 (d, J = 8.9 Hz, 1H), 8.02 (d, J = 6.6 Hz, 1H), 7.95 (d, J = 7.0 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H), 7.58 (m, 2H), 7.41 (dd, J = 8.9, 2.0 Hz, 1H), 7.18 (d, J = 9.2 Hz, 1H).
2-(2-methyl-4-nitrophenoxy)naphthalene (2e)
(43%); Rf = 0.54 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.27 (d, J = 2.6 Hz, 1H), 8.05 (dd, J = 8.8, 4.1 Hz, 2H), 7.97 (d, J = 7.2 Hz, 1H), 7.87 (d, J = 6.9 Hz, 1H), 7.57 (d, J = 1.9 Hz, 1H), 7.52 (m, 2H), 7.35 (dd, J = 8.9, 2.4 Hz, 1H), 6.93 (d, J = 9.0 Hz, 1H), 2.41 (s, 3H). = 0.65 (15% EtOAc/hexane); 1H NMR (300
2-chloro-4-nitro-1-(p-tolyloxy)benzene (2f)
(58%); Rf MHz, DMSO-d6) δ 8.42 (d, J = 2.7 Hz, 1H), 8.14 (dd, J = 9.2, 2.8 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 9.2 Hz, 1H), 2.33 (s, 3H). 2-chloro-4-nitro-1-phenoxybenzene (2g) (68%); Rf = 0.56 (15% EtOAc/hexane); 1H NMR (300 MHz, DMSO-d6) δ 8.46 (d, J = 2.7 Hz, 1H), 8.18 (dd, J = 9.1, 2.7 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 7.02 (d, J = 9.1 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H).
General procedure for the synthesis of compounds 3a-3g
To 2a-2g (3.0 mmol) dissolved in absolute ethanol and purged with nitrogen was added SnCl2 (15.0 mmol, 5 equiv) and left stirring at 70°C. Completion was monitored by TLC (CH2Cl2), and extra SnCl2 was added as needed. Once completed (usually 3 h), the solvent was removed in vacuo, and redissolved in EtOAc. The mixture was poured onto ice, and the pH adjusted to 9.0 with 6 M NaOH. This mixture was then filtered, and routine aqueous workup was performed. The organic phase was concentrated and purified by column chromatography (CH2Cl2) to obtain 3a-3g as yellow-orange oils.
3-chloro-4-(naphthalen-2-yloxy)aniline (3a)
(55%); Rf = 0.35 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.91 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.41 (dt, J = 14.7, 6.8 Hz, 2H), 7.24 (dd, J = 9.0, 1.8 Hz, 1H), 7.01 (d, J = 3.3 Hz, 1H), 6.99 (d, J = 5.0 Hz, 1H), 6.76 (d, J = 1.8 Hz, 1H), 6.60 (dd, J = 8.6, 1.8 Hz, 1H), 5.38 (s, 2H).
4-(naphthalen-2-yloxy)aniline (3b)
(45%); Rf = 0.40 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.87 (d, J = 9.2 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.39 (dt, J = 20.1, 6.7 Hz, 2H), 7.24 (dd, J = 8.9, 2.5 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 6.86 (d, J = 8.7 Hz, 2H), 6.66 (d, J = 8.7 Hz, 2H), 5.04 (s, 2H).
3-fluoro-4-(naphthalen-2-yloxy)aniline (3c)
(61%); Rf = 0.35 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.90 (d, J = 9.1 Hz, 1H), 7.87 (d, J = 10.4 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.41 (dt, J = 20.4, 6.8 Hz, 2H), 7.26 (dd, J = 8.9, 2.5 Hz, 1H), 7.07 (d, J = 2.2 Hz, 1H), 6.98 (d, J = 9.1 Hz, 1H), 6.53 (dd, J = 13.3, 2.5 Hz, 1H), 6.43 (dd, J = 8.7, 1.7 Hz, 1H), 5.39 (s, 2H).
4-(naphthalen-2-yloxy)-3-(trifluoromethyl)aniline (3d)
(55%); Rf = 0.44 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.90 (d, J = 9.3 Hz, 1H), 7.87 (d, J = 10.0 Hz, 1H), 7.77 (d, J = 6.7 Hz, 1H), 7.42 (m, 2H), 7.22 (dd, J = 9.1, 2.3 Hz, 1H), 7.14 (d, J = 2.0 Hz, 1H), 6.97 (d, J = 6.0 Hz, 1H), 6.95 (d, J = 2.2 Hz, 1H), 6.85 (dd, J = 8.8, 2.2 Hz, 1H), 5.50 (s, 2H).
2-(2-methyl-4-nitrophenoxy)naphthalene (3e)
(59%); Rf = 0.29 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.91 (d, J = 9.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 8.3 Hz, 1H), 7.40 (dt, J = 14.7, 6.9 Hz, 2H), 7.24 (dd, J = 8.8, 2.2 Hz, 1H), 7.00 (d, J = 1.6 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 2.0 Hz, 1H), 6.64 (dd, J = 8.2, 1.9 Hz, 1H), 6.54 (br s, 2H).
3-chloro-4-(p-tolyloxy)aniline (3f)
(47%); Rf = 0.40 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.09 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.3 Hz, 1H), 6.70 (d, J = 1.7 Hz, 1H), 6.69 (d, J = 8.1 Hz, 2H), 6.53 (dd, J = 8.7, 1.7 Hz, 1H), 5.29 (s, 2H), 2.23 (s, 3H).
3-chloro-4-phenoxyaniline (3g)
(52%); Rf = 0.27 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.30 (t, J = 7.2 Hz, 2H), 7.00 (t, J = 7.3 Hz, 1H), 6.90 (d, J = 8.7 Hz, 1H), 6.79 (d, J = 7.8 Hz, 2H), 6.72 (d, J = 2.6 Hz, 1H), 6.55 (dd, J = 8.7, 2.6 Hz, 1H), 5.32 (s, 2H).
3,5-dichloro-2-methoxybenzoic acid (4)
To 1.2 g (5.8 mmol) 3,5-dichloro salicylic acid and 3 g (21.7 mmol, 3.7 equiv) K2CO3 in 20 mL acetone was added 1 mL (17.7 mmol, 3 equiv) of iodomethane and refluxed overnight. After 24 hrs the solution was cooled to RT. 10 mL of methanol was added followed by 5 mL of 6 M NaOH and further refluxed for 3 hrs. The solution was acidified by the addition of 6 M HCl until pH 1 was reached. The precipitate was filtered and collected to obtain 4 as a pure white solid (80%). Rf = 0.72 (10% MeOH/CHCl3). 1H NMR (300 MHz, DMSO-d6) δ 13.58 (s, 1H), 7.88 (d, J = 2.6 Hz, 1H), 7.67 (d, J = 2.5 Hz, 1H), 3.82 (s, 3H).
General procedure for the synthesis of compounds 5a-5f
Corresponding benzoic acids were heated at 110°C in thionyl chloride for 1–2 h. After completion, excess thionyl chloride was removed in vacuo. The product mixture was dissolved in CH2Cl2 and filtered. Filtrates were concentrated and used without further purification.
General procedure for the synthesis of compounds 6a-6m
To a stirred solution of amine 3a-3g (0.2 mmol) in 5 mL of CH2Cl2 was added benzoic acid chlorides 5a-5f or phthalamic anhydride (0.2 mmol, 1 equiv) and the reaction mixture was stirred at RT for 0.5 h. The reaction was monitored by TLC (CH2Cl2) and once completed the mixture was concentrated and purified by column chromatography (CH2Cl2) to obtain 6a-6m as off-white solids.
3,5-dichloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxybenzamide (6a)
(65%); Rf = 0.78 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.49 (br s, 1H), 10.77 (s, 1H), 8.09 (d, J = 2.3 Hz, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 7.4 Hz, 1H), 7.81 (d, J = 2.5 Hz, 1H), 7.81 (d, J = 5.5, 1H), 7.70 (dd, J = 8.8, 2.3 Hz, 1H), 7.46 (m, 2H), 7.32 (dd, J = 9.0, 2.4 Hz, 1H), 7.28 (d, J = 9.3 Hz, 1H), 7.26 (d, J = 2.4, 1H); MS (ESI) m/z: 457.9 [M + H]+.
3,5-dichloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)benzamide (6b)
(57%); Rf = 0.78 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 10.63 (s, 1H), 8.14 (t, J = 2.1 Hz, 1H), 8.01 (d, J = 1.9 Hz, 1H), 8.00 (d, J = 1.9 Hz, 1H), 8.00 (d, J = 1.9 Hz, 1H), 7.95 (d, J = 11.0 Hz, 1H), 7.90 (d, J = 1.8 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.76 (dd, J = 8.9, 2.3 Hz, 1H), 7.46 (td, J = 14.7, 7.3 Hz, 2H), 7.32 (d, J = 9.1 Hz, 1H), 7.29 (dd, J = 8.9, 2.0 Hz, 1H), 7.23 (s, 1H); MS (ESI) m/z: 441.9 [M + H]+.
N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxybenzamide (6c)
(43%); Rf = 0.69 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.53 (s, 1H), 8.13 (s, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.92 (dd, J = 7.9, 1.5 Hz, 2H), 7.82 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 8.9 Hz, 1H), 7.46 (m, 3H), 7.33 (d, J = 8.9 Hz, 1H), 7.28 (dd, J = 8.9, 1.7 Hz, 1H), 7.22 (s, 1H), 7.01 (d, J = 9.6 Hz, 1H), 6.98 (d, J = 8.7 Hz, 1H); MS (ESI) m/z: 390.0 [M + H]+.
5-chloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxybenzamide (6d)
(67%); Rf = 0.72 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 11.73 (br s, 1H), 10.56 (s, 1H), 8.12 (d, J = 2.3 Hz, 1H), 7.95 (d, J = 9.1 Hz, 1H), 7.93 (d, J = 2.6 Hz, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.70 (dd, J = 8.8, 2.3 Hz, 1H), 7.47 (dd, J = 5.0, 3.8 Hz, 1H), 7.45 (m, 2H), 7.31 (dd, J = 8.9, 2.4 Hz, 1H), 7.26 (d, J = 8.9 Hz, 1H), 7.22 (d, J = 2.1 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H); MS (ESI) m/z: 424.0 [M + H]+.
3-chloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxybenzamide (6e)
(50%); Rf = 0.72 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.45 (s, 1H), 10.76 (s, 1H), 8.08 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 7.5 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 9.9 Hz, 2H), 7.47 (m, 2H), 7.33 (d, J = 10.4 Hz, 1H), 7.29 (d, J = 11.5 Hz, 1H), 7.26 (d, J = 5.1 Hz, 1H), 7.03 (t, J = 7.3 Hz, 1H); MS (ESI) m/z: 424.0 [M + H]+.
3,5-dichloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-methoxybenzamide (6f)
(55%); Rf = 0.67 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.12 (d, J = 2.5 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 2.6 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.67 (dd, J = 7.0, 2.5 Hz, 1H), 7.65 (d, J = 1.8 Hz, 1H), 7.45 (m, 2H), 7.32 (dd, J = 9.4, 2.0 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 7.22 (d, J = 2.4 Hz, 1H), 3.87 (s, 3H); MS (ESI) m/z: 470.1 [M - H]−.
2-(3-chloro-4-(naphthalen-2-yloxy)phenylcarbamoyl)benzoic acid (6g)
(92%); Rf = 0.22 (10% MeOH/CHCl3); 1H NMR (300 MHz, DMSO-d6) δ 13.12 (s, 1H), 10.64 (s, 1H), 8.10 (d, J = 2.4 Hz, 1H), 7.97 (d, J = 8.9 Hz, 1H), 7.91 (d, J = 7.8 Hz, 2H), 7.83 (d, J = 7.5 Hz, 1H), 7.68 (d, J = 7.3 Hz, 1H), 7.61 (m, 3H), 7.46 (m, 2H), 7.32 (dd, J = 9.6, 3.2 Hz, 1H), 7.28 (d, J = 8.9 Hz, 1H), 7.19 (d, J = 2.4 Hz, 1H); MS (ESI) m/z: 417.9 [M + H]+.
3,5-dichloro-2-hydroxy-N-(4-(naphthalen-2-yloxy)phenyl)benzamide (6h)
(47%); Rf = 0.60 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.82 (br s, 1H), 10.74 (s, 1H), 8.13 (d, J = 2.4 Hz, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.92 (d, J = 7.7 Hz, 1H), 7.84 (d, J = 5.6 Hz, 1H), 7.82 (d, J = 2.1 Hz, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.47 (m, 2H), 7.39 (d, J = 2.2 Hz, 1H), 7.32 (dd, J = 8.9, 2.3 Hz, 1H), 7.16 (d, J = 8.8 Hz, 2H); MS (ESI) m/z: 422.4 [M - H]−.
3,5-dichloro-N-(3-fluoro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxybenzamide (6i)
(57%); Rf = 0.72 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.45 (br s, 1H), 10.92 (s, 1H), 8.05 (d, J = 0.9 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 7.7 Hz, 1H), 7.83 (d, J = 1.8 Hz, 1H), 7.82 (d, J = 4.5 Hz, 1H), 7.55 (dd, J = 9.3, 1.7 Hz, 1H), 7.46 (m, 2H), 7.36 (dd, J = 2.5, 1.2 Hz, 1H), 7.32 (d, J = 9.0 Hz, 1H), 7.29 (d, J = 1.0 Hz, 1H); MS (ESI) m/z: 440.3 [M - H]−.
3,5-dichloro-2-hydroxy-N-(4-(naphthalen-2-yloxy)-3-(trifluoromethyl)phenyl)benzamide (6j)
(63%); Rf = 0.67 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.40 (br s, 1H), 10.90 (s, 1H), 8.22 (d, J = 2.2 Hz, 1H), 8.08 (d, J = 2.4 Hz, 1H), 8.01 (d, J = 9.1 Hz, 1H), 7.97 (dd, J = 2.3, 9.1 Hz, 1H), 7.95 (d, J = 7.1 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 7.83 (d, J = 2.4 Hz, 1H), 7.50 (td, J = 13.0, 6.0 Hz, 2H), 7.47 (d, J = 1.5 Hz, 1H), 7.33 (dd, J = 8.9, 2.5 Hz, 1H), 7.23 (d, J = 8.9 Hz, 1H); MS (ESI) m/z: 490.3 [M - H]−.
3,5-dichloro-2-hydroxy-N-(3-methyl-4-(naphthalen-2-yloxy)phenyl)benzamide (6k)
(64%); Rf = 0.64 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.80 (br s, 1H), 10.70 (s, 1H), 8.14 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.83 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 1.6 Hz, 1H), 7.58 (dd, J = 8.8, 2.3 Hz, 1H), 7.44 (dt, J = 15.9, 6.9 Hz, 2H), 7.29 (dd, J = 8.9, 2.4 Hz, 1H), 7.15 (d, J = 1.7 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H), 2.22 (s, 3H); MS (ESI) m/z: 436.2 [M - H]−.
3,5-dichloro-N-(3-chloro-4-(p-tolyloxy)phenyl)-2-hydroxybenzamide (6l)
(51%); Rf = 0.71 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.53 (br s, 1H), 10.69 (s, 1H), 8.07 (d, J = 2.6 Hz, 1H), 7.98 (d, J = 2.6 Hz, 1H), 7.78 (d, J = 2.5 Hz, 1H), 7.62 (dd, J = 8.9, 2.6 Hz, 1H), 7.18 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.9 Hz, 1H), 6.86 (d, J = 8.5 Hz, 2H), 2.27 (s, 3H); MS (ESI) m/z: 422.1 [M + H]+.
3,5-dichloro-N-(3-chloro-4-phenoxyphenyl)-2-hydroxybenzamide (6m)
(54%); Rf = 0.56 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.42 (br s, 1H), 10.79 (s, 1H), 8.06 (d, J = 2.9 Hz, 1H), 8.02 (d, J = 2.8 Hz, 1H), 7.83 (d, J = 2.8 Hz, 1H), 7.65 (dd, J = 8.9, 2.8 Hz, 1H), 7.39 (t, J = 7.7 Hz, 2H), 7.18 (d, J = 8.8 Hz, 1H), 7.13 (t, J = 7.0 Hz, 1H), 6.96 (d, J = 8.1 Hz, 2H); MS (ESI) m/z: 408.2 [M - H]−.
3,5-dichloro-2-hydroxybenzamide (7)
To a stirred solution of ammonium hydroxide (2 mmol, 5 equiv) in 5 mL CH2Cl2 was added salicylic acid chloride (0.4 mmol, 1 equiv), and the reaction mixture was stirred at RT for 30 min. The reaction was monitored by TLC (CH2Cl2) and once completed the mixture was concentrated in vacuo and purified by column chromatography (CH2Cl2) to obtain 7 as a white solid. (23%); Rf = 0.60 (10% MeOH/CHCl3); 1H NMR (300 MHz, DMSO-d6) δ 14.15 (s, 1H), 8.70 (s, 1H), 8.33 (s, 1H), 8.00 (d, J = 1.4 Hz, 1H), 7.77 (d, J = 1.3 Hz, 1H); MS (ESI) m/z: 204.1 [M – H]−.
General procedure for the synthesis of compounds 8a-8h
To a stirred solution of selected anilines (0.3 mmol) in 5 mL CH2Cl2 was added salicylic acid chloride (0.3 mmol, 1 equiv), and the reaction mixture was stirred at RT for 30 min. The reaction was monitored by TLC (CH2Cl2) and once completed the mixture was concentrated in vacuo and purified by column chromatography (CH2Cl2) to obtain compounds 8a-8h as off-white solids.
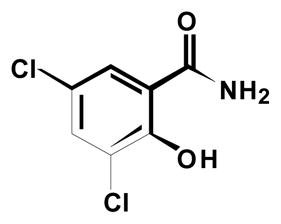
3,5-dichloro-N-(3-chloro-4-hydroxyphenyl)-2-hydroxybenzamide (8a)
(58%); Rf = 0.13 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.91 (br s, 1H), 10.59 (s, 1H), 8.15 (d, J = 2.5 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 2.4 Hz, 1H), 7.43 (dd, J = 8.8, 2.5 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 4.11 (s, 1H); MS (ESI): m/z 330.3 [M - H]−.
3,5-dichloro-N-(2-chlorophenyl)-2-hydroxybenzamide (8b)
(60%); Rf = 0.75 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.82 (br s, 1H), 10.92 (s, 1H), 8.13 (d, J = 2.5 Hz, 1H), 7.87 (d, J = 2.5 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H); MS (ESI) m/z: 314.2 [M - H]−.
3,5-dichloro-N-(3-chlorophenyl)-2-hydroxybenzamide (8c)
(62%); Rf = 0.70 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.39 (br s, 1H), 10.74 (s, 1H), 8.06 (d, J = 2.5 Hz, 1H), 7.87 (t, J = 2.0 Hz, 1H), 7.82 (d, J = 2.5 Hz, 1H), 7.63 (ddd, J = 8.2, 1.9, 0.9 Hz, 1H), 7.44 (t, J = 8.1 Hz, 1H), 7.26 (ddd, J = 8.0, 2.1, 0.9 Hz, 1H); MS (ESI) m/z: 314.2 [M - H]−.
3,5-dichloro-N-(4-chlorophenyl)-2-hydroxybenzamide (8d)
(73%); Rf = 0.70 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.53 (s, 1H), 10.72 (s, 1H), 8.11 (d, J = 2.5 Hz, 1H), 7.82 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 8.9 Hz, 2H), 7.47 (d, J = 8.9 Hz, 2H); MS (ESI) m/z: 314.2 [M - H]−. 1H NMR (300 MHz,
3,5-dichloro-2-hydroxy-N-o-tolylbenzamide (8e)
(57%); Rf = 0.69 (CH2Cl2); DMSO-d6) δ 13.10 (br s, 1H), 10.57 (s, 1H), 8.17 (d, J = 1.6 Hz, 1H), 7.85 (d, J = 1.0 Hz, 1H), 7.38 (d, J = 6.6 Hz, 1H), 7.32 (d, J = 6.7 Hz, 1H), 7.25 (m, 2H), 2.24 (s, 3H); MS (ESI) m/z: 294.3 [M - H]−. 1H NMR (300 MHz,
3,5-dichloro-2-hydroxy-N-m-tolylbenzamide (8f)
(71%); Rf = 0.74 (CH2Cl2); DMSO-d6) δ 12.81 (br s, 1H), 10.59 (s, 1H), 8.17 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 2.2 Hz, 1H), 7.53 (s, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.02 (d, J = 7.1 Hz, 1H), 2.33 (s, 3H); MS (ESI) m/z: 294.2 [M - H]−. 1H NMR (300 MHz,
3,5-dichloro-2-hydroxy-N-p-tolylbenzamide (8g)
(66%); Rf = 0.69 (CH2Cl2); DMSO-d6) δ 12.88 (br s, 1H), 10.56 (s, 1H), 8.18 (d, J = 2.2 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.38 (d, J = 6.6 Hz, 1H), 7.31 (d, J = 2.9 Hz, 1H), 7.25 (dd, J = 6.4, 3.4 Hz, 2H), 2.24 (s, 3H); MS (ESI) m/z: 294.3 [M - 1]−.
3,5-dichloro-2-hydroxy-N-benzamide (8h)
(66%); Rf = 0.69 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 12.78 (br s, 1H), 10.70 (s, 1H), 8.13 (d, J = 2.2 Hz, 1H), 7.82 (d, J = 2.3 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.41 (t, J = 7.8 Hz, 2H), 7.20 (t, J = 7.5 Hz, 1H); MS (ESI) m/z: 280.2 [M - 1]−.
2,4-dichloro-6-((3-chloro-4-(naphthalen-2-yloxy)phenylimino)methyl)phenol (9)
To a stirred solution of 100 mg (0.37 mmol) 3a in 10 mL ethanol was added 140 mg (0.74 mmol, 2 equiv) 3,5-dichloro-hydroxy benzaldehyde dissolved in 2 mL methanol and the reaction mixture was left stirring until a bright orange precipitate formed. The reaction was monitored by TLC (CH2Cl2) and once completed the mixture was chilled to 4°C. The precipitate was filtered and washed twice with 10 mL ethanol to obtain 9 as an orange solid. (90%); Rf = 0.83 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 13.98 (s, 1H), 9.09 (s, 1H), 8.01 (d, J = 9.7 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.90 (d, J = 2.5 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 2.6 Hz, 1H), 7.74 (d, J = 2.6 Hz, 1H), 7.55 (dd, J = 8.7, 2.5 Hz, 1H), 7.48 (m, 2H), 7.36 (dd, J = 6.6, 2.8 Hz, 1H), 7.35 (d, J = 3.1 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H); MS (ESI) m/z: 440.3 [M - H]−.
2,4-dichloro-6-((3-chloro-4-(naphthalen-2-yloxy)phenylamino)methyl)phenol (10)
To a stirred solution of 50 mg (0.113 mmol) 9 in 5 mL methanol was added 113 μL of 5 M sodium cyanoborohydride (0.565 mmol, 5 equiv) and left stirring for 24 h at 70°C. The reaction was monitored by TLC (CH2Cl2) and once completed routine aqueous workup was performed. The organic phase was concentrated and purified by column chromatography (CH2Cl2) to obtain 10 as an orange solid. (40%);Rf = 0.76 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 9.78 (s, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 2.5 Hz, 1H), 7.41 (m, 2H), 7.25 (d, J = 2.4 Hz, 1H), 7.23 (dd, J = 6.3, 2.6 Hz, 1H), 7.07 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 2.7 Hz, 1H), 6.62 (dd, J = 8.9, 2.7 Hz, 1H), 6.49 (t, J = 5.8 Hz, 1H), 4.29 (d, J = 5.6 Hz, 2H); MS (ESI) m/z: 442.1 [M - H]−.
3-chloro-N-methyl-4-(naphthalen-2-yloxy)aniline (11)
To 100 mg (0.37 mmol) 3a and 510 mg (3.7 mmol, 10 equiv) K2CO3 in 10 mL acetone was added 23 μL (0.37 mmol) iodomethane and refluxed for 1 h. Every hour 1 equivalent of iodomethane was added and monitored by TLC until the starting material was consumed. The solvent was removed in vacuo, and routine aqueous workup was performed. The mixture was concentrated and purified by column chromatography (CH2Cl2) to obtain 11 as a white solid. (29%); Rf = 0.58 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 7.91 (d, J = 9.1 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.41 (m, 2H), 7.25 (dd, J = 8.9, 2.5 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 2.7 Hz, 1H), 6.59 (dd, J = 8.8, 2.7 Hz, 1H), 5.99 (d, J = 5.0 Hz, 1H), 2.70 (d, J = 5.0 Hz, 3H).
3,5-dichloro-N-(3-chloro-4-(naphthalen-2-yloxy)phenyl)-2-hydroxy-N-methylbenzamide (12)
To a stirred solution of 30 mg (0.106 mmol) 11 in 5 mL CH2Cl2 was added 29 mg (0.127 mmol, 1.2 equiv) 5a and the mixture was left stirring at RT for 0.5 h. The reaction was monitored by TLC (CH2Cl2) and once completed routine aqueous workup was performed, and the organic phase was concentrated and purified by column chromatography (CH2Cl2) to obtain 9 as a white solid. (48%); Rf = 0.42 (CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.51 (d, J = 2.4 Hz, 1H), 7.48 (dt, J = 19.9, 6.7 Hz, 2H), 7.24 (d, J = 3.3 Hz, 1H), 7.22 (dd, J = 11.1, 2.5 Hz, 1H), 7.20 (d, J = 1.7 Hz, 1H), 7.13 (dd, J = 8.6, 2.4 Hz, 1H), 7.08 (d, J = 1.3 Hz, 1H), 3.35 (s, 3H); MS (ESI): m/z 470.1 [M - H]−.
Acknowledgments
This research was supported in part by NIH grants CA43894, CA27502, and CA090085 (MKJ), a grant from Telik Inc., and a pre-doctoral fellowship from the ACS Division of Medicinal Chemistry sponsored by Amgen (to JDS). The authors thank Trevigen Inc. for providing PARP-1 and PAR ELISA kits. MKJ serves as a consultant to Trevigen.
Abbreviations
- ADP
adenosine diphosphate
- PAR
poly(ADP-ribose)
- PARP
poly(ADP-ribose) polymerase
- BRCA1/2
breast cancer susceptibility protein 1/2
- PARG
poly(ADP-ribose) glycohydrolase
- NAD+
the oxidized form of nicotinamide adenine dinucleotide
- NAD(H)
the total cellular pool of nicotinamide adenine dinucleotide, both oxidized and reduced forms
- TRAP
Target-related Affinity Profiling
- IC50
half maximal inhibitory concentration
- HBD
hydrogen bond donor
- HBA
hydrogen bond acceptor
- SAR
structure-activity relationship
- ADP-HPD
adenosine 5′-diphosphate (hydroxymethyl) pyrrolidinediol
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
References
- 1.Diefenbach J, Burkle A. Introduction to poly(ADP-ribose) metabolism. Cell Mol Life Sci. 2005;62(7–8):721–730. doi: 10.1007/s00018-004-4503-3. [DOI] [PubMed] [Google Scholar]
- 2.Juarez-Salinas H, Sims JL, Jacobson MK. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature. 1979;282(5740):740–741. doi: 10.1038/282740a0. [DOI] [PubMed] [Google Scholar]
- 3.Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268(1):7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 5.Braun SA, Panzeter PL, Collinge MA, Althaus FR. Endoglycosidic cleavage of branched polymers by poly(ADP-ribose) glycohydrolase. Eur J Biochem. 1994;220(2):369–375. doi: 10.1111/j.1432-1033.1994.tb18633.x. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez-Gonzalez R, Jacobson MK. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry. 1987;26(11):3218–3224. doi: 10.1021/bi00385a042. [DOI] [PubMed] [Google Scholar]
- 7.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342 ( Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson MK, Levi V, Juarez-Salinas H, Barton RA, Jacobson EL. Effect of carcinogenic N-alkyl-N-nitroso compounds on nicotinamide adenine dinucleotide metabolism. Cancer Res. 1980;40(6):1797–1802. [PubMed] [Google Scholar]
- 9.Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101(1):4–15. [PubMed] [Google Scholar]
- 10.Curtin NJ. PARP inhibitors for cancer therapy. Expert Rev Mol Med. 2005;7(4):1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 11.Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem. 2010;53(12):4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 12.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunderland PT, Dhami A, Mahon MF, Jones LA, Tully SR, Lloyd MD, Thompson AS, Javaid H, Martin NM, Threadgill MD. Synthesis of 4-alkyl-, 4-aryl- and 4-arylamino-5-aminoisoquinolin-1-ones and identification of a new PARP-2 selective inhibitor. Org Biomol Chem. 2011;9(3):881–891. doi: 10.1039/c0ob00665c. [DOI] [PubMed] [Google Scholar]
- 14.Plummer ER. Inhibition of poly(ADP-ribose) polymerase in cancer. Curr Opin Pharmacol. 2006;6(4):364–368. doi: 10.1016/j.coph.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 16.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 17.Min W, Wang ZQ. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci. 2009;14:1619–1626. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 18.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004;101(51):17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, Wang ZQ. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol. 2004;24(16):7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao H, Coyle DL, Meyer-Ficca ML, Meyer RG, Jacobson EL, Wang ZQ, Jacobson MK. Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp Cell Res. 2007;313(5):984–996. doi: 10.1016/j.yexcr.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 21.Blenn C, Althaus FR, Malanga M. Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J. 2006;396(3):419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohausz O, Blenn C, Malanga M, Althaus FR. The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol Life Sci. 2008;65(4):644–655. doi: 10.1007/s00018-008-7516-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burns DM, Ying W, Kauppinen TM, Zhu K, Swanson RA. Selective down-regulation of nuclear poly(ADP-ribose) glycohydrolase. PLoS One. 2009;4(3):e4896. doi: 10.1371/journal.pone.0004896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, Hamilton RL, Banze LA, Brown AR, Sobol RW. N-methylpurine DNA glycosylase and DNA polymerase {beta} modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011;13(5):471–486. doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanuma S, Sakagami H, Endo H. Inhibitory effect of tannin on poly(ADP-ribose) glycohydrolase from human placenta. Biochem Int. 1989;18(4):701–708. [PubMed] [Google Scholar]
- 26.Bakondi E, Bai P, Erdelyi K, Szabo C, Gergely P, Virag L. Cytoprotective effect of gallotannin in oxidatively stressed HaCaT keratinocytes: the role of poly(ADP-ribose) metabolism. Exp Dermatol. 2004;13(3):170–178. doi: 10.1111/j.0906-6705.2004.0150.x. [DOI] [PubMed] [Google Scholar]
- 27.Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci U S A. 2001;98(21):12227–12232. doi: 10.1073/pnas.211202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tavassoli M, Tavassoli MH, Shall S. Effect of DNA intercalators on poly(ADP-ribose) glycohydrolase activity. Biochim Biophys Acta. 1985;827(3):228–234. doi: 10.1016/0167-4838(85)90207-9. [DOI] [PubMed] [Google Scholar]
- 29.Ramsinghani S, Koh DW, Ame JC, Strohm M, Jacobson MK, Slama JT. Syntheses of photoactive analogues of adenosine diphosphate (hydroxymethyl)pyrrolidinediol and photoaffinity labeling of poly(ADP-ribose) glycohydrolase. Biochemistry. 1998;37(21):7801–7812. doi: 10.1021/bi9730386. [DOI] [PubMed] [Google Scholar]
- 30.Slama JT, Aboul-Ela N, Goli DM, Cheesman BV, Simmons AM, Jacobson MK. Specific inhibition of poly(ADP-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J Med Chem. 1995;38(2):389–393. doi: 10.1021/jm00002a021. [DOI] [PubMed] [Google Scholar]
- 31.Slama JT, Aboul-Ela N, Jacobson MK. Mechanism of inhibition of poly(ADP-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J Med Chem. 1995;38(21):4332–4336. doi: 10.1021/jm00021a023. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Ferraris D, Kletzly P, Li W, Wang E, Xing A, Xu W, Zhang J. Symmetrically disubstituted aromatic compounds and pharmaceutical compositions for inhibiting poly(ADP-ribose) glycohydrolase, and methods for their use. WO. 2002:02057211.
- 33.Falsig J, Christiansen SH, Feuerhahn S, Burkle A, Oei SL, Keil C, Leist M. Poly(ADP-ribose) glycohydrolase as a target for neuroprotective intervention: assessment of currently available pharmacological tools. Eur J Pharmacol. 2004;497(1):7–16. doi: 10.1016/j.ejphar.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 34.Labieniec M, Gabryelak T, Falcioni G. Antioxidant and pro-oxidant effects of tannins in digestive cells of the freshwater mussel Unio tumidus. Mutat Res. 2003;539(1–2):19–28. doi: 10.1016/s1383-5718(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 35.Blenn C, Wyrsch P, Althaus FR. The Ups and Downs of Tannins as Inhibitors of Poly(ADP-Ribose)glycohydrolase. Molecules. 2011;16(2):1854–1877. doi: 10.3390/molecules16021854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beroza P, Damodaran K, Lum RT. Target-related affinity profiling: Telik’s lead discovery technology. Curr Top Med Chem. 2005;5(4):371–381. doi: 10.2174/1568026053828394. [DOI] [PubMed] [Google Scholar]
- 37.Suezawa H, Hirota M, Yuzuri T, Hamada Y, Takeuchi I, Sugiura M. Studies on the Conformations of Antimicrobial Salicylanilide Derivatives by Spectroscopy. Bull Chem Soc Jpn. 2000;73:2335–2339. [Google Scholar]
- 38.Botta D, Jacobson MK. Identification of a regulatory segment of poly(ADP-ribose) glycohydrolase. Biochemistry. 2010;49(35):7674–7682. doi: 10.1021/bi100973m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem. 1989;264(8):4312–4317. [PubMed] [Google Scholar]
- 40.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 41.Berger NA, Berger SJ. Metabolic consequences of DNA damage: the role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 1986;38:357–363. doi: 10.1007/978-1-4615-9462-8_39. [DOI] [PubMed] [Google Scholar]
- 42.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103(48):18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 44.Szabo C. Pharmacological inhibition of poly(ADP-ribose) polymerase in cardiovascular disorders: future directions. Curr Vasc Pharmacol. 2005;3(3):301–303. doi: 10.2174/1570161054368553. [DOI] [PubMed] [Google Scholar]
- 45.Sodhi RK, Singh N, Jaggi AS. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol. 2010;53(3–4):77–87. doi: 10.1016/j.vph.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Genovese T, Di Paola R, Catalano P, Li JH, Xu W, Massuda E, Caputi AP, Zhang J, Cuzzocrea S. Treatment with a novel poly(ADP-ribose) glycohydrolase inhibitor reduces development of septic shock-like syndrome induced by zymosan in mice. Crit Care Med. 2004;32(6):1365–1374. doi: 10.1097/01.ccm.0000127775.70867.0c. [DOI] [PubMed] [Google Scholar]
- 47.Lu XC, Massuda E, Lin Q, Li W, Li JH, Zhang J. Post-treatment with a novel PARG inhibitor reduces infarct in cerebral ischemia in the rat. Brain Res. 2003;978(1–2):99–103. doi: 10.1016/s0006-8993(03)02774-4. [DOI] [PubMed] [Google Scholar]
- 48.Lin W, Ame JC, Aboul-Ela N, Jacobson EL, Jacobson MK. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J Biol Chem. 1997;272(18):11895–11901. doi: 10.1074/jbc.272.18.11895. [DOI] [PubMed] [Google Scholar]
- 49.Menard L, Poirier GG. Rapid assay of poly(ADP-ribose) glycohydrolase. Biochem Cell Biol. 1987;65(7):668–673. doi: 10.1139/o87-088. [DOI] [PubMed] [Google Scholar]
- 50.Jacobson EL, Jacobson MK. Pyridine nucleotide levels as a function of growth in normal and transformed 3T3 cells. Arch Biochem Biophys. 1976;175(2):627–634. doi: 10.1016/0003-9861(76)90553-1. [DOI] [PubMed] [Google Scholar]